
Risk Factors from Pregnancy to Adulthood in Multiple Sclerosis Outcome

 ,
,  ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
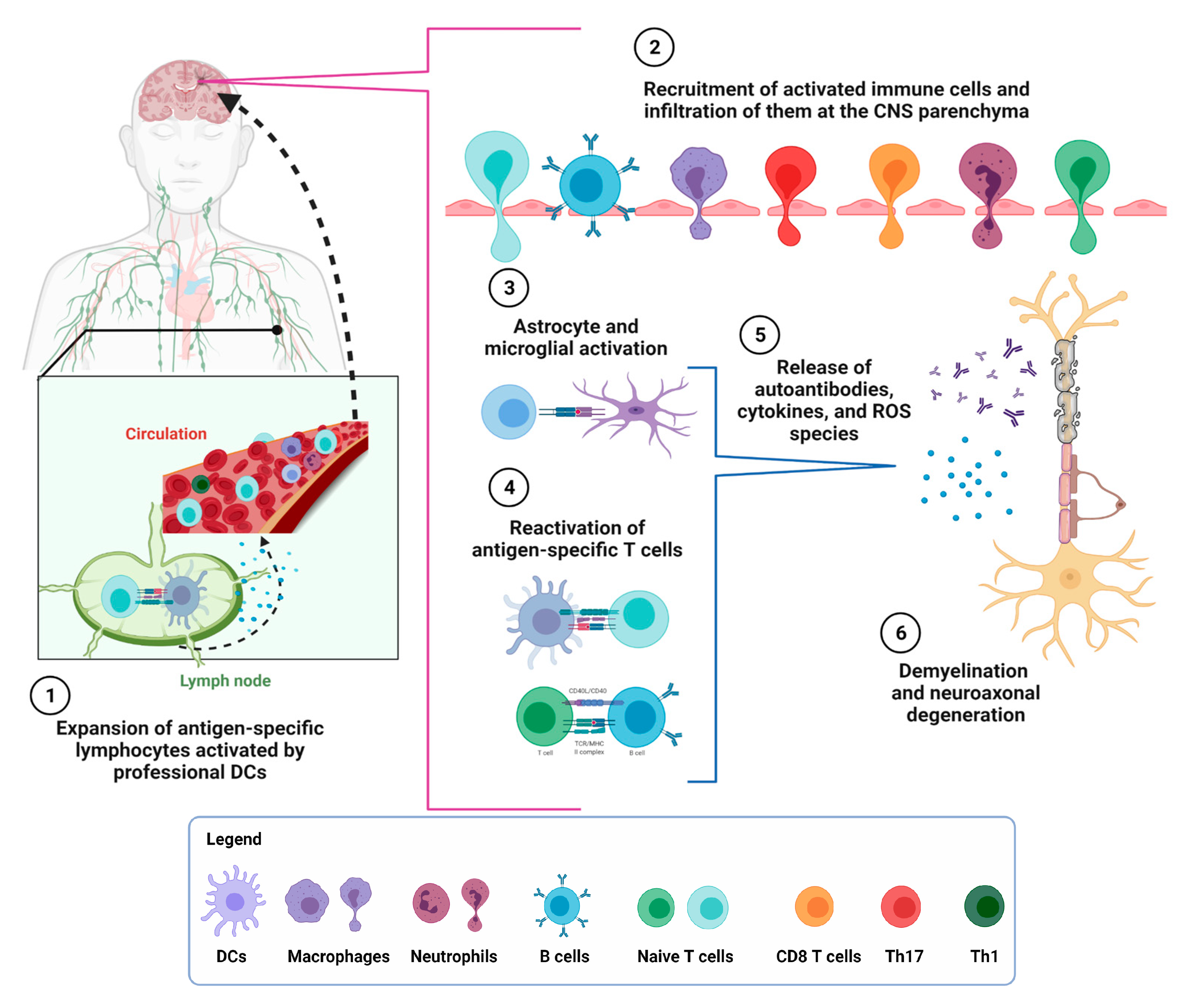
2. Multiple Sclerosis
3. Interplay between the Adaptative and Innate Immune Response in MS
4. Genetic Susceptibility
5. Epigenetics Associated with MS Susceptibility
6. Risk Factors in Postnatal Life Related to MS Pathogenesis
6.1. Conditions of Birth and Newborn Feeding
6.2. Reduced Antioxidant Capacity
6.3. Vitamin D Intake in Adulthood
6.4. Intestinal Microbiota
6.5. Nutritional Factors Involved in MS Development
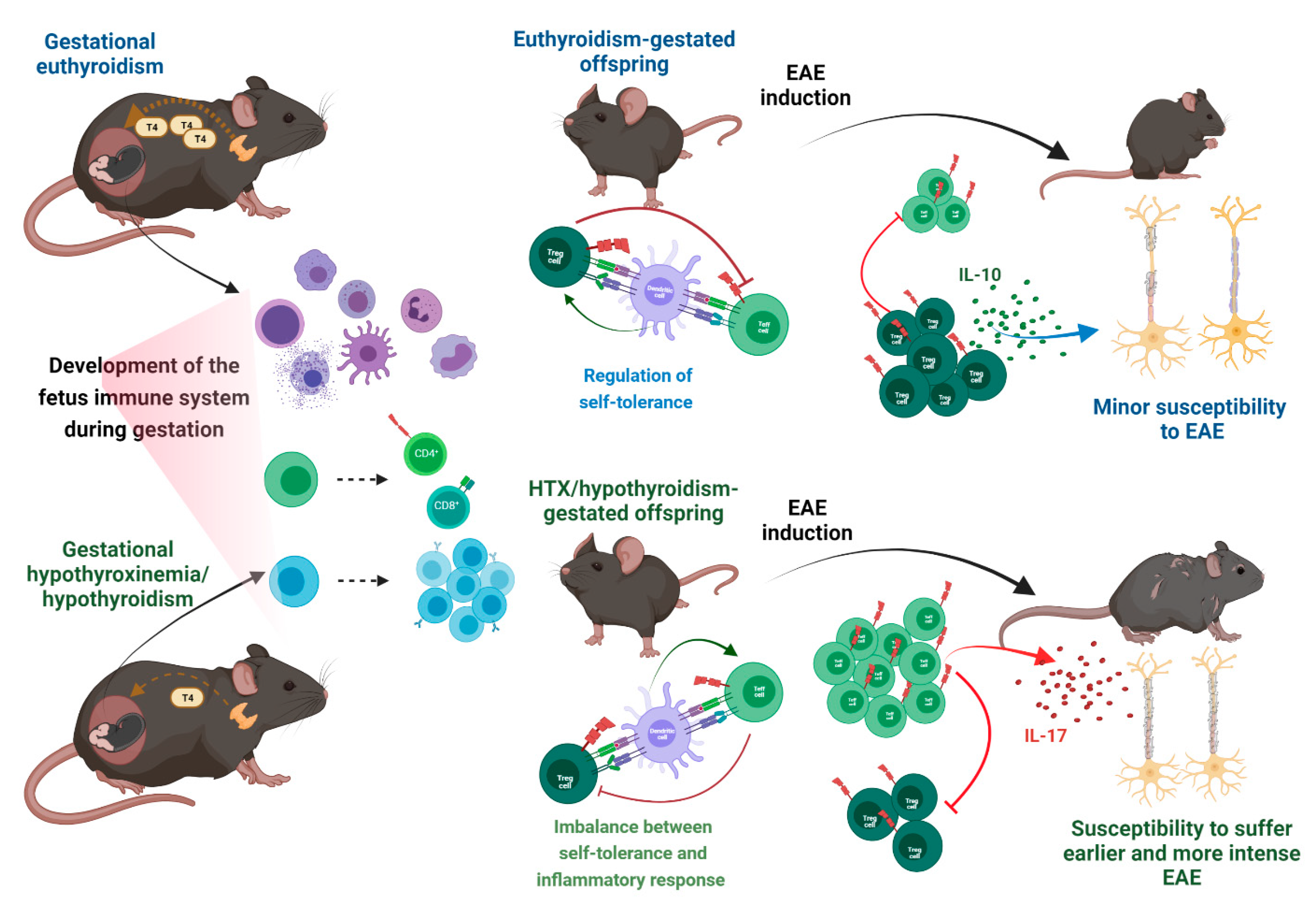
7. Risk Factors in Pregnancy for the Offspring to Suffer MS in Adult Life
7.1. Adaptative Immune System Development during Gestation
7.2. Vitamin D
7.3. Maternal Glucocorticoids Related to Fetal Development
7.4. Thyroid Hormones in the Development of the Immune System
7.5. Maternal Thyroid Hormones during Pregnancy
7.6. Gestational Hypothyroidism and Hypothyroxinemia Increase Susceptibility to Suffering MS
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamińska, J.; Koper, O.M.; Piechal, K.; Kemona, H. Multiple Sclerosis—Etiology and Diagnostic Potential. Postepy Hig. I Med. Dosw. (Online) 2017, 71, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple Sclerosis: Clinical Aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; la Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights from the Atlas of MS, Third Edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Kimura, K. Regulatory T Cells in Multiple Sclerosis. Clin. Exp. Neuroimmunol. 2020, 11, 148–155. [Google Scholar] [CrossRef]
- Donzelli, G.; Carnesecchi, G.; Amador, C.; di Tommaso, M.; Filippi, L.; Caporali, R.; Codullo, V.; Riccieri, V.; Valesini, G.; Gabrielli, A.; et al. Fetal Programming and Systemic Sclerosis. Am. J. Obstet. Gynecol. 2015, 213, 839.e1–839.e8. [Google Scholar] [CrossRef] [Green Version]
- Sand, I.K. Classification, Diagnosis, and Differential Diagnosis of Multiple Sclerosis. Curr. Opin. Neurol. 2015, 28, 193–205. [Google Scholar] [CrossRef]
- Kearney, H.; Altmann, D.R.; Samson, R.S.; Yiannakas, M.C.; Wheeler-Kingshott, C.A.M.; Ciccarelli, O.; Miller, D.H. Cervical Cord Lesion Load Is Associated with Disability Independently from Atrophy in MS. Neurology 2014, 84, 367–373. [Google Scholar] [CrossRef]
- Popescu, B.F.G.; Lucchinetti, C.F. Pathology of Demyelinating Diseases. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 185–217. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Leary, S.M. Primary-Progressive Multiple Sclerosis. Lancet Neurol. 2007, 6, 903–912. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple Sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Browne, P.; Chandraratna, D.; Angood, C.; Tremlett, H.; Baker, C.; Taylor, B.V.; Thompson, A.J. Atlas of Multiple Sclerosis 2013: A Growing Global Problem with Widespread Inequity. Neurology 2014, 83, 1022–1024. [Google Scholar] [CrossRef] [Green Version]
- Ventura, R.E.; Antezana, A.O.; Bacon, T.; Kister, I. Hispanic Americans and African Americans with Multiple Sclerosis Have More Severe Disease Course than Caucasian Americans. Mult. Scler. 2017, 23, 1554–1557. [Google Scholar] [CrossRef]
- Laroni, A.; Signori, A.; Maniscalco, G.T.; Lanzillo, R.; Russo, C.V.; Binello, E.; lo Fermo, S.; Repice, A.; Annovazzi, P.; Bonavita, S.; et al. Assessing Association of Comorbidities with Treatment Choice and Persistence in MS: A Real-Life Multicenter Study. Neurology 2017, 89, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.P.; Alschuler, K.N.; Hughes, A.J.; Beier, M.; Haselkorn, J.K.; Sloan, A.P.; Ehde, D.M. Mental Health Comorbidity in MS: Depression, Anxiety, and Bipolar Disorder. Curr. Neurol. Neurosci. Rep. 2016, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [Green Version]
- Beutler, B. Innate Immunity: An Overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [Green Version]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in Inflammatory Multiple Sclerosis Lesions Have an Intermediate Activation Status. J. Neuroinflamm. 2013, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranzini, S.E.; Elfstrom, C.; Chang, S.-Y.; Butunoi, C.; Murray, R.; Higuchi, R.; Oksenberg, J.R. Transcriptional Analysis of Multiple Sclerosis Brain Lesions Reveals a Complex Pattern of Cytokine Expression. J. Immunol. 2000, 165, 6576–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, P.; Kempuraj, D. Important Role of Mast Cells in Multiple Sclerosis. Mult. Scler. Relat. Disord. 2016, 5, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-Microarray Analysis of Multiple Sclerosis Lesions Yields New Targets Validated in Autoimmune Encephalomyelitis. Nat Med. 2002, 8, 500–508. [Google Scholar] [CrossRef]
- Lan, M.; Tang, X.; Zhang, J.; Yao, Z. Insights in Pathogenesis of Multiple Sclerosis: Nitric Oxide May Induce Mitochondrial Dysfunction of Oligodendrocytes. Rev. Neurosci. 2017, 29, 39–53. [Google Scholar] [CrossRef]
- Sakuishi, K.; Miyake, S.; Yamamura, T. Role of NK Cells and Invariant NKT Cells in Multiple Sclerosis. Results Probl. Cell Differ. 2010, 51, 127–147. [Google Scholar] [CrossRef]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the Innate and Adaptive Immune Responses in the Course of Multiple Sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef]
- Van Langelaar, J.; Rijvers, L.; Smolders, J.; van Luijn, M.M. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 2020, 11, 760. [Google Scholar] [CrossRef]
- Kaskow, B.J.; Baecher-Allan, C. Effector t Cells in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a029025. [Google Scholar] [CrossRef]
- Salou, M.; Nicol, B.; Garcia, A.; Laplaud, D.A. Involvement of CD8+ T Cells in Multiple Sclerosis. Front. Immunol. 2015, 6, 604. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal Vulnerability and Multilineage Diversity in Multiple Sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Huseby, E.S.; Huseby, P.G.; Shah, S.; Smith, R.; Stadinski, B.D. Pathogenic CD8T Cells in Multiple Sclerosis and Its Experimental Models. Front. Immunol. 2012, 3, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T Cells in Multiple Sclerosis and Myasthenia Gravis. J. Neuroinflamm. 2017, 14, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregori, S.; Goudy, K.S.; Roncarolo, M.G. The Cellular and Molecular Mechanisms of Immuno-Suppression by Human Type 1 Regulatory T Cells. Front. Immunol. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Patterson, K.R.; Bar-Or, A. Reassessing B Cell Contributions in Multiple Sclerosis. Nat. Immunol. 2018, 19, 696–707. [Google Scholar] [CrossRef]
- Montalban, X.; Hayser, L.; Kappos, L.; Arnold, D.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. J. Fur Neurol. Neurochir. Und Psychiatr. 2017, 18, 30–31. [Google Scholar] [CrossRef]
- Vasileiadis, G.K.; Dardiotis, E.; Mavropoulos, A.; Tsouris, Z.; Tsimourtou, V.; Bogdanos, D.P.; Sakkas, L.I.; Hadjigeorgiou, G.M. Regulatory B and T Lymphocytes in Multiple Sclerosis: Friends or Foes? Autoimmun. Highlights 2018, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Yanaba, K.; Bouaziz, J.D.; Fujimoto, M.; Tedder, T.F. Regulatory B Cells Inhibit EAE Initiation in Mice While Other B Cells Promote Disease Progression. J. Clin. Investig. 2008, 118, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Pennati, A.; Ng, S.; Wu, Y.; Murphy, J.R.; Deng, J.; Rangaraju, S.; Asress, S.; Blanchfield, J.L.; Evavold, B.; Galipeau, J. Regulatory B Cells Induce Formation of IL-10-Expressing T Cells in Mice with Autoimmune Neuroinflammation. J. Neurosci. 2016, 36, 12598–12610. [Google Scholar] [CrossRef] [Green Version]
- Mavropoulos, A.; Simopoulou, T.; Varna, A.; Liaskos, C.; Katsiari, C.G.; Bogdanos, D.P.; Sakkas, L.I. Breg Cells Are Numerically Decreased and Functionally Impaired in Patients with Systemic Sclerosis. Arthritis Rheumatol. 2016, 68, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Jiusheng Deng, J.H. Blood B Cell and Regulatory Subset Content in Multiple Sclerosis Patients. J. Mult. Scler. 2015, 2, 1000139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Andrés, C.; Tejera-Alhambra, M.; Alonso, B.; Valor, L.; Teijeiro, R.; Ramos-Medina, R.; Mateos, D.; Faure, F.; Sánchez-Ramón, S. New Regulatory CD19+CD25+ B-Cell Subset in Clinically Isolated Syndrome and Multiple Sclerosis Relapse. Changes after Glucocorticoids. J. Neuroimmunol. 2014, 270, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Qin, Y.; Duquette, P.; Zhang, Y.; Talbot, P.; Poole, R.; Antel, J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J. Clin. Investig. 1998, 102, 1045–1050. [Google Scholar] [CrossRef]
- Lou, Y.-H.; Park, K.-K.; Agersborg, S.; Alard, P.; Tung, K.S.K. Retargeting T Cell-Mediated Inflammation: A New Perspective on Autoantibody Action. J. Immunol. 2000, 164, 5251–5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scolding, N.J.; Compston, D.A.S. Oligodendrocyte-Macrophage Interactions in Vitro Triggered by Specific Antibodies. Immunology 1991, 72, 127–132. [Google Scholar] [PubMed]
- Kuerten, S.; Pauly, R.; Rottlaender, A.; Rodi, M.; Gruppe, T.L.; Addicks, K.; Tary-Lehmann, M.; Lehmann, P.V. Myelin-Reactive Antibodies Mediate the Pathology of MBP-PLP Fusion Protein MP4-Induced EAE. Clin. Immunol. 2011, 140, 54–62. [Google Scholar] [CrossRef]
- Weber, M.S.; Derfuss, T.; Metz, I.; Brück, W. Defining Distinct Features of Anti-MOG Antibody Associated Central Nervous System Demyelination. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418762083. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.; Iglesias, E.; Nasrallah, M.; González-Álvarez, V.; Sunõl, M.; Anton, J.; Saiz, A.; Lancaster, E.; Armangue, T. Anti-MOG Encephalitis Mimicking Small Vessel CNS Vasculitis. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e538. [Google Scholar] [CrossRef] [Green Version]
- Goodin, D.S.; Khankhanian, P.; Gourraud, P.A.; Vince, N. The Nature of Genetic and Environmental Susceptibility to Multiple Sclerosis. PLoS ONE 2021, 16, e0246157. [Google Scholar] [CrossRef]
- Shepard, C.J.; Cline, S.G.; Hinds, D.; Jahanbakhsh, S.; Prokop, J.W.; Cj, S.; Sg, C.; Prokop, J.S. Breakdown of Multiple Sclerosis Genetics to Identify an Integrated Disease Network and Potential Variant Mechanisms. Physiol. Genom. 2019, 51, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Cotsapas, C.; Mitrovic, M. Genome-Wide Association Studies of Multiple Sclerosis. Clin. Transl. Immunol. 2018, 7, e1018. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hou, X.; Liang, Y.; Xu, F.; Zhang, X.; Cui, P.; Xing, G.; Wang, X.; Jiang, W. Gene-Based Tests of a Genome-Wide Association Study Dataset Highlight Novel Multiple Sclerosis Risk Genes. Front. Neurosci. 2021, 15, 614528. [Google Scholar] [CrossRef] [PubMed]
- Gresle, M.M.; Jordan, M.A.; Stankovich, J.; Spelman, T.; Johnson, L.J.; Laverick, L.; Hamlett, A.; Smith, L.D.; Jokubaitis, V.G.; Baker, J.; et al. Multiple Sclerosis Risk Variants Regulate Gene Expression in Innate and Adaptive Immune Cells. Life Sci. Alliance 2020, 3, e202000650. [Google Scholar] [CrossRef] [PubMed]
- Mokry, L.E.; Ross, S.; Ahmad, O.S.; Forgetta, V.; Smith, G.D.; Leong, A.; Greenwood, C.M.T.; Thanassoulis, G.; Richards, J.B. Vitamin D and Risk of Multiple Sclerosis: A Mendelian Randomization Study. PLoS Med. 2015, 12, e1001866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scazzone, C.; Agnello, L.; Ragonese, P.; lo Sasso, B.; Bellia, C.; Bivona, G.; Schillaci, R.; Salemi, G.; Ciaccio, M. Association of CYP2R1 Rs10766197 with MS Risk and Disease Progression. J. Neurosci. Res. 2018, 96, 297–304. [Google Scholar] [CrossRef]
- Mimpen, M.; Rolf, L.; Poelmans, G.; van den Ouweland, J.; Hupperts, R.; Damoiseaux, J.; Smolders, J. Vitamin D Related Genetic Polymorphisms Affect Serological Response to High-Dose Vitamin D Supplementation in Multiple Sclerosis. PLoS ONE 2021, 16, e26186. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Genes, T.M. Obesity and Multiple Sclerosis—A Multifaceted Association. J. Clin. Med. 2021, 10, 2689. [Google Scholar] [CrossRef]
- Guerrero-García, J.D.J.; Carrera-Quintanar, L.; López-Roa, R.I.; Márquez-Aguirre, A.L.; Rojas-Mayorquín, A.E.; Ortuño-Sahagún, D. Multiple Sclerosis and Obesity: Possible Roles of Adipokines. Mediat. Inflamm. 2016, 2016, 4036232. [Google Scholar] [CrossRef] [Green Version]
- Gianfrancesco, M.A.; Glymour, M.M.; Walter, S.; Rhead, B.; Shao, X.; Shen, L.; Quach, H.; Hubbard, A.; Jónsdóttir, I.; Stefánsson, K.; et al. Causal Effect of Genetic Variants Associated with Body Mass Index on Multiple Sclerosis Susceptibility. Am. J. Epidemiol. 2017, 185, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Al-Serri, A.; Alroughani, R.; Al-Temaimi, R.A. The FTO Gene Polymorphism Rs9939609 Is Associated with Obesity and Disability in Multiple Sclerosis Patients. Sci. Rep. 2019, 9, 19071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.M. Fat Mass and Obesity Associated (FTO) Gene and Hepatic Glucose and Lipid Metabolism. Nutrients 2018, 10, 1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Serri, A.; Al-Bustan, S.A.; Kamkar, M.; Thomas, D.; Alsmadi, O.; Al-Temaimi, R.; Mojiminiyi, O.A.; Abdella, N.A. Association of FTO Rs9939609 with Obesity in the Kuwaiti Population: A Public Health Concern? Med. Princ. Pract. 2018, 27, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedström, A.K.; Katsoulis, M.; Hössjer, O.; Bomfim, I.L.; Oturai, A.; Sondergaard, H.B.; Sellebjerg, F.; Ullum, H.; Thørner, L.W.; Gustavsen, M.W.; et al. The Interaction between Smoking and HLA Genes in Multiple Sclerosis: Replication and Refinement. Eur. J. Epidemiol. 2017, 32, 909–919. [Google Scholar] [CrossRef]
- Schmidt, H.; Williamson, D.; Ashley-Koch, A. HLA-DR15 Haplotype and Multiple Sclerosis: A HuGE Review. Am. J. Epidemiol. 2007, 165, 1097–1109. [Google Scholar] [CrossRef]
- Fogdell, A.; Hillert, J.; Sachs, C. The Multiple Sclerosis-and Narcolepsy-Associated-HLA Class I1 Haplotype-Includes the DRB.5 *0101 Allele. Tissue Anrigens 1995, 46, 333–336. [Google Scholar] [CrossRef]
- Wang, J.; Jelcic, I.; Mühlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20. [Google Scholar] [CrossRef]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple Sclerosis Genomic Map Implicates Peripheral Immune Cells and Microglia in Susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [Green Version]
- Frazzi, R. BIRC3 and BIRC5: Multi-faceted Inhibitors in Cancer. Cell Biosci. 2021, 11, 8. [Google Scholar] [CrossRef]
- Sharief, M.K.; Semra, Y.K. Heightened Expression of Survivin in Activated T Lymphocytes from Patients with Multiple Sclerosis. J. Neuroimmunol. 2001, 119, 358–364. [Google Scholar] [CrossRef]
- Ebrahimiyan, H.; Aslani, S.; Rezaei, N.; Jamshidi, A.; Mahmoudi, M. Survivin and Autoimmunity; the Ins and Outs. Immunol. Lett. 2018, 193, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.; Wasén, C.; Garcia-Bonete, M.J.; Turkkila, M.; Erlandsson, M.C.; Töyrä Silfverswärd, S.; Brisslert, M.; Pullerits, R.; Andersson, K.M.; Katona, G.; et al. Survivin in Autoimmune Diseases. Autoimmun. Rev. 2017, 16, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Fenstermaker, R.A.; Figel, S.A.; Qiu, J.; Barone, T.A.; Dharma, S.S.; Winograd, E.K.; Galbo, P.M.; Wiltsie, L.M.; Ciesielski, M.J. Survivin Monoclonal Antibodies Detect Survivin Cell Surface Expression and Inhibit Tumor Growth in Vivo. Clin. Cancer Res. 2018, 24, 2642–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, J.L.; Oliveira, E.M.; Pontillo, A. Variants in NLRP3 and NLRC4 Inflammasome Associate with Susceptibility and Severity of Multiple Sclerosis. Mult. Scler. Relat. Disord. 2019, 29, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Zamzam, D.; Foad, M.; Swelam, M.; AbdelHafez, M.; AbdelNasser, A.; Mahmoud, R.; Aref, H.; Zakaria, M. Vitamin D and Body Mass Index in Egyptian Multiple Sclerosis Patients. Mult. Scler. Relat. Disord. 2019, 28, 313–316. [Google Scholar] [CrossRef]
- Jamebozorgi, K.; Rostami, D.; Pormasoumi, H.; Taghizadeh, E.; Barreto, G.E.; Sahebkar, A. Epigenetic Aspects of Multiple Sclerosis and Future Therapeutic Options. Int. J. Neurosci. 2021, 131, 56–64. [Google Scholar] [CrossRef]
- Chomyk, A.M.; Volsko, C.; Tripathi, A.; Deckard, S.A.; Trapp, B.D.; Fox, R.J.; Dutta, R. DNA Methylation in Demyelinated Multiple Sclerosis Hippocampus. Sci. Rep. 2017, 7, 8696. [Google Scholar] [CrossRef] [Green Version]
- Rito, Y.; Torre-Villalvazo, I.; Flores, J.; Rivas, V.; Corona, T. Epigenetics in Multiple Sclerosis: Molecular Mechanisms and Dietary Intervention. Cent. Nerv. Syst. Agents Med. Chem. 2016, 18, 8–15. [Google Scholar] [CrossRef]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High Density DNA Methylation Array with Single CpG Site Resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Kular, L.; Jagodic, M. Epigenetic Insights into Multiple Sclerosis Disease Progression. J. Intern. Med. 2020, 288, 82–102. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.W.; Metz, L.M.; Kovalchuk, O. Epigenetic Changes in Patients with Multiple Sclerosis. Nat. Rev. Neurol. 2013, 9, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Maltby, V.E.; Lea, R.A.; Sanders, K.A.; White, N.; Benton, M.C.; Scott, R.J.; Lechner-Scott, J. Differential Methylation at MHC in CD4+ T Cells Is Associated with Multiple Sclerosis Independently of HLA-DRB1. Clin. Epigenetics 2017, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.S.; Beck, S. Advances in Epigenome-Wide Association Studies for Common Diseases. Trends Mol. Med. 2014, 20, 541–543. [Google Scholar] [CrossRef] [Green Version]
- Belbasis, L.; Bellou, V.; Tzoulaki, I.; Evangelou, E. Early-Life Factors and Risk of Multiple Sclerosis: An MR-EWAS. Neuroepidemiology 2020, 54, 433–445. [Google Scholar] [CrossRef]
- Maltby, V.E.; Graves, M.C.; Lea, R.A.; Benton, M.C.; Sanders, K.A.; Tajouri, L.; Scott, R.J.; Lechner-Scott, J. Genome-Wide DNA Methylation Profiling of CD8+ T Cells Shows a Distinct Epigenetic Signature to CD4+ T Cells in Multiple Sclerosis Patients. Clin. Epigenetics 2015, 7, 118. [Google Scholar] [CrossRef] [Green Version]
- Maltby, V.E.; Lea, R.A.; Graves, M.C.; Sanders, K.A.; Benton, M.C.; Tajouri, L.; Scott, R.J.; Lechner-Scott, J. Genome-Wide DNA Methylation Changes in CD19+ B Cells from Relapsing-Remitting Multiple Sclerosis Patients. Sci. Rep. 2018, 8, 17418. [Google Scholar] [CrossRef]
- He, H.; Hu, Z.; Xiao, H.; Zhou, F.; Yang, B. The Tale of Histone Modifications and Its Role in Multiple Sclerosis. Hum. Genom. 2018, 12, 31. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of Histone Modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Karlić, R.; Chung, H.R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone Modification Levels Are Predictive for Gene Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [Green Version]
- Mei, F.; Christin Chong, S.Y.; Chan, J.R. Myelin-Based Inhibitors of Oligodendrocyte Myelination: Clues from Axonal Growth and Regeneration. Neurosci. Bull. 2013, 29, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.; Wang, J.; Richard Lu, Q. Epigenetic Regulation of Oligodendrocyte Myelination in Developmental Disorders and Neurodegenerative Diseases. F1000Res 2020, 9, (F1000 Faculty Rev):105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedre, X.; Mastronardi, F.; Bruck, W.; López-Rodas, G.; Kuhlmann, T.; Casaccia, P. Changed Histone Acetylation Patterns in Normal-Appearing White Matter and Early Multiple Sclerosis Lesions. J. Neurosci. 2011, 31, 3435–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The Roles of MicroRNAs in Epigenetic Regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimiyan, H.; Gharibdoost, F.; Aslani, S.; Kavosi, H.; Farsad, F.; Jamshidi, A.; Mahmoudi, M. MicroRNAs Are Potentially Regulating the Survivin Gene in PBMCs from Systemic Sclerosis Patients. Mod. Rheumatol. 2020, 30, 862–869. [Google Scholar] [CrossRef]
- Alizadeh-Fanalou, S.; Alian, F.; Mohammadhosayni, M.; Rahban, D.; Abbasi Ghasem Kheyli, P.; Ahmadi, M. Dysregulation of MicroRNAs Regulating Survivin in CD4+ T Cells in Multiple Sclerosis. Mult. Scler. Relat. Disord. 2020, 44, 102303. [Google Scholar] [CrossRef]
- Basak, J.; Majsterek, I. MiRNA-Dependent CD4+ T Cell Differentiation in the Pathogenesis of Multiple Sclerosis. Mult. Scler. Int. 2021, 2021, 8825588. [Google Scholar] [CrossRef]
- Venkatesha, S.H.; Dudics, S.; Song, Y.; Mahurkar, A.; Moudgil, K.D. The MiRNA Expression Profile of Experimental Autoimmune Encephalomyelitis Reveals Novel Potential Disease Biomarkers. Int. J. Mol. Sci. 2018, 19, 3990. [Google Scholar] [CrossRef] [Green Version]
- Sohan Forooshan Moghadam, A.; Ataei, M.; Arabzadeh, G.; Falahati, K.; Roshani, F.; Sanati, M.H. Analysis of MicroRNA-18a Expression in Multiple Sclerosis Patients. Rep. Biochem. Mol. Biol. 2020, 8, 429–437. [Google Scholar]
- Liguori, M.; Nuzziello, N.; Licciulli, F.; Consiglio, A.; Simone, M.; Viterbo, R.G.; Creanza, T.M.; Ancona, N.; Tortorella, C.; Margari, L.; et al. Combined MicroRNA and MRNA Expression Analysis in Pediatric Multiple Sclerosis: An Integrated Approach to Uncover Novel Pathogenic Mechanisms of the Disease. Hum. Mol. Genet. 2018, 27, 66–79. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dasgupta, S.; Mishra, P.K.; Chaudhury, K. Air Pollution-Induced Epigenetic Changes: Disease Development and a Possible Link with Hypersensitivity Pneumonitis. Environ. Sci. Pollut. Res. 2021, 28, 55981–56002. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Herrera-Soto, A.; Jury, N.; Maher, B.A.; González-Maciel, A.; Reynoso-Robles, R.; Ruiz-Rudolph, P.; van Zundert, B.; Varela-Nallar, L. Reduced Repressive Epigenetic Marks, Increased DNA Damage and Alzheimer’s Disease Hallmarks in the Brain of Humans and Mice Exposed to Particulate Urban Air Pollution. Environ. Res. 2020, 183, 109226. [Google Scholar] [CrossRef] [PubMed]
- Sakoda, A.; Matsushita, T.; Nakamura, Y.; Watanabe, M.; Shinoda, K.; Masaki, K.; Isobe, N.; Yamasaki, R.; Kira, J. ichi Environmental Risk Factors for Multiple Sclerosis in Japanese People. Mult. Scler. Relat. Disord. 2020, 38, 101872. [Google Scholar] [CrossRef]
- Marabita, F.; Almgren, M.; Sjöholm, L.K.; Kular, L.; Liu, Y.; James, T.; Kiss, N.B.; Feinberg, A.P.; Olsson, T.; Kockum, I.; et al. Smoking Induces DNA Methylation Changes in Multiple Sclerosis Patients with Exposure-Response Relationship. Sci. Rep. 2017, 7, 14589. [Google Scholar] [CrossRef] [Green Version]
- Langer-Gould, A.; Smith, J.B.; Hellwig, K.; Gonzales, E.; Haraszti, S.; Koebnick, C.; Xiang, A. Breastfeeding, Ovulatory Years, and Risk of Multiple Sclerosis. Neurology 2017, 89, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Weström, B.; Arévalo Sureda, E.; Pierzynowska, K.; Pierzynowski, S.G.; Pérez-Cano, F.J. The Immature Gut Barrier and Its Importance in Establishing Immunity in Newborn Mammals. Front. Immunol. 2020, 11, 1153. [Google Scholar] [CrossRef]
- Graves, J.S.; Chitnis, T.; Weinstock-Guttman, B.; Rubin, J.; Zelikovitch, A.S.; Nourbakhsh, B.; Simmons, T.; Waltz, M.; Casper, T.C.; Waubant, E.; et al. Maternal and Perinatal Exposures Are Associated with Risk for Pediatric-Onset Multiple Sclerosis. Pediatrics 2017, 139, e20162838. [Google Scholar] [CrossRef] [Green Version]
- Hedström, A.K.; Adams, C.; Shao, X.; Schaefer, C.; Olsson, T.; Barcellos, L.F.; Alfredsson, L. Breastfeeding Is Associated with Reduced Risk of Multiple Sclerosis in Males, Predominantly among HLA-DRB1*15:01 Carriers. Mult. Scler. J. Exp. Transl. Clin. 2020, 6, 2055217320928101. [Google Scholar] [CrossRef]
- Luetic, G.G.; Menichini, M.L.; Deri, N.; Steinberg, J.; Carrá, A.; Cristiano, E.; Patrucco, L.; Curbelo, M.C.; Rojas, J.I. High Birth Weight and Risk of Multiple Sclerosis: A Multicentre Study in Argentina. Mult. Scler. Relat. Disord. 2021, 47, 102628. [Google Scholar] [CrossRef]
- Wood, H.; Acharjee, A.; Pearce, H.; Quraishi, M.N.; Powell, R.; Rossiter, A.; Beggs, A.; Ewer, A.; Moss, P.; Toldi, G. Breastfeeding Promotes Early Neonatal Regulatory T-Cell Expansion and Immune Tolerance of Non-Inherited Maternal Antigens. Allergy Eur. J. Allergy Clin. Immunol. 2021, 76, 2447–2460. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.Y.; Gui, L.N.; Liu, Y.Y.; Shi, S.; Cheng, Y. Oxidative Stress Marker Aberrations in Multiple Sclerosis: A Meta-Analysis Study. Front. Neurosci. 2020, 14, 823. [Google Scholar] [CrossRef] [PubMed]
- Padureanu, R.; Albu, C.V.; Mititelu, R.R.; Bacanoiu, M.V.; Docea, A.O.; Calina, D.; Padureanu, V.; Olaru, G.; Sandu, R.E.; Malin, R.D.; et al. Oxidative Stress and Inflammation Interdependence in Multiple Sclerosis. J. Clin. Med. 2019, 8, 1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siotto, M.; Filippi, M.M.; Simonelli, I.; Landi, D.; Ghazaryan, A.; Vollaro, S.; Ventriglia, M.; Pasqualetti, P.; Rongioletti, M.C.A.; Squitti, R.; et al. Oxidative Stress Related to Iron Metabolism in Relapsing Remitting Multiple Sclerosis Patients with Low Disability. Front. Neurosci. 2019, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimonti, A.; Ristori, G.; Giubilei, F.; Stazi, M.A.; Pino, A.; Visconti, A.; Brescianini, S.; Monti, M.S.; Forte, G.; Stanzione, P.; et al. Serum Chemical Elements and Oxidative Status in Alzheimer’s Disease, Parkinson Disease and Multiple Sclerosis. NeuroToxicology 2007, 28, 450–456. [Google Scholar] [CrossRef]
- Previte, D.M.; O’connor, E.C.; Novak, E.A.; Martins, C.P.; Mollen, K.P.; Piganelli, J.D. Reactive Oxygen Species Are Required for Driving Efficient and Sustained Aerobic Glycolysis during CD4+ T Cell Activation. PLoS ONE 2017, 12, e0175549. [Google Scholar] [CrossRef]
- Gonzalo, H.; Nogueras, L.; Gil-Sánchez, A.; Hervás, J.V.; Valcheva, P.; González-Mingot, C.; Martin-Gari, M.; Canudes, M.; Peralta, S.; Solana, M.J.; et al. Impairment of Mitochondrial Redox Status in Peripheral Lymphocytes of Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 938. [Google Scholar] [CrossRef]
- Ravelli, K.G.; Santos, G.D.; dos Santos, N.B.; Munhoz, C.D.; Azzi-Nogueira, D.; Campos, A.C.; Pagano, R.L.; Britto, L.R.; Hernandes, M.S. Nox2-Dependent Neuroinflammation in an EAE Model of Multiple Sclerosis. Transl. Neurosci. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Bhargava, P.; Fitzgerald, K.C.; Calabresi, P.A.; Mowry, E.M. Metabolic Alterations in Multiple Sclerosis and the Impact of Vitamin D Supplementation. JCI Insight 2017, 2, e95302. [Google Scholar] [CrossRef] [Green Version]
- Gianfrancesco, M.A.; Stridh, P.; Rhead, B.; Shao, X.; Xu, E.; Graves, J.S.; Chitnis, T.; Waldman, A.; Lotze, T.; Schreiner, T.; et al. Evidence for a Causal Relationship between Low Vitamin D, High BMI, and Pediatric-Onset MS. Neurology 2017, 88, 1623–1629. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.; Razzitte, G. Helminth Infections Associated with Multiple Sclerosis Induce Regulatory B Cells. Ann. Neurol. 2008, 64, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and Multiple Sclerosis: A Comprehensive Review. Neurol. Ther. 2018, 7, 59–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killick, J.; Hay, J.; Morandi, E.; Vermeren, S.; Kari, S.; Angles, T.; Williams, A.; Damoiseaux, J.; Astier, A.L. Vitamin D/CD46 Crosstalk in Human T Cells in Multiple Sclerosis. Front. Immunol. 2020, 11, 598727. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, T.; Aznar, P.; Soriano, L.; Olaskoaga, A.; Roldán, M.; Otano, M.; Ajuria, I.; Soriano, G.; Lacruz, F.; Mendioroz, M. Vitamin D Receptor Gene Isepigenetically Altered and Transcriptionally Up-Regulated in Multiple Sclerosis. PLoS ONE 2017, 12, e0174726. [Google Scholar] [CrossRef]
- Hashemi, R.; Morshedi, M.; Jafarabadi, M.A.; Altafi, D.; Hosseini-Asl, S.S.; Rafie-Arefhosseini, S. Anti-Inflammatory Effects of Dietary Vitamin D 3 in Patients with Multiple Sclerosis. Neurol. Genet. 2018, 4, e278. [Google Scholar] [CrossRef] [Green Version]
- Berlanga-Taylor, A.J.; Plant, K.; Dahl, A.; Lau, E.; Hill, M.; Sims, D.; Heger, A.; Emberson, J.; Armitage, J.; Clarke, R.; et al. Genomic Response to Vitamin D Supplementation in the Setting of a Randomized, Placebo-Controlled Trial. EBioMedicine 2018, 31, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Dalla Costa, G.; Romeo, M.; Esposito, F.; Sangalli, F.; Colombo, B.; Radaelli, M.; Moiola, L.; Comi, G.; Martinelli, V. Caesarean Section and Infant Formula Feeding Are Associated with an Earlier Age of Onset of Multiple Sclerosis. Mult. Scler. Relat. Disord. 2019, 33, 75–77. [Google Scholar] [CrossRef]
- Wendel-Haga, M.; Celius, E.G. Is the Hygiene Hypothesis Relevant for the Risk of Multiple Sclerosis? Acta Neurol. Scand. 2017, 136, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, A.; Cicero, C.E.; Giuliano, L.; Todaro, V.; lo Fermo, S.; Chisari, C.; D’Amico, E.; Paradisi, V.; Mantella, A.; Bartoloni, A.; et al. Toxoplasma Gondii and Multiple Sclerosis: A Population-Based Case–Control Study. Sci. Rep. 2020, 10, 18855. [Google Scholar] [CrossRef]
- Wasko, N.J.; Nichols, F.; Clark, R.B. Multiple Sclerosis, the Microbiome, TLR2, and the Hygiene Hypothesis. Autoimmun. Rev. 2020, 19, 102430. [Google Scholar] [CrossRef]
- Bach, J.F. The Hygiene Hypothesis in Autoimmunity: The Role of Pathogens and Commensals. Nat. Rev. Immunol. 2018, 18, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Soldan, M.M.P.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple Sclerosis Patients Have a Distinct Gut Microbiota Compared to Healthy Controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzo, P.; Capri, F.C.; Vecchioni, L.; Realmuto, S.; Scalisi, L.; Cottone, S.; Nuzzo, D.; Alduina, R. Comparison of the Intestinal Microbiome of Italian Patients with Multiple Sclerosis and Their Household Relatives. Life 2021, 11, 620. [Google Scholar] [CrossRef]
- Lin, X.; Liu, Y.; Ma, L.; Ma, X.; Shen, L.; Ma, X.; Chen, Z.; Chen, H.; Li, D.; Su, Z.; et al. Constipation Induced Gut Microbiota Dysbiosis Exacerbates Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice. J. Transl. Med. 2021, 19, 317. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.; Gustavsen, S.; Langkilde, A.R.; Hansen, T.H.; Sellebjerg, F.; Bach Søndergaard, H.; Oturai, A.B. Circulating Levels of Tight Junction Proteins in Multiple Sclerosis: Association with Inflammation and Disease Activity before and after Disease Modifying Therapy. Mult. Scler. Relat. Disord. 2021, 54, 103136. [Google Scholar] [CrossRef]
- Haase, S.; Haghikia, A.; Wilck, N.; Müller, D.N.; Linker, R.A. Impacts of Microbiome Metabolites on Immune Regulation and Autoimmunity. Immunology 2018, 154, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Suez, J.; Elinav, E. You Are What You Eat: Diet, Health and the Gut Microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut Microbial Metabolites as Multi-Kingdom Intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of Functional Suppression by CD4+CD25+ Regulatory T Cells in Patients with Multiple Sclerosis. J. Exp. Med. 2004, 199, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Trend, S.; Leffler, J.; Jones, A.P.; Cha, L.; Gorman, S.; Brown, D.A.; Breit, S.N.; Kermode, A.G.; French, M.A.; Ward, N.C.; et al. Associations of Serum Short-Chain Fatty Acids with Circulating Immune Cells and Serum Biomarkers in Patients with Multiple Sclerosis. Sci. Rep. 2021, 11, 5244. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.; Vige, R.; Calder, P.C. Review: The Nutritional Management of Multiple Sclerosis with Propionate. Front. Immunol. 2021, 12, 676016. [Google Scholar] [CrossRef] [PubMed]
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080.e16. [Google Scholar] [CrossRef]
- Shahi, S.K.; Freedman, S.N.; Mangalam, A.K. Gut Microbiome in Multiple Sclerosis: The Players Involved and the Roles They Play. Gut Microbes 2017, 8, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Habek, M.; Hojsak, I.; Brinar, V. v. Nutrition in Multiple Sclerosis. Clin. Neurol. Neurosurg. 2010, 112, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagur, M.J.; Antonia Murcia, M.; Jiménez-Monreal, A.M.; Tur, J.A.; Mar Bibiloni, M.; Alonso, G.L.; Martínez-Tomé, M. Influence of Diet in Multiple Sclerosis: A Systematic Review. Adv. Nutr. 2017, 8, 463–472. [Google Scholar] [CrossRef] [Green Version]
- von Geldern, G.; Mowry, E.M. The Influence of Nutritional Factors on the Prognosis of Multiple Sclerosis. Nat. Rev. Neurol. 2012, 8, 678–689. [Google Scholar] [CrossRef]
- Payne, A. Nutrition and Diet in the Clinical Management of Multiple Sclerosis. J. Hum. Nutr. Diet 2001, 14, 349–357. [Google Scholar] [CrossRef]
- Hedman, A.; Breithaupt, L.; Hübel, C.; Thornton, L.M.; Tillander, A.; Norring, C.; Birgegård, A.; Larsson, H.; Ludvigsson, J.F.; Sävendahl, L.; et al. Bidirectional Relationship between Eating Disorders and Autoimmune Diseases. J. Child Psychol. Psychiatry Allied Discip. 2019, 60, 803–812. [Google Scholar] [CrossRef]
- Wotton, C.J.; James, A.; Goldacre, M.J. Coexistence of Eating Disorders and Autoimmune Diseases: Record Linkage Cohort Study, UK. Int. J. Eat. Disord. 2016, 49, 663–672. [Google Scholar] [CrossRef]
- Zerwas, S.; Tidselbak Larsen, J.; Petersen, L.; Thornton, L.M.; Quaranta, M.; Vinkel Koch, S.; Pisetsky, D.; Bo Mortensen, P.; Bulik, C.M. Eating Disorders, Autoimmune, and Autoinflammatory Disease. Pediatrics 2017, 140, e20162089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raevuori, A.; Haukka, J.; Vaarala, O.; Suvisaari, J.M.; Gissler, M.; Grainger, M.; Linna, M.S.; Suokas, J.T. The Increased Risk for Autoimmune Diseases in Patients with Eating Disorders. PLoS ONE 2014, 9, e104845. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the Immune System in Humans from Infancy to Old Age. Proc. R. Soc. B Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, J.C.; Patisaul, H.B. Endocrine Disruptors and the Developing Immune System. Curr. Opin. Toxicol. 2018, 10, 31–36. [Google Scholar] [CrossRef]
- Holladay, S.D.; Smialowicz, R.J. Development of the Murine and Human Immune System: Differential Effects of Immunotoxicants Depend on Time of Exposure. Environ. Health Perspect. 2000, 108, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Jardine, L.; Gottgens, B.; Teichmann, S.A.; Haniffa, M. Prenatal Development of Human Immunity. Science 2020, 368, 600–603. [Google Scholar] [CrossRef]
- Marques, A.H.; O’Connor, T.G.; Roth, C.; Susser, E.; Bjørke-Monsen, A.L. The Influence of Maternal Prenatal and Early Childhood Nutrition and Maternal Prenatal Stress on Offspring Immune System Development and Neurodevelopmental Disorders. Front. Neurosci. 2013, 7, 120. [Google Scholar] [CrossRef] [Green Version]
- Munger, K.L.; Hongell, K.; Åivo, J.; Soilu-Hänninen, M.; Surcel, H.M.; Ascherio, A. 25-Hydroxyvitamin D Deficiency and Risk of MS among Women in the Finnish Maternity Cohort. Neurology 2017, 89, 1578–1583. [Google Scholar] [CrossRef]
- Cyprian, F.; Lefkou, E.; Varoudi, K.; Girardi, G. Immunomodulatory Effects of Vitamin D in Pregnancy and Beyond. Front. Immunol. 2019, 10, 2739. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, N.M.; Munger, K.L.; Koch-Henriksen, N.; Hougaard, D.M.; Magyari, M.; Jørgensen, K.T.; Lundqvist, M.; Simonsen, J.; Jess, T.; Cohen, A.; et al. Neonatal Vitamin D Status and Risk of Multiple Sclerosis: A Population-Based Case-Control Study. Neurology 2017, 88, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Salle, B.L.; Delvin, E.E.; Lapillonne, A.; Bishop, N.J.; Glorieux, F.H. Perinatal Metabolism of Vitamin D. Am. J. Clin. Nutr. 2000, 71, 1317S–1324S. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, F.; Michels, K.B.; Munger, K.; O’Reilly, E.; Chitnis, T.; Forman, M.R.; Giovannucci, E.; Rosner, B.; Ascherio, A. Gestational Vitamin D and the Risk of Multiple Sclerosis in Offspring. Ann. Neurol. 2011, 70, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of Glucocorticoid Negative Feedback in the Regulation of HPA Axis Pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.L. Plasma Steroid-Binding Proteins: Primary Gatekeepers of Steroid Hormone Action. J. Endocrinol. 2016, 230, R13–R25. [Google Scholar] [CrossRef] [Green Version]
- Chapman, K.; Holmes, M.; Seckl, J. 11-Hydroxysteroid Dehydrogenases: Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [Green Version]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid Receptor Control of Transcription: Precision and Plasticity via Allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef]
- Edwards, P.D.; Boonstra, R. Glucocorticoids and CBG during Pregnancy in Mammals: Diversity, Pattern, and Function. Gen. Comp. Endocrinol. 2018, 259, 122–130. [Google Scholar] [CrossRef]
- Solano, M.E.; Arck, P.C. Steroids, Pregnancy and Fetal Development. Front. Immunol. 2020, 10, 3017. [Google Scholar] [CrossRef]
- Panton, K.K.; Mikkelsen, G.; Irgens, W.Ø.; Hovde, A.K.; Killingmo, M.W.; Øien, M.A.; Thorsby, P.M.; Åsberg, A. New Reference Intervals for Cortisol, Cortisol Binding Globulin and Free Cortisol Index in Women Using Ethinyl Estradiol. Scand. J. Clin. Lab. Investig. 2019, 79, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.A.; Bales, N.J.; Myers, S.A.; Bautista, A.I.; Roueinfar, M.; Hale, T.M.; Handa, R.J. The Hypothalamic-Pituitary-Adrenal Axis: Development, Programming Actions of Hormones, and Maternal-Fetal Interactions. Front. Behav. Neurosci. 2021, 14, 601939. [Google Scholar] [CrossRef] [PubMed]
- Duthie, L.; Reynolds, R.M. Changes in the Maternal Hypothalamic-Pituitary-Adrenal Axis in Pregnancy and Postpartum: Influences on Maternal and Fetal Outcomes. Neuroendocrinology 2013, 98, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune Regulation by Glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Chen, T.; Liu, H.X.; Yan, H.Y.; Wu, D.M.; Ping, J. Developmental Origins of Inflammatory and Immune Diseases. Mol. Hum. Reprod. 2016, 22, 558–565. [Google Scholar] [CrossRef]
- Solano, M.E.; Holmes, M.C.; Mittelstadt, P.R.; Chapman, K.E.; Tolosa, E. Antenatal Endogenous and Exogenous Glucocorticoids and Their Impact on Immune Ontogeny and Long-Term Immunity. Semin. Immunopathol. 2016, 38, 739–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, H.; Siebler, T.; Shalet, S.M.; Williams, G.R. Interactions between GH, IGF-I, Glucocorticoids, and Thyroid Hormones during Skeletal Growth. Pediatric Res. 2002, 52, 137–147. [Google Scholar] [CrossRef]
- Hoshiro, M.; Ohno, Y.; Masaki, H.; Iwase, H.; Aoki, N. Comprehensive Study of Urinary Cortisol Metabolites in Hyperthyroid and Hypothyroid Patients. Clin. Endocrinol. 2006, 64, 37–45. [Google Scholar] [CrossRef]
- Klecha, A.J.; Genaro, A.M.; Lysionek, A.E.; Caro, R.A.; Coluccia, A.G.; Cremaschi, G.A. Experimental Evidence Pointing to the Bidirectional Interaction between the Immune System and the Thyroid Axis. Int J Immunopharmacol. 2000, 22, 491–500. [Google Scholar] [CrossRef]
- Foster, M.P.; Montecino-Rodriguez, E.; Dorshkind, K. Proliferation of Bone Marrow Pro-B Cells Is Dependent on Stimulation by the Pituitary/Thyroid Axis. J. Immunol. 1999, 163, 5883–5890. [Google Scholar]
- Arpin, C.; Pihlgren, M.; Fraichard, A.; Aubert, D.; Samarut, J.; Chassande, O.; Marvel, J. Effects of T3Rα1 and T3Rα2 Gene Deletion on T and B Lymphocyte Development. J. Immunol. 2000, 164, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Zakharova, L.A. Cross-Regulation in Development of Neuroendocrine and Immune Systems. Russ. J. Dev. Biol. 2010, 41, 347–356. [Google Scholar] [CrossRef]
- Lam, S.H.; Sin, Y.M.; Gong, Z.; Lam, T.J. Effects of Thyroid Hormone on the Development of Immune System in Zebrafish. Gen. Comp. Endocrinol. 2005, 142, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Jara, E.L.; Muñoz-Durango, N.; Llanos, C.; Fardella, C.; González, P.A.; Bueno, S.M.; Kalergis, A.M.; Riedel, C.A. Modulating the Function of the Immune System by Thyroid Hormones and Thyrotropin. Immunol. Lett. 2017, 184, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, M.; Pellizas, C. Thyroid Hormone Action on Innate Immunity. Front. Endocrinol. 2019, 10, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, A.A.; Fournier, M.; Bernier, J.; Cyr, D.G. Neonatal Exposure to Propylthiouracil Induces a Shift in Lymphoid Cell Sub-Populations in the Developing Postnatal Male Rat Spleen and Thymus. Cell. Immunol. 2003, 223, 91–102. [Google Scholar] [CrossRef]
- de Vito, P.; Incerpi, S.; Pedersen, J.Z.; Luly, P.; Davis, F.B.; Davis, P.J. Thyroid Hormones as Modulators of Immune Activities at the Cellular Level. Thyroyd 2011, 21, 879–890. [Google Scholar] [CrossRef]
- Pirahanchi, Y.; Tariq, M.A.; Jialal, I. Physiology, Thyroid; StatPearls: Treasure Island, FL, USA, 2021; pp. 6–11. [Google Scholar]
- Ortiga-Carvalho, T.M.; Chiamolera, M.I.; Pazos-Moura, C.C.; Wondisford, F.E. Hypothalamus-Pituitary-Thyroid Axis. Compr. Physiol. 2016, 6, 1387–1428. [Google Scholar] [CrossRef]
- Walter, K.M.; Dach, K.; Hayakawa, K.; Giersiefer, S.; Heuer, H.; Lein, P.J.; Fritsche, E. Ontogenetic Expression of Thyroid Hormone Signaling Genes: An in Vitro and in Vivo Species Comparison. PLoS ONE 2019, 14, e0221230. [Google Scholar] [CrossRef]
- Schroeder AC, P.ML. Thyroid Hormones, T3 AndT4, in the Brain. Front. Endocrinol. 2014, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Rousset, B.; Dupuy, C.; Miot, F.; Dumont, J. Chapter 2 Thyroid Hormone Synthesis and Secretion. In Endotex; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; pp. 1–52. [Google Scholar]
- Liu, Y.Y.; Milanesi, A.; Brent, G.A. Thyroid Hormones. In Hormonal Signaling in Biology and Medicine: Comprehensive Modern Endocrinology, 1st ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 487–506. [Google Scholar] [CrossRef]
- Giammanco, M.; Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. Int. J. Mol. Sci. 2020, 21, 4140. [Google Scholar] [CrossRef]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic Actions of Thyroid Hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Springer, D.; Jiskra, J.; Limanova, Z.; Zima, T.; Potlukova, E. Thyroid in Pregnancy: From Physiology to Screening. Crit. Rev. Clin. Lab. Sci. 2017, 54, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Jameson, J.; Fauci, A.; Kasper, D.; Hauser, S.; Longo, D.; Loscalzo, J. Harrison’s Principles of Internal Medicine; McGraw Hill: New York, NY, USA, 2018. [Google Scholar]
- Calvo, R.M.; Jauniaux, E.; Gulbis, B.; Asunción, M.; Gervy, C.; Contempré, B.; de Escobar, G.M. Fetal Tissues Are Exposed to Biologically Relevant Free Thyroxine Concentrations during Early Phases of Development. J. Clin. Endocrinol. Metab. 2002, 87, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Negro, R.; Soldin, O.P.; Obregon, M.J.; Stagnaro-Green, A. Hypothyroxinemia and Pregnancy. Endocr. Pract. 2011, 17, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Kwon, E.J.; Kim, Y.J. What Is Fetal Programming?: A Lifetime Health Is under the Control of in Utero Health. Obstet. Gynecol. Sci. 2017, 60, 506–519. [Google Scholar] [CrossRef]
- Moog, N.K.; Entringer, S.; Heim, C.; Wadhwa, P.D.; Kathmann, N. Influence of Maternal Thyroid Hormones during Gestation on Fetal Brain Development. Neuroscience 2017, 342, 68–100. [Google Scholar] [CrossRef] [Green Version]
- Prezioso, G.; Giannini, C.; Chiarelli, F. Effect of Thyroid Hormones on Neurons and Neurodevelopment. Horm. Res. Paediatr. 2018, 90, 73–81. [Google Scholar] [CrossRef]
- Rossi, C.; Cicalini, I.; Zucchelli, M.; di Ioia, M.; Onofrj, M.; Federici, L.; del Boccio, P.; Pieragostino, D. Metabolomic Signature in Sera of Multiple Sclerosis Patients during Pregnancy. Int. J. Mol. Sci. 2018, 19, 3589. [Google Scholar] [CrossRef] [Green Version]
- Benvenga, S.; Elia, G.; Ragusa, F.; Paparo, S.R.; Sturniolo, M.M.; Ferrari, S.M.; Antonelli, A.; Fallahi, P. Endocrine Disruptors and Thyroid Autoimmunity. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101377. [Google Scholar] [CrossRef]
- Decallonne, B.; Bartholomé, E.; Delvaux, V.; D’haeseleer, M.; el Sankari, S.; Seeldrayers, P.; van Wijmeersch, B.; Daumerie, C. Thyroid Disorders in Alemtuzumab-Treated Multiple Sclerosis Patients: A Belgian Consensus on Diagnosis and Management. Acta Neurol. Belg. 2018, 118, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Sahay RK, N.VS. Hypothyroidism in Pregnancy. Indian J. Endocrinol. Metab. 2021, 16, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Chaker, L.; Bianco, A.C.; Jonklaas, J.; Peeters, R.P. Hypothyroidism. Lancet 2017, 390, 1550–1562. [Google Scholar] [CrossRef]
- López-Muñoz, E.; Mateos-Sánchez, L.; Mejía-Terrazas, G.E.; Bedwell-Cordero, S.E. Hypothyroidism and Isolated Hypothyroxinemia in Pregnancy, from Physiology to the Clinic. Taiwan. J. Obstet. Gynecol. 2019, 58, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.O.; Loureiro, S.M.A.; Alves, I.G.N.; de Jesus, C.S.; dos Santos, P.R.; dos Santos, M.R.V.; Dias, D.P.M.; Santana-Filho, V.J.; Badauê-Passos, D. Experimental Gestational Hypothyroidism Evokes Hypertension in Adult Offspring Rats. Auton. Neurosci. Basic Clin. 2012, 170, 36–41. [Google Scholar] [CrossRef]
- Albornoz, E.A.; Carreño, L.J.; Cortes, C.M.; Gonzalez, P.A.; Cisternas, P.A.; Cautivo, K.M.; Catalán, T.P.; Opazo, M.C.; Eugenin, E.A.; Berman, J.W.; et al. Gestational Hypothyroidism Increases the Severity of Experimental Autoimmune Encephalomyelitis in Adult Offspring. Thyroid 2013, 23, 1627–1637. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.G. Maternal Hypothyroidism and Multiple Sclerosis: Disruption the Developing Neuroendocrine System. Austin J. Mult. Scler. Neuroimmunol. 2018, 4, 1030. [Google Scholar]
- Alexander, E.K.; Pearce, E.N.; Brent, G.A.; Brown, R.S.; Chen, H.; Dosiou, C.; Grobman, W.A.; Laurberg, P.; Lazarus, J.H.; Mandel, S.J.; et al. 2017 Guidelines of the American Thyroid Association for the Diagnosis and Management of Thyroid Disease during Pregnancy and the Postpartum. Thyroid 2017, 27, 315–389. [Google Scholar] [CrossRef] [Green Version]
- Min, H.; Dong, J.; Wang, Y.; Wang, Y.; Teng, W.; Xi, Q.; Chen, J. Maternal Hypothyroxinemia-Induced Neurodevelopmental Impairments in the Progeny. Mol. Neurobiol. 2016, 53, 1613–1624. [Google Scholar] [CrossRef]
- Furnica, R.M.; Lazarus, J.H.; Gruson, D.; Daumerie, C. Update on a New Controversy in Endocrinology: Isolated Maternal Hypothyroxinemia. J. Endocrinol. Invest. 2015, 38, 117–123. [Google Scholar] [CrossRef]
- Haensgen, H.; Albornoz, E.; Opazo, M.C.; Bugueño, K.; Jara Fernández, E.L.; Binzberger, R.; Rivero-Castillo, T.; Venegas Salas, L.F.; Simon, F.; Cabello-Verrugio, C.; et al. Gestational Hypothyroxinemia Affects Its Offspring with a Reduced Suppressive Capacity Impairing the Outcome of the Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 1257. [Google Scholar] [CrossRef] [Green Version]
- Nieto, P.A.; Peñaloza, H.F.; Salazar-Echegarai, F.J.; Castellanos, R.M.; Opazo, M.C.; Venegas, L.; Padilla, O.; Kalergis, A.M.; Riedel, C.A.; Bueno, S.M. Gestational Hypothyroidism Improves the Ability of the Female Offspring to Clear Streptococcus Pneumoniae Infection and to Recover from Pneumococcal Pneumonia. Endocrinology 2016, 157, 2217–2228. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Madrid, E.; Rangel-Ramírez, M.A.; Mendoza-León, M.J.; Álvarez-Mardones, O.; González, P.A.; Kalergis, A.M.; Opazo, M.C.; Riedel, C.A. Risk Factors from Pregnancy to Adulthood in Multiple Sclerosis Outcome. Int. J. Mol. Sci. 2022, 23, 7080. https://doi.org/10.3390/ijms23137080
González-Madrid E, Rangel-Ramírez MA, Mendoza-León MJ, Álvarez-Mardones O, González PA, Kalergis AM, Opazo MC, Riedel CA. Risk Factors from Pregnancy to Adulthood in Multiple Sclerosis Outcome. International Journal of Molecular Sciences. 2022; 23(13):7080. https://doi.org/10.3390/ijms23137080
Chicago/Turabian StyleGonzález-Madrid, Enrique, Ma. Andreina Rangel-Ramírez, María José Mendoza-León, Oscar Álvarez-Mardones, Pablo A. González, Alexis M. Kalergis, Ma. Cecilia Opazo, and Claudia A. Riedel. 2022. "Risk Factors from Pregnancy to Adulthood in Multiple Sclerosis Outcome" International Journal of Molecular Sciences 23, no. 13: 7080. https://doi.org/10.3390/ijms23137080
APA StyleGonzález-Madrid, E., Rangel-Ramírez, M. A., Mendoza-León, M. J., Álvarez-Mardones, O., González, P. A., Kalergis, A. M., Opazo, M. C., & Riedel, C. A. (2022). Risk Factors from Pregnancy to Adulthood in Multiple Sclerosis Outcome. International Journal of Molecular Sciences, 23(13), 7080. https://doi.org/10.3390/ijms23137080