
Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer


Abstract
:1. Introduction
1.1. Overview of the PI3K and MAPK Signaling Pathways and Their Inhibitors
1.2. Compensatory MAPK Signaling in PI3K/AKT/mTOR Inhibition Resistance and Dual Inhibition Therapies
1.3. The rationale for Combined Immune Checkpoint Blockade and Targeted Therapy
1.4. Treatment Sequence of Combined Immunotherapy and Targeted BRAF/MEK Inhibition
2. Quadruplet Combination of PI3K, RAF/MEK Inhibition, and Immunotherapy
2.1. Novel Pharmacological Inhibitors of PI3K and MAPK Pathways
2.2. Biomarkers and Genetic Characteristics for Patient Selection and Stratification
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Flaherty, K.T.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Update on tolerability and overall survival in COLUMBUS: Landmark analysis of a randomised phase 3 trial of encorafenib plus binimetinib vs. vemurafenib or encorafenib in patients with BRAFV600-mutant melanoma. Eur. J. Cancer 2020, 126, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K inhibitors in cancer: Clinical implications and adverse effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef] [PubMed]
- Bergholz, J.S.; Zhao, J.J. How compensatory mechanisms and adaptive rewiring have shaped our understanding of therapeutic resistance in cancer. Cancer Res. 2021, 81, 6074–6077. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Cornu, M.; Albert, V.; Hall, M.N. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 2013, 23, 53–62. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Saez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; Gonzalez-Farre, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Braso-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef]
- Vasan, N.; Cantley, L.C. At a crossroads: How to translate the roles of PI3K in oncogenic and metabolic signalling into improvements in cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 471–485. [Google Scholar] [CrossRef]
- Solit, D.B.; Rosen, N. Resistance to BRAF inhibition in melanomas. N. Engl. J. Med. 2011, 364, 772–774. [Google Scholar] [CrossRef]
- Chien, A.J.; Tripathy, D.; Albain, K.S.; Symmans, W.F.; Rugo, H.S.; Melisko, M.E.; Wallace, A.M.; Schwab, R.; Helsten, T.; Forero-Torres, A.; et al. MK-2206 and standard neoadjuvant chemotherapy improves response in patients with human epidermal growth factor receptor 2-positive and/or hormone receptor-negative breast cancers in the I-SPY 2 trial. J. Clin. Oncol. 2020, 38, 1059–1069. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Chambard, J.C.; Lefloch, R.; Pouyssegur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S. Paradoxical oncogenesis—The long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Chandarlapaty, S.; Scaltriti, M.; Angelini, P.; Ye, Q.; Guzman, M.; Hudis, C.A.; Norton, L.; Solit, D.B.; Arribas, J.; Baselga, J.; et al. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene 2010, 29, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Leroy, C.; Ramos, P.; Cornille, K.; Bonenfant, D.; Fritsch, C.; Voshol, H.; Bentires-Alj, M. Activation of IGF1R/p110beta/AKT/mTOR confers resistance to alpha-specific PI3K inhibition. Breast Cancer Res. 2016, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yan, H.; Frost, P.; Gera, J.; Lichtenstein, A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer Ther. 2005, 4, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Clement, E.; Inuzuka, H.; Nihira, N.T.; Wei, W.; Toker, A. Skp2-dependent reactivation of AKT drives resistance to PI3K inhibitors. Sci. Signal. 2018, 11, eaao3810. [Google Scholar] [CrossRef] [Green Version]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Mundt, F.; Rajput, S.; Li, S.; Ruggles, K.V.; Mooradian, A.D.; Mertins, P.; Gillette, M.A.; Krug, K.; Guo, Z.; Hoog, J.; et al. Mass spectrometry-based proteomics reveals potential roles of NEK9 and MAP2K4 in resistance to PI3K inhibition in triple-negative breast cancers. Cancer Res. 2018, 78, 2732–2746. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Atefi, M.; von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Merchant, M.; Orr, C.; Chan, J.; Den Otter, D.; Berry, L.; Kasman, I.; Koeppen, H.; Rice, K.; Yang, N.Y.; et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012, 72, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Hernandez, I.; Baquero, P.; Calleros, L.; Chiloeches, A. Dual inhibition of V600EBRAF and the PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma cells through a MEK-independent mechanism. Cancer Lett. 2012, 314, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Algazi, A.P.; Rotow, J.; Posch, C.; Ortiz-Urda, S.; Pelayo, A.; Munster, P.N.; Daud, A. A dual pathway inhibition strategy using BKM120 combined with vemurafenib is poorly tolerated in BRAF V600E/K mutant advanced melanoma. Pigment Cell Melanoma Res. 2019, 32, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Kurzrock, R.; Valero, V.; Gonzalez, R.; Heist, R.S.; Tan, A.R.; Means-Powell, J.; Werner, T.L.; Becerra, C.; Wang, C.; et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother. Pharmacol. 2020, 85, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Sen, S.; Hess, K.R.; Janku, F.; Hong, D.S.; Khatua, S.; Karp, D.D.; Munoz, J.; Falchook, G.S.; Groisberg, R.; et al. Phase I study of the BRAF inhibitor vemurafenib in combination with the mammalian target of rapamycin inhibitor everolimus in patients with BRAF-mutated malignancies. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A. The role of PI3K inhibition in the treatment of breast cancer, alone or combined with immune checkpoint inhibitors. Front. Mol. Biosci. 2021, 8, 648663. [Google Scholar] [CrossRef]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef]
- Schmid, P.; Im, S.-A.; Armstrong, A.; Park, Y.H.; Chung, W.-P.; Nowecki, Z.; Lord, S.; Wysocki, P.J.; Lu, Y.-S.; Dry, H.; et al. BEGONIA: Phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)—Initial results from arm 1, d+paclitaxel (P), and arm 6, d+trastuzumab deruxtecan (T-DXd). J. Clin. Oncol. 2021, 39 (Suppl. 15), 1023. [Google Scholar] [CrossRef]
- Hatem, S.; Hargis, J.; Elias, A.; Lee, A.; Swart, R.; Dahkil, S.; Drakaki, A.; Phan, V.; Kass, F.; Cobleigh, M.; et al. Abstract P5-16-02: Updated efficacy, safety and translational data from MARIO-3, a phase II open-label study evaluating a novel triplet combination of eganelisib (IPI-549), atezolizumab (atezo), and nab-paclitaxel (nab-pac) as first-line (1L) therapy for locally advanced or metastatic triple-negative breast cancer (TNBC). Cancer Res. 2022, 82 (Suppl. 4), P5-16-02. [Google Scholar]
- Ho, P.C.; Meeth, K.M.; Tsui, Y.C.; Srivastava, B.; Bosenberg, M.W.; Kaech, S.M. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNγ. Cancer Res. 2014, 74, 3205–3217. [Google Scholar] [CrossRef] [Green Version]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Knight, D.A.; Ngiow, S.F.; Li, M.; Parmenter, T.; Mok, S.; Cass, A.; Haynes, N.M.; Kinross, K.; Yagita, H.; Koya, R.C.; et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J. Clin. Investig. 2016, 126, 402–403. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Henry, T.; Baranda, S.J.; Frleta, D.; Manches, O.; Bogunovic, D.; Bhardwaj, N. Inhibition of both BRAF and MEK in BRAFV600E mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol. Immunother. 2013, 62, 811–822. [Google Scholar] [CrossRef]
- Homet Moreno, B.; Mok, S.; Comin-Anduix, B.; Hu-Lieskovan, S.; Ribas, A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology 2016, 5, e1052212. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef] [Green Version]
- Minor, D.R.; Puzanov, I.; Callahan, M.K.; Hug, B.A.; Hoos, A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015, 28, 611–612. [Google Scholar] [CrossRef]
- Ribas, A.; Hodi, F.S.; Callahan, M.; Konto, C.; Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 2013, 368, 1365–1366. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Di Giacomo, A.M.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Stephens, R.; Svane, I.M.; et al. KEYNOTE-022 part 3: A randomized, double-blind, phase 2 study of pembrolizumab, dabrafenib, and trametinib in BRAF-mutant melanoma. J. Immunother. Cancer 2020, 8, e001806. [Google Scholar] [CrossRef]
- Dummer, R.; Long, G.V.; Robert, C.; Tawbi, H.A.; Flaherty, K.T.; Ascierto, P.A.; Nathan, P.D.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; et al. Randomized phase III trial evaluating spartalizumab plus dabrafenib and trametinib for BRAFV600-mutant unresectable or metastatic melanoma. J. Clin. Oncol. 2022, 40, 1428–1438. [Google Scholar] [CrossRef]
- Kakavand, H.; Wilmott, J.S.; Menzies, A.M.; Vilain, R.; Haydu, L.E.; Yearley, J.H.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Long, G.V.; et al. PD-L1 expression and tumor-infiltrating lymphocytes define different subsets of MAPK inhibitor-treated melanoma patients. Clin. Cancer Res. 2015, 21, 3140–3148. [Google Scholar] [CrossRef] [Green Version]
- Phadke, M.S.; Chen, Z.; Li, J.; Mohamed, E.; Davies, M.A.; Smalley, I.; Duckett, D.R.; Palve, V.; Czerniecki, B.J.; Forsyth, P.A.; et al. Targeted therapy given after anti-PD-1 leads to prolonged responses in mouse melanoma models through sustained antitumor immunity. Cancer Immunol. Res. 2021, 9, 554–567. [Google Scholar] [CrossRef]
- Haas, L.; Elewaut, A.; Gerard, C.L.; Umkehrer, C.; Leiendecker, L.; Pedersen, M.; Krecioch, I.; Hoffmann, D.; Novatchkova, M.; Kuttke, M.; et al. Acquired resistance to anti-MAPK targeted therapy confers an immune-evasive tumor microenvironment and cross-resistance to immunotherapy in melanoma. Nat. Cancer 2021, 2, 693–708. [Google Scholar] [CrossRef]
- Yan, C.; Saleh, N.; Yang, J.; Nebhan, C.A.; Vilgelm, A.E.; Reddy, E.P.; Roland, J.T.; Johnson, D.B.; Chen, S.C.; Shattuck-Brandt, R.L.; et al. Novel induction of CD40 expression by tumor cells with RAS/RAF/PI3K pathway inhibition augments response to checkpoint blockade. Mol. Cancer 2021, 20, 85. [Google Scholar] [CrossRef]
- Yan, C.; Richmond, A. Hiding in the dark: Pan-cancer characterization of expression and clinical relevance of CD40 to immune checkpoint blockade therapy. Mol. Cancer 2021, 20, 146. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Ribas, A.; Tarhini, A.A.; Truong, T.-G.; Davar, D.; O’Rourke, M.A.; Curti, B.D.; Brell, J.M.; et al. DREAMseq (doublet, randomized evaluation in advanced melanoma sequencing): A phase III trial—ECOG-ACRIN EA6134. J. Clin. Oncol. 2021, 39 (Suppl. 36). [Google Scholar] [CrossRef]
- Dimitriou, F.; Matter, A.V.; Mangana, J.; Urosevic-Maiwald, M.; Micaletto, S.; Braun, R.P.; French, L.E.; Dummer, R. Cytokine release syndrome during sequential treatment with immune checkpoint inhibitors and kinase inhibitors for metastatic melanoma. J. Immunother. 2019, 42, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Deken, M.A.; Gadiot, J.; Jordanova, E.S.; Lacroix, R.; van Gool, M.; Kroon, P.; Pineda, C.; Geukes Foppen, M.H.; Scolyer, R.; Song, J.Y.; et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology 2016, 5, e1238557. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, C.; Vilgelm, A.E.; Chen, S.C.; Ayers, G.D.; Johnson, C.A.; Richmond, A. Targeted deletion of CXCR2 in myeloid cells alters the tumor immune environment to improve antitumor immunity. Cancer Immunol. Res. 2021, 9, 200–213. [Google Scholar] [CrossRef]
- Song, K.W.; Edgar, K.A.; Hanan, E.J.; Hafner, M.; Oeh, J.; Merchant, M.; Sampath, D.; Nannini, M.A.; Hong, R.; Phu, L.; et al. RTK-dependent inducible degradation of mutant PI3Kα drives GDC-0077 (inavolisib) efficacy. Cancer Discov. 2022, 12, 204–219. [Google Scholar] [CrossRef]
- Xu, J.; Yu, X.; Martin, T.C.; Bansal, A.; Cheung, K.; Lubin, A.; Stratikopoulos, E.; Cahuzac, K.M.; Wang, L.; Xie, L.; et al. AKT degradation selectively inhibits the growth of PI3K/PTEN pathway-mutant cancers with wild-type KRAS and BRAF by destabilizing aurora kinase B. Cancer Discov. 2021, 11, 3064–3089. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, L.; Xu, Q.; Wang, K.; Xie, D.; Yu, Z.; Jiang, K.; Liao, L.; Yates, J.R.; Lee, J.D.; et al. Targeting BMK1 impairs the drug resistance to combined inhibition of BRAF and MEK1/2 in melanoma. Sci. Rep. 2017, 7, 46244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubita, A.; Lombardi, Z.; Tusa, I.; Lazzeretti, A.; Sgrignani, G.; Papini, D.; Menconi, A.; Gagliardi, S.; Lulli, M.; Dello Sbarba, P.; et al. Inhibition of ERK5 elicits cellular senescence in melanoma via the cyclin-dependent kinase inhibitor p21. Cancer Res. 2022, 82, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Ruiz, M.J.; Alvarez-Fernandez, S.; Parrott, T.; Zaknoen, S.; Burrows, F.J.; Ocana, A.; Pandiella, A.; Esparis-Ogando, A. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget 2014, 5, 11308–11318. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Lewis, K.D.; Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Biomarkers of treatment benefit with atezolizumab plus vemurafenib plus cobimetinib in BRAFV600 mutation-positive melanoma. Ann. Oncol. 2022, 33, 544–555. [Google Scholar] [CrossRef]
- Dummer, R.; Brase, J.C.; Garrett, J.; Campbell, C.D.; Gasal, E.; Squires, M.; Gusenleitner, D.; Santinami, M.; Atkinson, V.; Mandala, M.; et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAFV600-mutant, stage III melanoma (COMBI-AD): Exploratory biomarker analyses from a randomised, phase 3 trial. Lancet Oncol. 2020, 21, 358–372. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- Shi, Z.; Wulfkuhle, J.; Nowicka, M.; Gallagher, R.I.; Saura, C.; Nuciforo, P.G.; Calvo, I.; Andersen, J.; Passos-Coelho, J.L.; Gil-Gil, M.J.; et al. Functional mapping of AKT signaling and biomarkers of response from the FAIRLANE trial of neoadjuvant ipatasertib plus paclitaxel for triple-negative breast cancer. Clin. Cancer Res. 2022, 28, 993–1003. [Google Scholar] [CrossRef]
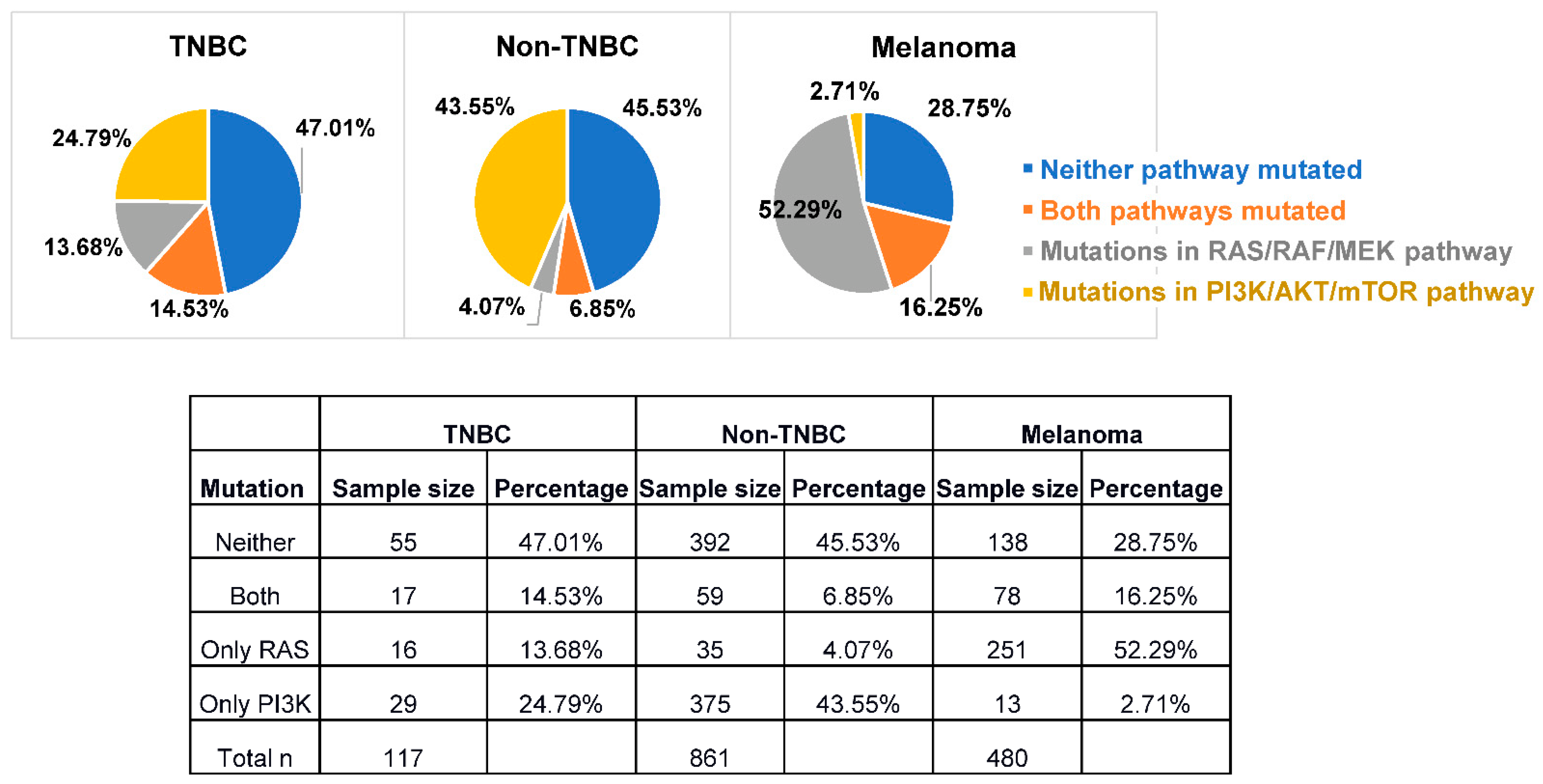

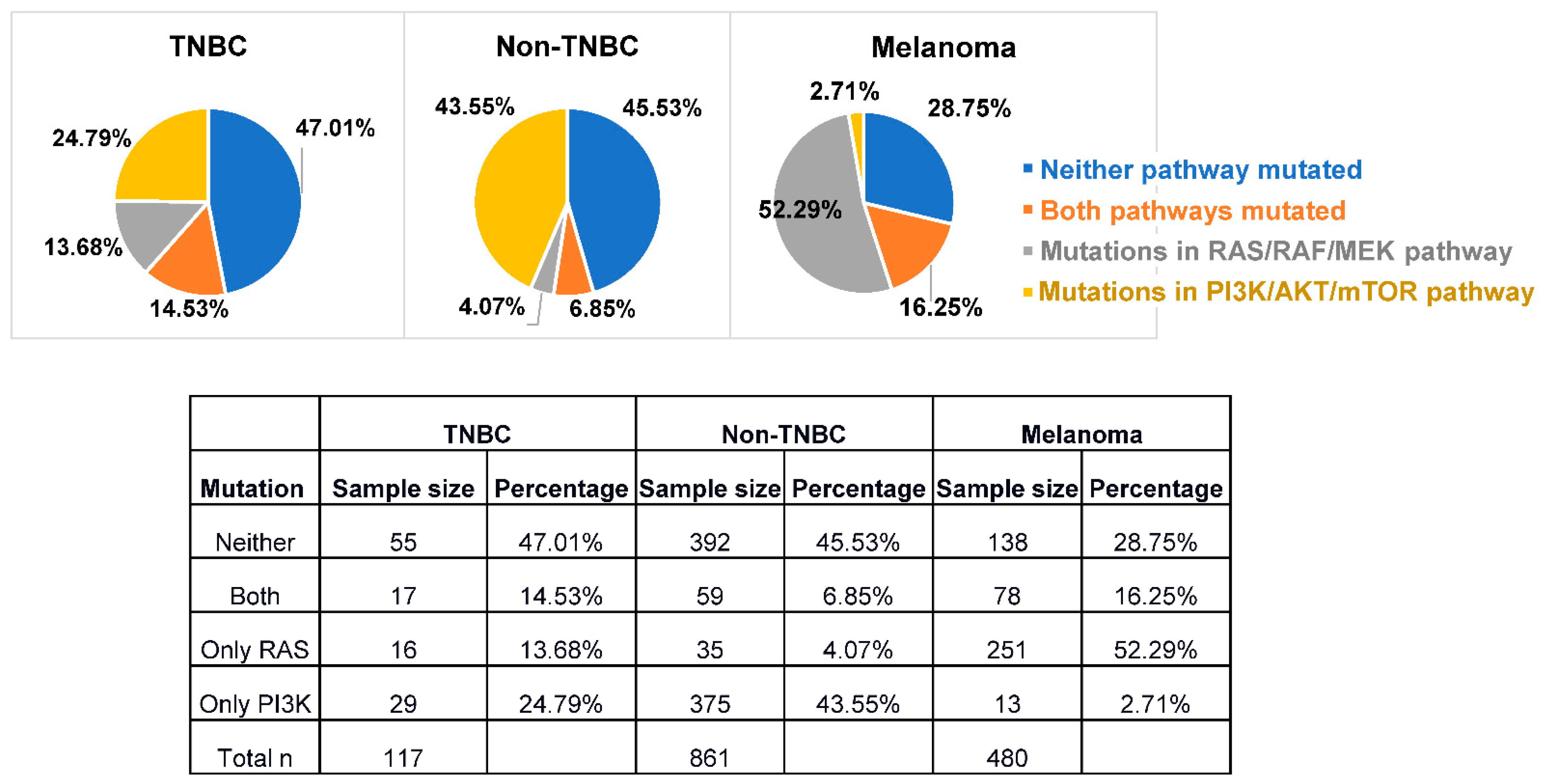{kind=link}
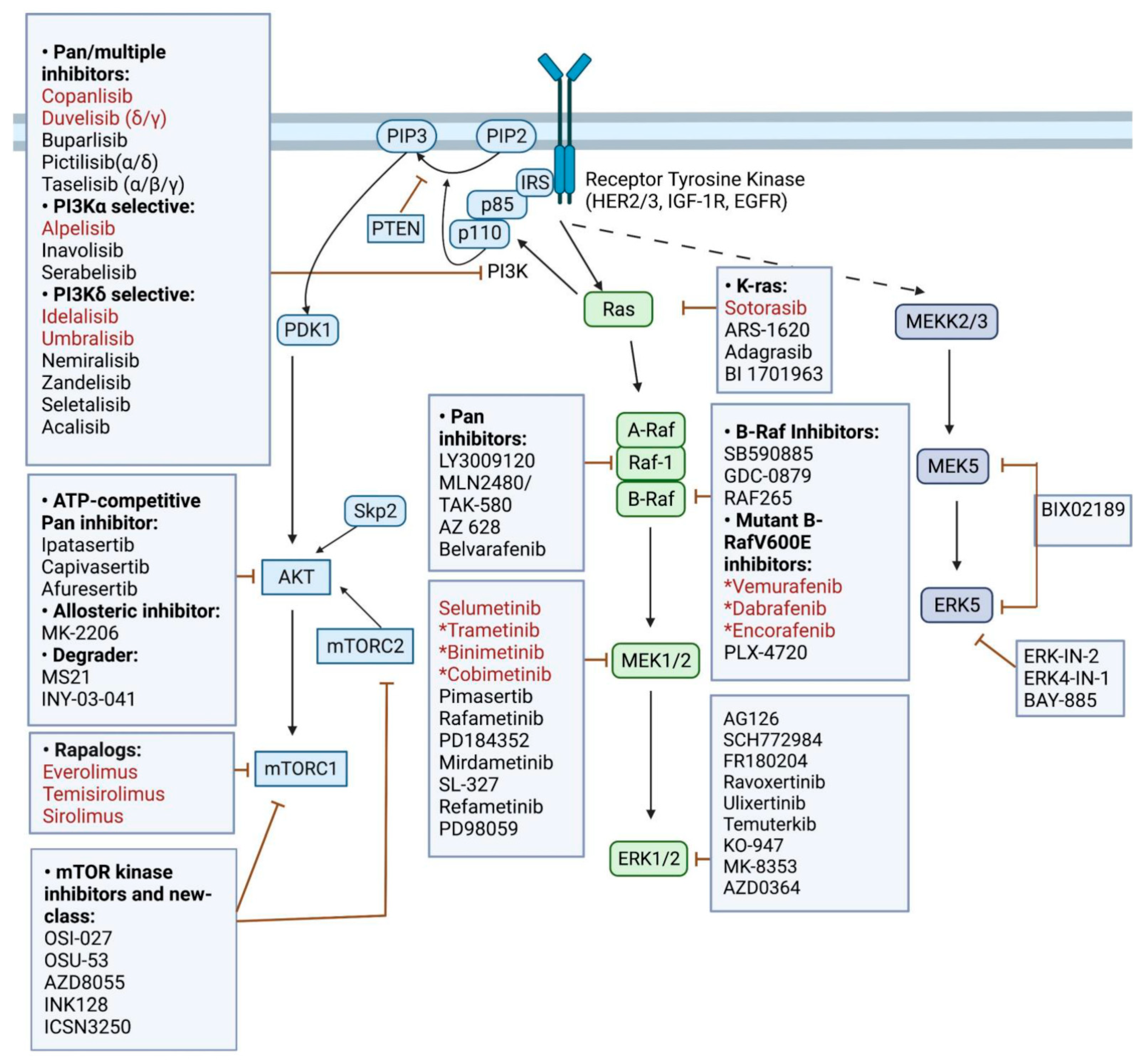
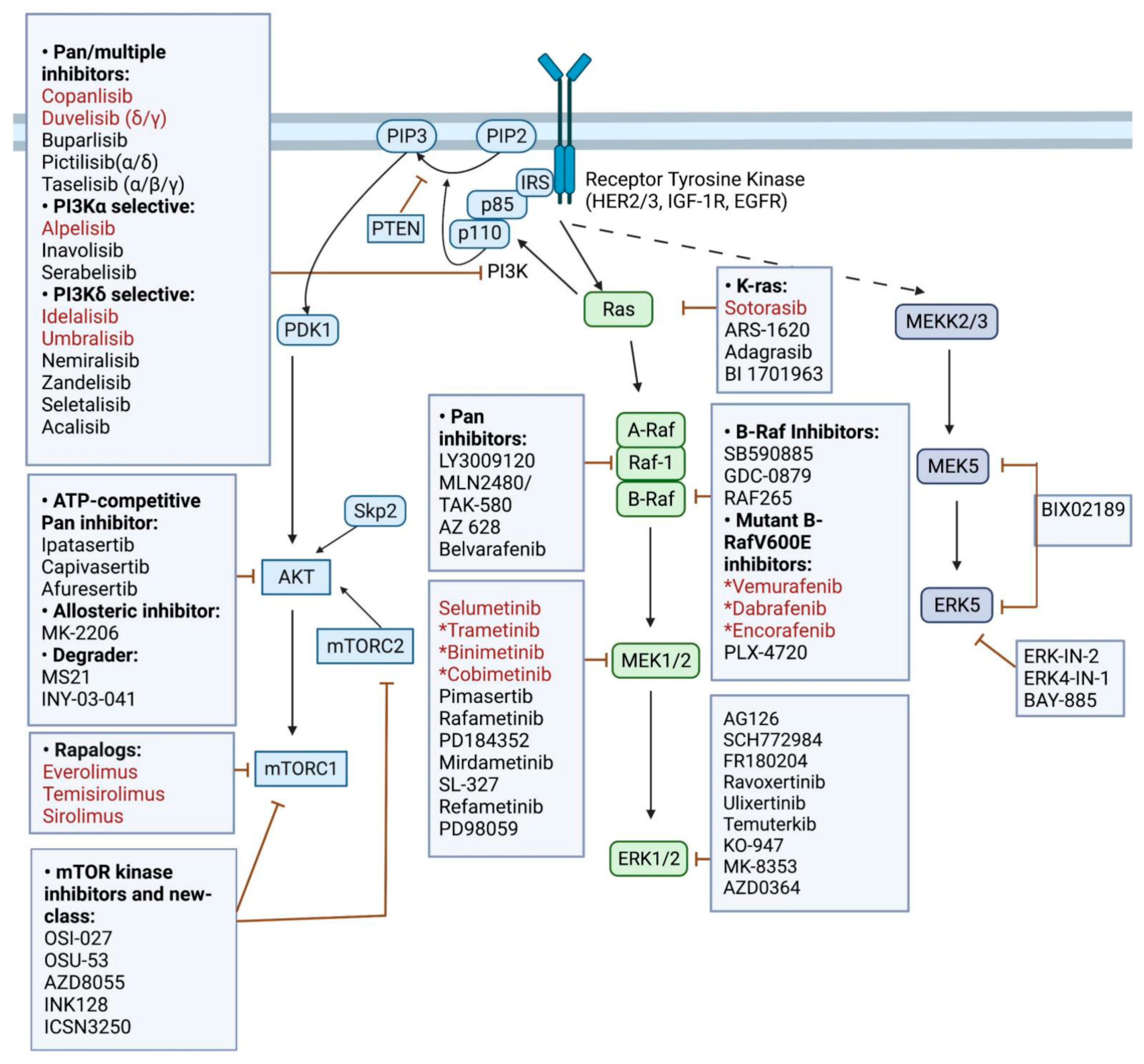{kind=link}
| Identifier | Phase | Combination | Drug Names | Indications |
|---|---|---|---|---|
| NCT01902173 | I/II | AKTi + RAFi + MEK1/2i | Uprosertib Dabrafenib Trametinib | Stage IIIC-IV BRAF mutant Cancer |
| NCT04177108 | III | AKTi + α-PDL1 + Chemo | Ipatasertib Atezolizumab Paclitaxel | Locally advanced unresectable or metastatic TNBC |
| NCT03395899 | II | AKTi + α-PDL1, orMEK1/2i + α-PDL1 | Ipatasertib Atezolizumb Cobimetinib | Untreated operable ER+ HER2- breast cancer |
| NCT03424005 | Ib/II | AKTi + α-PDL1 | Ipatasertib Atezolizumab | Locally advanced unresectable or metastatic TNBC |
| NCT03742102 | Ib/II | AKTi + α-PDL1 + Chemo | Capivasertib Durvalumab Paclitaxel | Metastatic TNBC |
| NCT02858921 | II | B-RAFi + MEK1/2i +α-PD1 | Dabrafenib Trametinib Pembrolizumab | BRAF mutant resectable stage II melanoma |
| NCT04835805 | Ib | pan-RAFi + MEK1/2i +α-PDL1 | Belvarafenib Cobimetinib Atezolizumab | NRAS-mutant advanced melanoma |
| NCT03625141 | II | MEK1/2i + α-PDL1 +BRAFi | Cobimetinib Atezolizumab Vemurafenib | BRAFV600 wild-type or mutant melanoma with central nervous system metastases |
| NCT04722575 | II | Neoadjuvant BRAFi +MEK1/2i + combinationor adjuvant α-PDL1 | Vermurafenib Cobimetinib Atezolizumab (neoadjuvant vs. adjuvant) | High-risk, surgically resectable BRAF-mutated melanoma |
| NCT03554083 | I | BRAFi + MEK1/2i +αPD-L1 | Vemurafenib Cobimetinib Atezolizumab (neoadjuvant + adjuvant) | High-risk, stage III melanoma |
| NCT02910700 | II | BRAFi + MEKi + αPD1 | Dabrafenib Nivolumab Trametinib Binimetinib Encorafenib | Metastatic melanoma |
| Identifier | Phase | Combination | Drug Names | Indications | Results |
|---|---|---|---|---|---|
| NCT03742102 (BEGONIA) | Ib/II | AKTi + αPDL1 + Chemo | Capivasertib Durvalumab Paclitaxel | Metastatic PD-L1+ TNBC | ORR = 16/30 (53.3%) G3/4 trAE =22/30 (73%) |
| NCT03961698 (Mario-3) | II | PI3Kγi + α-PDL1 + Chemo | Eganelisib Atezolizumab Nab-paclitaxel | Locally advanced unresectable or metastatic TNBC | ORR = 21/38 (55.3%) |
| NCT02908672 (IMspire150) | III | B-RAFi + MEK1/2i → B-RAFi + MEK1/2i + α-PDL1 | Vemurafenib Cobimetinib Atezolizumab | Advanced unresectable BRAFV600E melanoma | PFS = 15.1mo vs. 10.6mo G3/4 trAE = 79% vs. 73% n = 514 |
| NCT02130466 (KEYNOTE-22) | I/II | B-RAFi +MEK1/2i + α-PD1 | Dabrafenib Trametinib Pembrolizumab | Unresectable or metastatic BRAFV600E melanoma | PFS = 16.9mo vs. 10.7mo G3-5 trAE = 58% vs. 25% n =120 |
| NCT02967692 (COMBI-i) | III | B-RAFi + MEK1/2i + α-PD1 | Dabrafenib Trametinib Spartalizumab | Unresectable or metastatic BRAFV600E melanoma | PFS = 16.2mo vs. 12.0mo G3-5 trAE = 55% vs. 33% n = 532 |
| NCT02224781 (DREAMSeq) | III | α-PD1 + α-CTLA4 (IT) or BRAFi + MEK1/2i (TT) first, switch treatment upon progression | Nivolumab-Ipilimumab Dabrafenib-trametinib | Metastatic BRAFV600E melanoma | 2-yr OS = 72% vs. 52% n = 265 |
| NCT02631447 (SECOMBIT) | II | B-RAFi MEK1/2i (TT) or α-PDL1 + α-CTLA4 (IT) first, switch treatment upon progression, or TT(8wks) + IT until progression + TT | Nivolumab-Ipilimumab Encorafenib-Binimetinib | Metastatic BRAFV600E melanoma | 2-yr OS = 62% vs. 73% vs. 69% 3-yr OS = 53% vs. 63% vs. 60% G3/4 trAE = 28% vs. 54% vs. 32% n = 251 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Richmond, A.; Yan, C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7353. https://doi.org/10.3390/ijms23137353
Zhang Z, Richmond A, Yan C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. International Journal of Molecular Sciences. 2022; 23(13):7353. https://doi.org/10.3390/ijms23137353
Chicago/Turabian StyleZhang, Zhizhu, Ann Richmond, and Chi Yan. 2022. "Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer" International Journal of Molecular Sciences 23, no. 13: 7353. https://doi.org/10.3390/ijms23137353
APA StyleZhang, Z., Richmond, A., & Yan, C. (2022). Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. International Journal of Molecular Sciences, 23(13), 7353. https://doi.org/10.3390/ijms23137353