
Draft Genome of Tanacetum Coccineum: Genomic Comparison of Closely Related Tanacetum-Family Plants


Abstract
:1. Introduction
2. Results and Discussion
2.1. Sequencing of the T. coccineum Genome
2.2. Size Estimation of the T. coccineum Genome
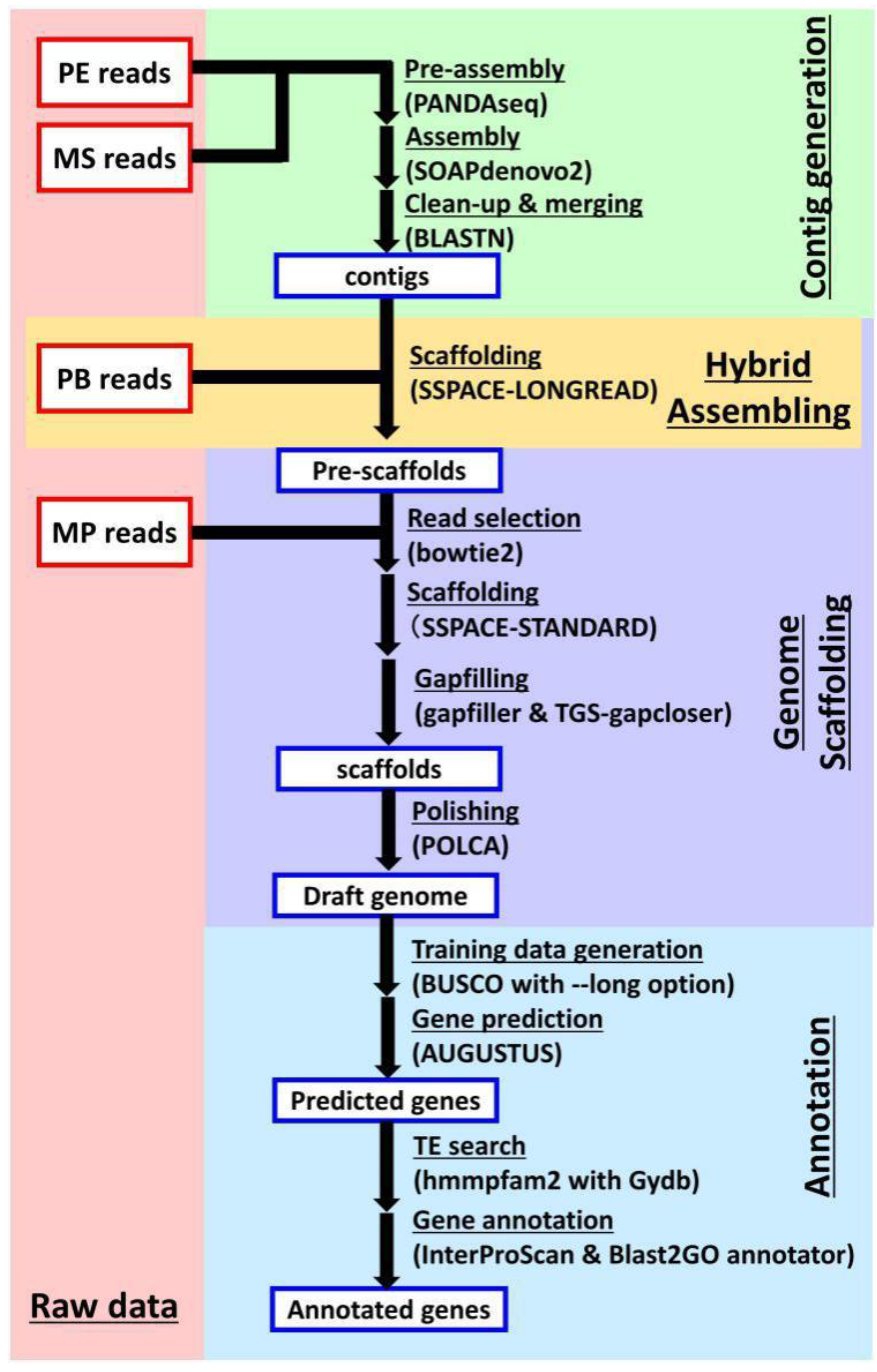
2.3. Sequence Assembly and Annotation of the T. coccineum Genome
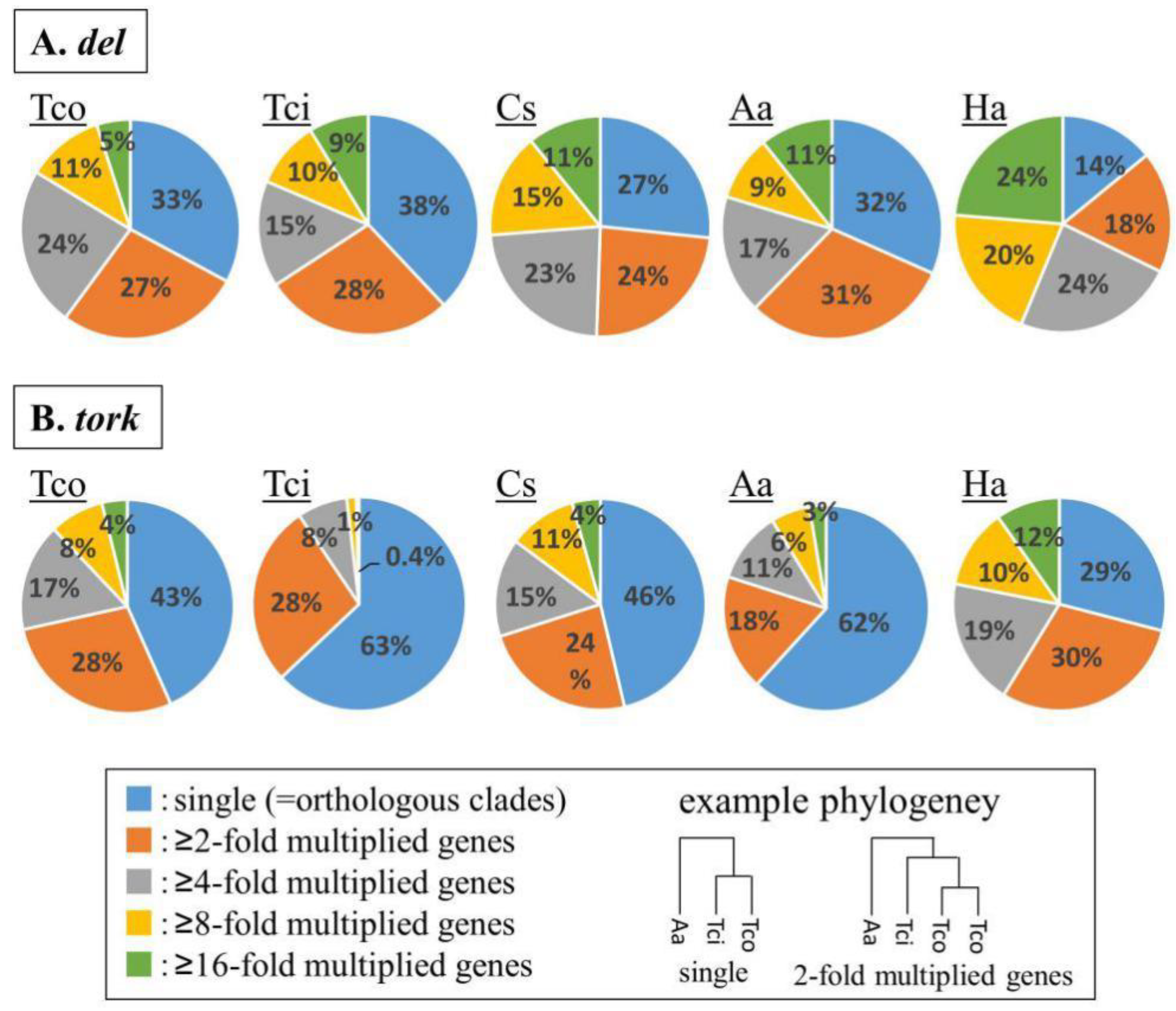
2.4. Inter-Genus Comparative Analysis of TE Classification
2.5. Pyrethrin-Related Enzymes Encoded in the T. coccineum Genome
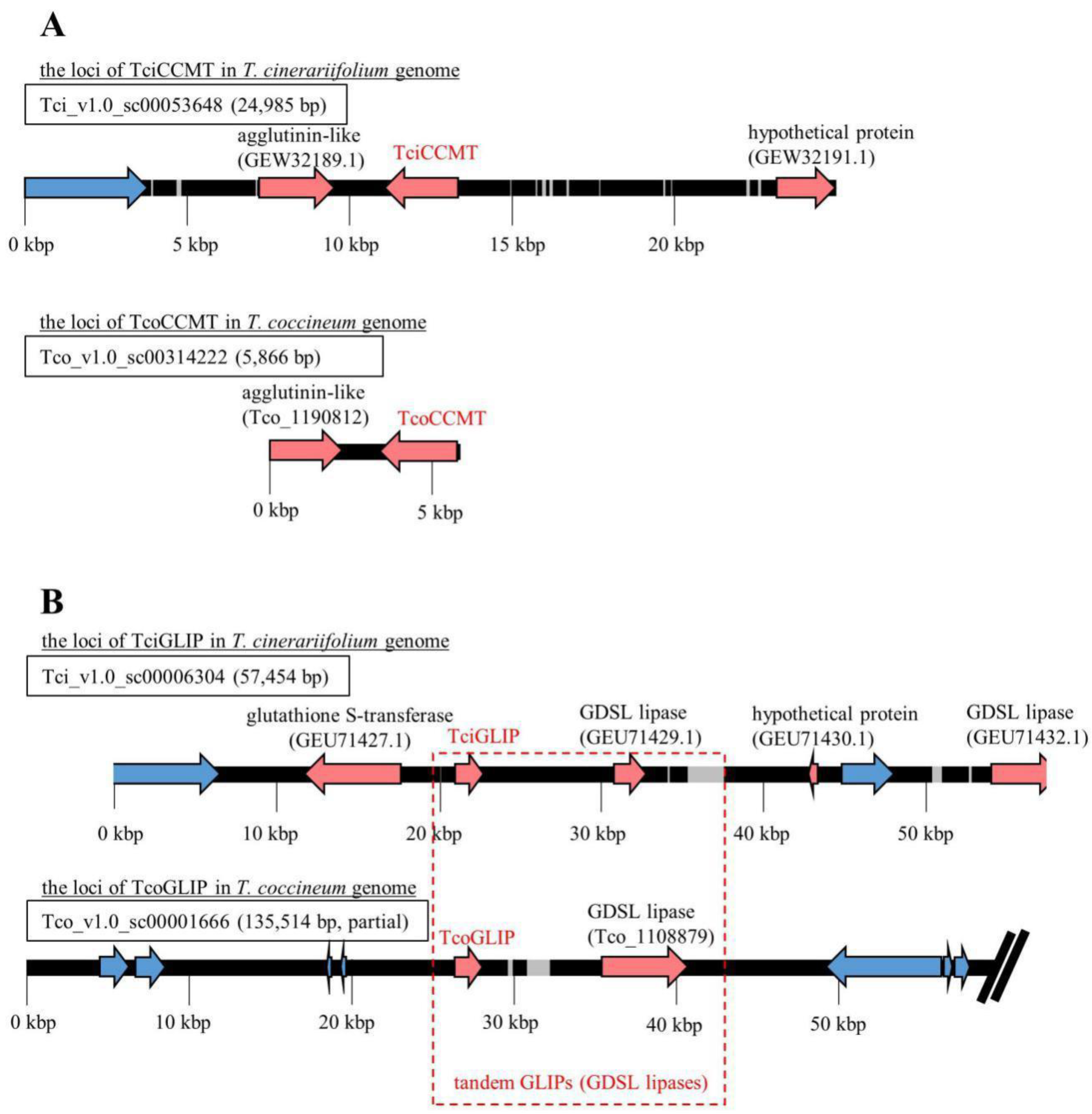
2.6. Synteny Analysis of Genes Encoding Pyrethrin-Related Enzymes
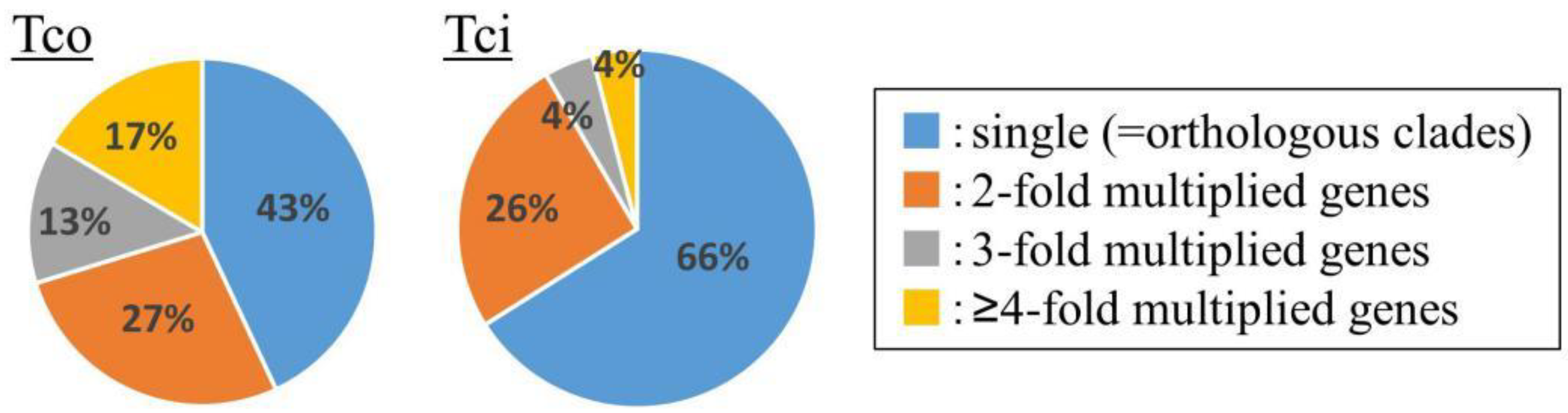
2.7. Functional Annotation of the T. coccineum Genes and Inter-Genus Comparative Analysis
3. Materials and Methods
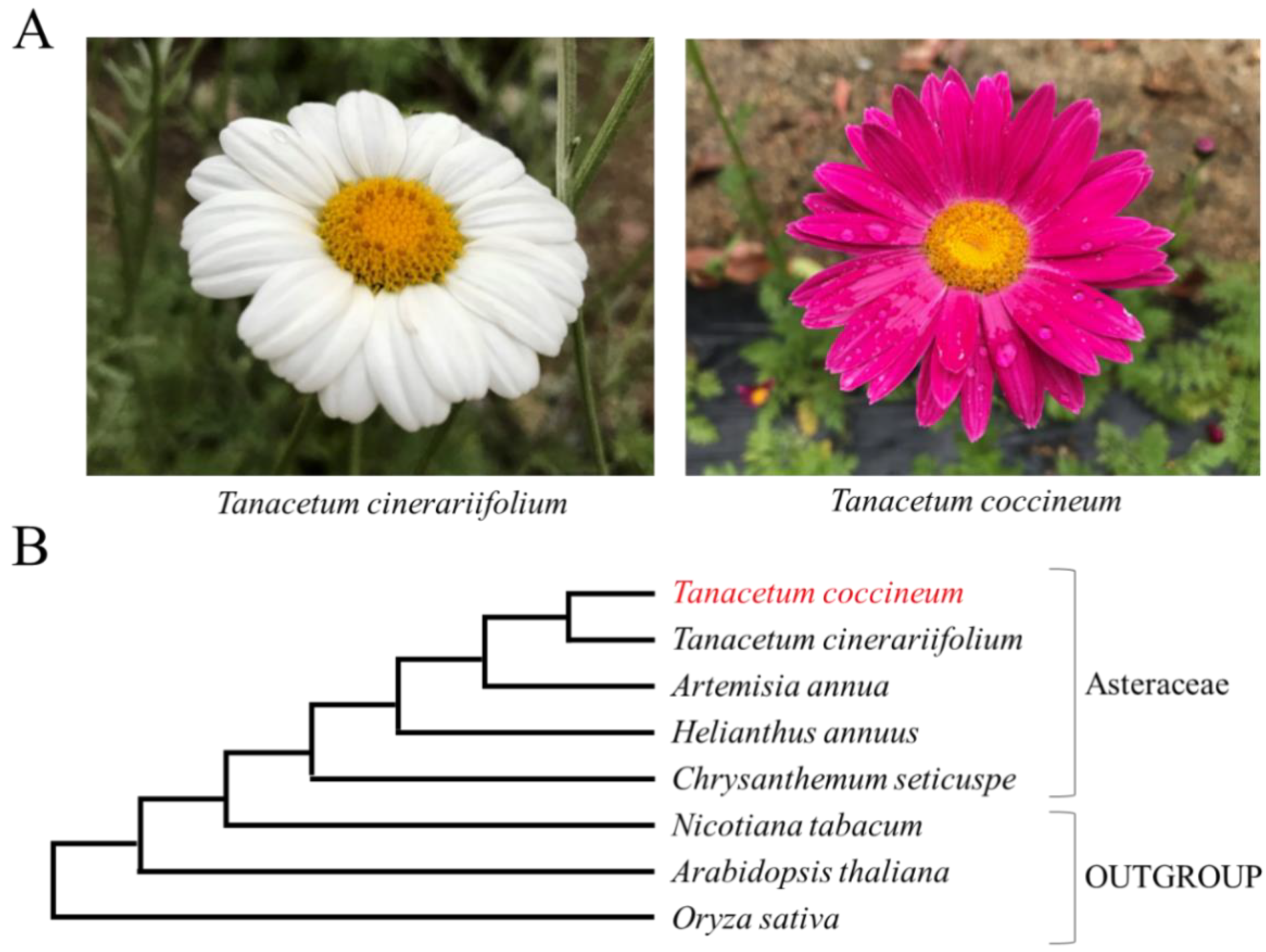
3.1. Phylogenetic Analysis of Plants in This Paper
3.2. Plant Materials and Genome Sequencing
3.3. Genome Size Estimation Using Flow Cytometry
3.4. Genome Size Estimation Using K-Mer Depth Information
3.5. De Novo Assembly of Genome Sequences
3.6. Gene Prediction and Annotation
3.7. Comparative Analysis of TE Content Versus That in Other Plants
3.8. Homology Search and Synteny Analysis of Genes Encoding Pyrethrin-Related Enzymes
3.9. Comparative Analysis of Protein Superfamily Content Versus That in Other Plants
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lybrand, D.B.; Xu, H.; Last, R.L.; Pichersky, E. How Plants Synthesize Pyrethrins: Safe and Biodegradable Insecticides. Trends Plant Sci. 2020, 25, 1240–1251. [Google Scholar] [CrossRef]
- Yamashiro, T.; Shiraishi, A.; Satake, H.; Nakayama, K. Draft genome of Tanacetum cinerariifolium, the natural source of mosquito coil. Sci. Rep. 2019, 9, 18249. [Google Scholar] [CrossRef]
- Kikuta, Y.; Ueda, H.; Nakayama, K.; Katsuda, Y.; Ozawa, R.; Takabayashi, J.; Hatanaka, A.; Matsuda, K. Specific regulation of pyrethrin biosynthesis in Chrysanthemum cinerariaefolium by a blend of volatiles emitted from artificially damaged conspecific plants. Plant Cell Physiol. 2011, 52, 588–596. [Google Scholar] [CrossRef]
- Katsuda, Y. Progress and future of pyrethroids. Top. Curr. Chem. 2012, 314, 1–30. [Google Scholar] [CrossRef]
- Nakano, M.; Taniguchi, K.; Masuda, Y.; Kozuka, T.; Aruga, Y.; Han, J.; Motohara, K.; Nakata, M.; Sumitomo, K.; Hisamatsu, T.; et al. A pure line derived from a self-compatible Chrysanthemum seticuspe mutant as a model strain in the genus Chrysanthemum. Plant Sci. 2019, 287, 110174. [Google Scholar] [CrossRef]
- Bennett, M.D.; Smith, J.B.; Heslop-Harrison, J.S. Nuclear DNA amounts in angiosperms. Proc. R. Soc. Lond B 1982, 216, 179–199. [Google Scholar] [CrossRef]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Alm Rosenblad, M.; Abramova, A.; Lind, U.; Olason, P.; Giacomello, S.; Nystedt, B.; Blomberg, A. Genomic Characterization of the Barnacle Balanus improvisus Reveals Extreme Nucleotide Diversity in Coding Regions. Mar. Biotechnol. 2021, 23, 402–416. [Google Scholar] [CrossRef]
- Williams, D.; Trimble, W.L.; Shilts, M.; Meyer, F.; Ochman, H. Rapid quantification of sequence repeats to resolve the size, structure and contents of bacterial genomes. BMC Genom. 2013, 14, 537. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Boetzer, M.; Pirovano, W. SSPACE-LongRead: Scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform. 2014, 15, 211. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13 (Suppl. 14), S8. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L.; et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 2020, 9, giaa094. [Google Scholar] [CrossRef]
- Zimin, A.V.; Salzberg, S.L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 2020, 16, e1007981. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Llorens, C.; Futami, R.; Covelli, L.; Dominguez-Escriba, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodriguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, D70–D74. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Moghe, G.D.; Wiegert-Rininger, K.; Schilmiller, A.L.; Barry, C.S.; Last, R.L.; Pichersky, E. Coexpression Analysis Identifies Two Oxidoreductases Involved in the Biosynthesis of the Monoterpene Acid Moiety of Natural Pyrethrin Insecticides in Tanacetum cinerariifolium. Plant Physiol. 2018, 176, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Li, W.; Schilmiller, A.L.; van Eekelen, H.; de Vos, R.C.H.; Jongsma, M.A.; Pichersky, E. Pyrethric acid of natural pyrethrin insecticide: Complete pathway elucidation and reconstitution in Nicotiana benthamiana. New Phytol. 2019, 223, 751–765. [Google Scholar] [CrossRef]
- Rivera, S.B.; Swedlund, B.D.; King, G.J.; Bell, R.N.; Hussey, C.E., Jr.; Shattuck-Eidens, D.M.; Wrobel, W.M.; Peiser, G.D.; Poulter, C.D. Chrysanthemyl diphosphate synthase: Isolation of the gene and characterization of the recombinant non-head-to-tail monoterpene synthase from Chrysanthemum cinerariaefolium. Proc. Natl. Acad. Sci. USA 2001, 98, 4373–4378. [Google Scholar] [CrossRef] [Green Version]
- Kikuta, Y.; Ueda, H.; Takahashi, M.; Mitsumori, T.; Yamada, G.; Sakamori, K.; Takeda, K.; Furutani, S.; Nakayama, K.; Katsuda, Y.; et al. Identification and characterization of a GDSL lipase-like protein that catalyzes the ester-forming reaction for pyrethrin biosynthesis in Tanacetum cinerariifolium—A new target for plant protection. Plant J. 2012, 71, 183–193. [Google Scholar] [CrossRef]
- Li, W.; Zhou, F.; Pichersky, E. Jasmone Hydroxylase, a Key Enzyme in the Synthesis of the Alcohol Moiety of Pyrethrin Insecticides. Plant Physiol. 2018, 177, 1498–1509. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.M.; Yang, T.; Bouwmeester, H.J.; Jongsma, M.A. A trichome-specific linoleate lipoxygenase expressed during pyrethrin biosynthesis in pyrethrum. Lipids 2013, 48, 1005–1015. [Google Scholar] [CrossRef]
- Li, W.; Lybrand, D.B.; Zhou, F.; Last, R.L.; Pichersky, E. Pyrethrin Biosynthesis: The Cytochrome P450 Oxidoreductase CYP82Q3 Converts Jasmolone To Pyrethrolone. Plant Physiol. 2019, 181, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Bolognesi, A.; Bortolotti, M.; Maiello, S.; Battelli, M.G.; Polito, L. Ribosome-Inactivating Proteins from Plants: A Historical Overview. Molecules 2016, 21, 1627. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Zhou, Y.K.; Ji, Z.L.; Chen, X.R. The Plant Ribosome-Inactivating Proteins Play Important Roles in Defense against Pathogens and Insect Pest Attacks. Front. Plant Sci. 2018, 9, 146. [Google Scholar] [CrossRef] [Green Version]
- Shahidi-Noghabi, S.; Van Damme, E.J.; Smagghe, G. Carbohydrate-binding activity of the type-2 ribosome-inactivating protein SNA-I from elderberry (Sambucus nigra) is a determining factor for its insecticidal activity. Phytochemistry 2008, 69, 2972–2978. [Google Scholar] [CrossRef]
- Shakeel, S.N.; Wang, X.; Binder, B.M.; Schaller, G.E. Mechanisms of signal transduction by ethylene: Overlapping and non-overlapping signalling roles in a receptor family. AoB Plants 2013, 5, plt010. [Google Scholar] [CrossRef] [Green Version]
- Li, K.B. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics 2003, 19, 1585–1586. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Sumitomo, K.; Hisamatsu, T.; Nagano, S.; Shirasawa, K.; Higuchi, Y.; Kusaba, M.; Koshioka, M.; Nakano, Y.; Yagi, M.; et al. De novo whole-genome assembly in Chrysanthemum seticuspe, a model species of Chrysanthemums, and its application to genetic and gene discovery analysis. DNA Res. 2019, 26, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Sierro, N.; Battey, J.N.; Ouadi, S.; Bakaher, N.; Bovet, L.; Willig, A.; Goepfert, S.; Peitsch, M.C.; Ivanov, N.V. The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 2014, 5, 3833. [Google Scholar] [CrossRef]
- Rice Annotation, P.; Tanaka, T.; Antonio, B.A.; Kikuchi, S.; Matsumoto, T.; Nagamura, Y.; Numa, H.; Sakai, H.; Wu, J.; Itoh, T.; et al. The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008, 36, D1028–D1033. [Google Scholar] [CrossRef] [Green Version]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Briere, C.; Owens, G.L.; Carrere, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Zhang, L.; Liao, Z.; Wang, S.; Yan, T.; Shi, P.; Liu, M.; Fu, X.; Pan, Q.; Wang, Y.; et al. The Genome of Artemisia annua Provides Insight into the Evolution of Asteraceae Family and Artemisinin Biosynthesis. Mol. Plant 2018, 11, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Inoue, J.; Satoh, N. ORTHOSCOPE: An Automatic Web Tool for Phylogenetically Inferring Bilaterian Orthogroups with User-Selected Taxa. Mol. Biol. Evol. 2019, 36, 621–631. [Google Scholar] [CrossRef]
- Shiraishi, A.; Okuda, T.; Miyasaka, N.; Osugi, T.; Okuno, Y.; Inoue, J.; Satake, H. Repertoires of G protein-coupled receptors for Ciona-specific neuropeptides. Proc. Natl. Acad. Sci. USA 2019, 116, 7847–7856. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library | Insert Size (bp) | Read Length (Bases) | Number of Reads | Total Read Length (Bases) |
|---|---|---|---|---|
| PE | 350 | 151 | 5,732,398,372 | 854,270,829,961 |
| MP-3kb | 3000 | 151 | 698,859,570 | 99,096,491,543 |
| MP-5kb | 5000 | 151 | 750,513,382 | 107,931,325,410 |
| MP-8kb | 8000 | 151 | 762,709,822 | 109,359,355,641 |
| MS | 550 | 301 | 97,731,712 | 26,503,089,921 |
| PB | Ave. 10,738 | 8,670,092 | 93,100,193,428 |
| Contigs | Scaffolds (before Gapfilling) | Draft Genome | |
|---|---|---|---|
| Total number of sequence fragments | 6,500,576 | 3,061,809 | 2,836,647 |
| Total length (bp) | 8,565,698,618 | 9,395,951,224 | 9,463,677,832 |
| N50 (bp) | 8465 | 25,397 | 27,784 |
| Length of longest contig (bp) | 149,916 | 329,693 | 331,286 |
| Gaps (bp) | 0 | 777,041,487 | 724,210,424 |
| GC content (%) | 34.9 | 35.1 | 35.1 |
| Number of Predicted Genes | 1,582,136 |
| BUSCO v5 | * C: 92.7% (Single: 70.8%, Duplicated: 21.9%) F: 5.1% M: 2.2% |
| Number of predicted TEs | 772,794 |
| Number of predicted genes encoding products with known protein signatures | 103,680 |
| Rank | Tco | Tci | Cs | Aa | Ha | Nt | Os | At |
|---|---|---|---|---|---|---|---|---|
| 1 | sire (25.7) | sire (33.0) | sire (32.0) | sire (21.8) | del (37.7) | del (40.4) | tat (11.4) | athila (9.54) |
| 2 | del (15.3) | athila (17.0) | athila (10.9) | athila (19.6) | sire (9.85) | tat (20.5) | retroviridae (8.97) | retroviridae (4.89) |
| 3 | athila (12.5) | del (12.0) | oryco (5.11) | del (6.57) | lentiviridae (8.72) | athila (9.87) | del (8.39) | caulimovirus (4.15) |
| 4 | oryco (7.25) | oryco (6.34) | lentiviridae (5.06) | oryco (4.59) | tat (6.76) | sire (3.02) | tork (4.73) | badnavirus (4.05) |
| 5 | tork (4.70) | lentiviridae (4.92) | del (5.03) | tork (4.01) | athila (5.17) | tork (2.80) | alpharetroviridae (4.66) | tork (3.08) |
| Known Pyrethrin-Related Enzymes | Corresponding Proteins of T. coccineum | Protein Sequence Similarity |
|---|---|---|
| TciADH2 | Tco_0487905 | Identities = 340/378 (90%), Positives = 359/378 (95%), Gaps = 2/378 (1%) |
| TciALDH1 | Tco_0682217 | Identities = 448/499 (90%), Positives = 471/499 (94%), Gaps = 1/499 (0%) |
| TciCCH | Tco_0360514 | Identities = 470/498 (94%), Positives = 484/498 (97%), Gaps = 1/498 (0%) |
| TciCCMT | Tco_1190813 | Identities = 358/374 (96%), Positives = 361/374 (97%), Gaps = 5/374 (1%) |
| TciCDS | Tco_1315810 | Identities = 358/395 (91%), Positives = 374/395 (95%), Gaps = 0/395 (0%) |
| TciGLIP | Tco_1108878 | Identities = 337/365 (92%), Positives = 348/365 (95%), Gaps = 0/365 (0%) |
| TciJMH | Tco_0572988 | Identities = 450/512 (88%), Positives = 479/512 (94%), Gaps = 2/512 (0%) |
| TciLOX1 | Tco_0863779 | Identities = 847/861 (98%), Positives = 853/861 (99%), Gaps = 0/861 (0%) |
| TciPYS | Tco_1240348 | Identities = 465/488 (95%), Positives = 475/488 (97%), Gaps = 0/488 (0%) |
| Category | IPR ID | Superfamily Name | Tco | Tci | Cs | Aa | Ha | Nt | Os | At |
|---|---|---|---|---|---|---|---|---|---|---|
| Biodefense | IPR036041 | Ribosome-inactivating protein | 1.96 (159) | 1.29 (98) | −1.81 (7) | −1.00 (16) | −3.07 (0) | −3.07 (0) | −0.94 (17) | −3.07 (0) |
| Metabolism | IPR005848 | Urease, alpha subunit | 2.36 (108) | −0.14 (15) | −1.87 (1) | −1.46 (3) | −1.87 (1) | −0.87 (7) | −2.14 (0) | −1.87 (1) |
| Metabolism | IPR036226 | Lipoxygenase, C-terminal domain | 1.86 (232) | 0.48 (86) | −0.22 (51) | −0.82 (32) | −1.12 (25) | −0.67 (36) | −1.86 (13) | −2.44 (7) |
| Metabolism | IPR033966 | RuBisCO | 1.60 (42) | −0.25 (8) | 0.05 (11) | −0.15 (9) | −1.37 (1) | −0.95 (3) | −0.25 (8) | −1.15 (2) |
| Metabolism | IPR032466 | Metal-dependent hydrolase | 1.48 (166) | 0.69 (94) | −1.13 (23) | −0.89 (28) | −0.32 (44) | −0.05 (54) | −1.61 (15) | −0.98 (26) |
| Metabolism | IPR036849 | Enolase-like, C-terminal domain | 1.38 (71) | 0.91 (50) | −0.62 (14) | −0.55 (15) | −0.78 (12) | −0.29 (19) | −1.41 (6) | −1.29 (7) |
| Metabolism | IPR036396 | Cytochrome P450 | 0.90 (1220) | 0.19 (745) | 0.16 (732) | 0.07 (688) | −0.20 (568) | −0.12 (600) | −1.05 (314) | −0.85 (361) |
| Signaling | IPR035983 | HECT, E3 ligase catalytic domain | 1.22 (95) | 0.84 (72) | 0.10 (41) | −0.52 (25) | −0.90 (18) | −0.21 (32) | −1.84 (7) | −1.25 (13) |
| Category | IPR ID | Superfamily Name | Tco | Tci | Cs | Aa | Ha | Nt | Os | At |
|---|---|---|---|---|---|---|---|---|---|---|
| Signaling | IPR039512 | RCHY1, zinc-ribbon | −1.18 (3) | −0.48 (8) | −0.09 (12) | −0.09 (12) | −0.01 (13) | 1.28 (39) | −0.72 (6) | −0.09 (12) |
| Category | IPR ID | Superfamily Name | Tco | Tci | Cs | Aa | Ha | Nt | Os | At |
|---|---|---|---|---|---|---|---|---|---|---|
| Biodefense | IPR035992 | Ricin B-like lectins | 0.81 (44) | 1.41 (69) | −0.34 (17) | −0.05 (22) | −1.22 (7) | −0.80 (11) | −1.10 (8) | −1.48 (5) |
| Biodefense | IPR036861 | Endochitinase-like | −0.13 (7) | −1.13 (1) | −0.39 (5) | −0.25 (6) | 0.53 (14) | 0.29 (11) | 0.09 (9) | 0.37 (12) |
| Signaling | IPR036097 | Signal transduction histidine kinase, dimerization/phosphoacceptor domain | −0.11 (32) | 1.41 (101) | −0.62 (21) | −0.37 (26) | −0.28 (28) | 0.35 (46) | −1.74 (7) | −0.74 (19) |
| Signaling | IPR024792 | Rho GDP-dissociation inhibitor domain | 0.48 (18) | 1.24 (34) | −0.14 (10) | −0.58 (6) | −0.34 (8) | −0.14 (10) | −1.04 (3) | −1.04 (3) |
| Metabolism | IPR012347 | Ferritin-like | 0.72 (22) | 1.29 (35) | −0.03 (11) | −0.71 (5) | −0.57 (6) | −0.86 (4) | −1.23 (2) | −0.57 (6) |
| Metabolism | IPR036909 | Cytochrome c-like domain | 0.40 (21) | 1.16 (39) | −0.50 (9) | −0.84 (6) | −0.22 (12) | −0.16 (17) | −0.82 (7) | −0.82 (7) |
| Metabolism | IPR037069 | Acyl-CoA dehydrogenase/oxidase, N-terminal domain | 0.45 (22) | 1.05 (36) | −0.60 (8) | −0.30 (11) | −0.22 (12) | 0.22 (18) | −0.98 (5) | −0.84 (6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashiro, T.; Shiraishi, A.; Nakayama, K.; Satake, H. Draft Genome of Tanacetum Coccineum: Genomic Comparison of Closely Related Tanacetum-Family Plants. Int. J. Mol. Sci. 2022, 23, 7039. https://doi.org/10.3390/ijms23137039
Yamashiro T, Shiraishi A, Nakayama K, Satake H. Draft Genome of Tanacetum Coccineum: Genomic Comparison of Closely Related Tanacetum-Family Plants. International Journal of Molecular Sciences. 2022; 23(13):7039. https://doi.org/10.3390/ijms23137039
Chicago/Turabian StyleYamashiro, Takanori, Akira Shiraishi, Koji Nakayama, and Honoo Satake. 2022. "Draft Genome of Tanacetum Coccineum: Genomic Comparison of Closely Related Tanacetum-Family Plants" International Journal of Molecular Sciences 23, no. 13: 7039. https://doi.org/10.3390/ijms23137039
APA StyleYamashiro, T., Shiraishi, A., Nakayama, K., & Satake, H. (2022). Draft Genome of Tanacetum Coccineum: Genomic Comparison of Closely Related Tanacetum-Family Plants. International Journal of Molecular Sciences, 23(13), 7039. https://doi.org/10.3390/ijms23137039