
Genomic Analysis of Waterpipe Smoke-Induced Lung Tumor Autophagy and Plasticity

 ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
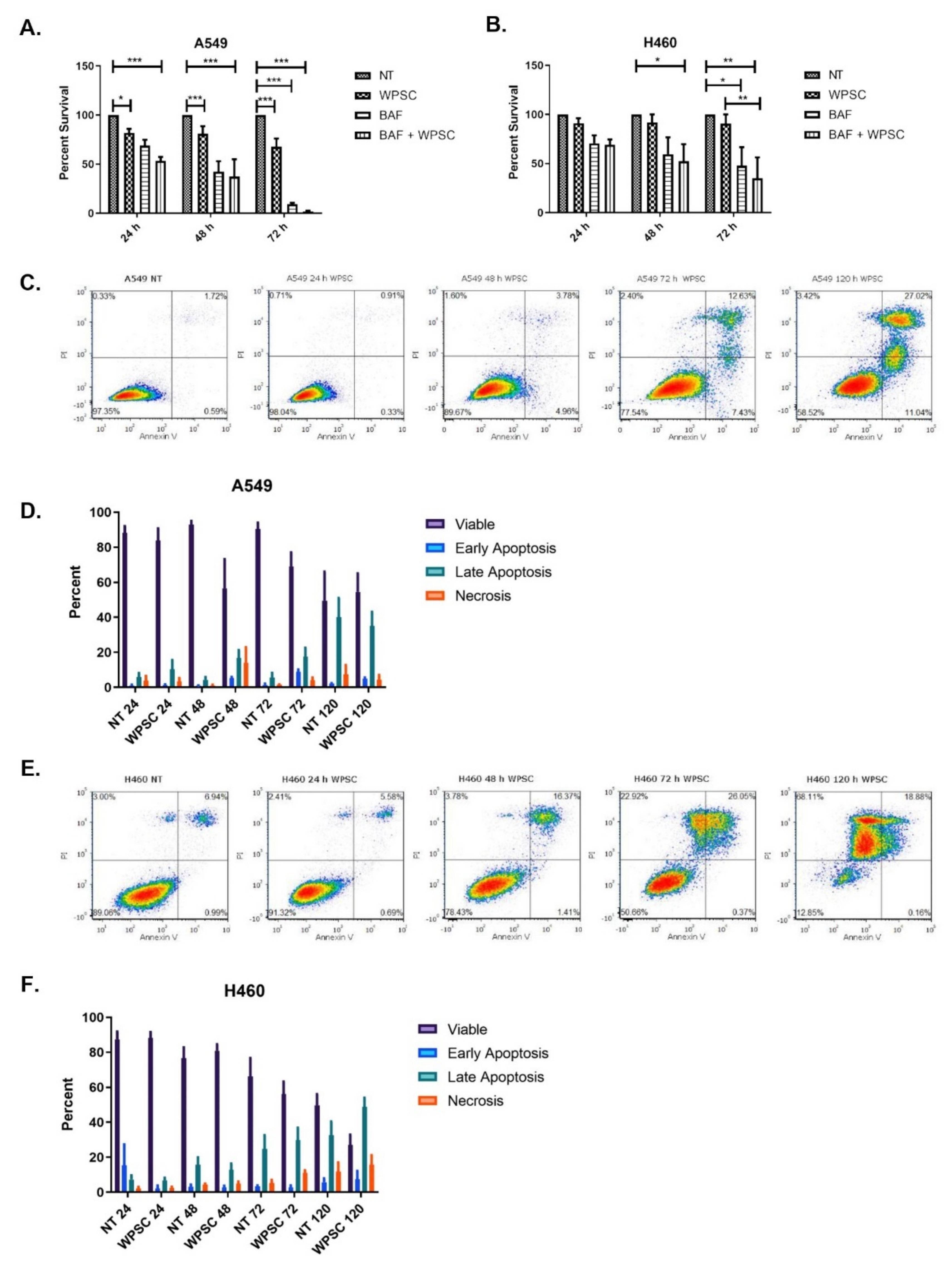
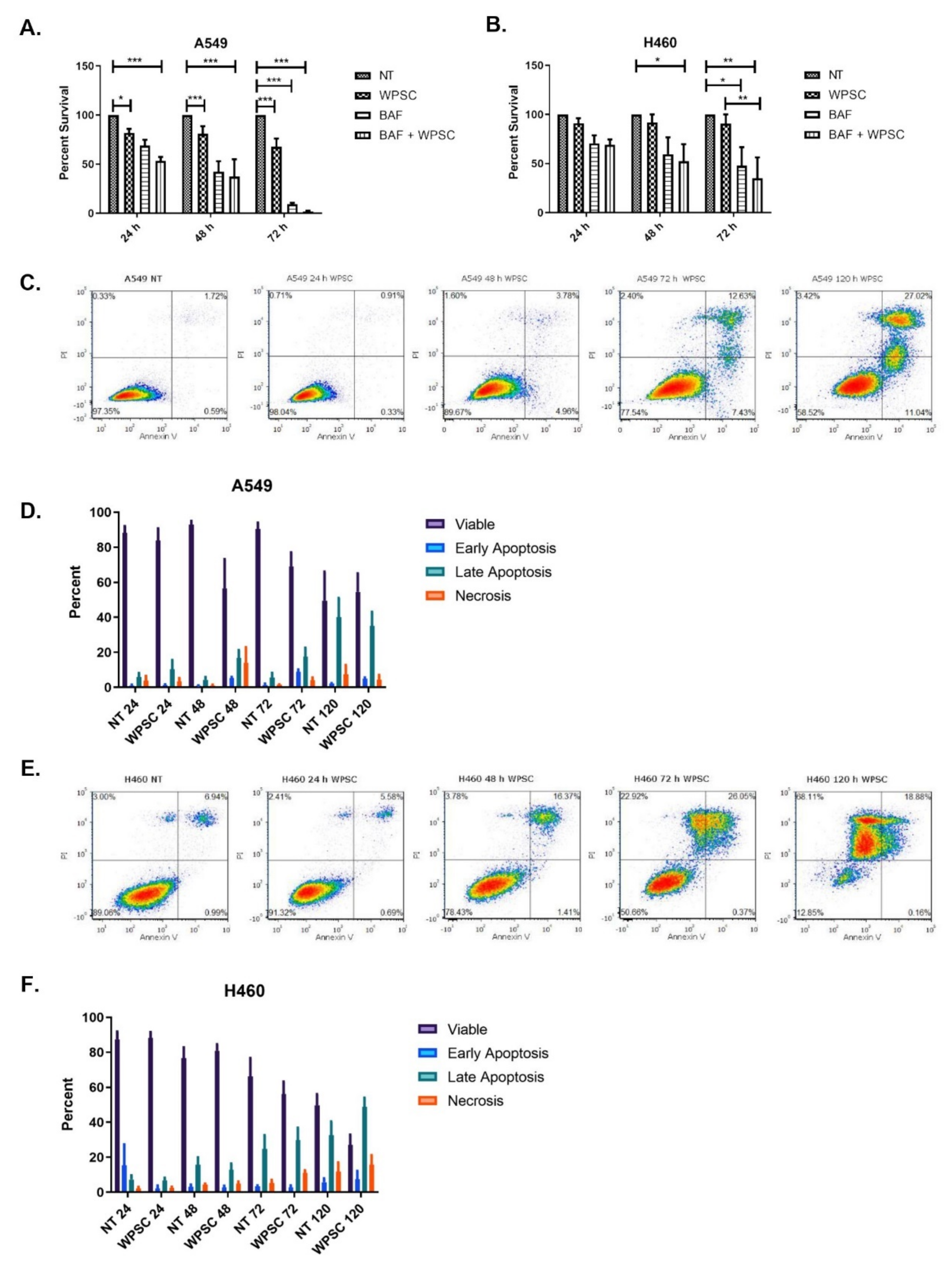
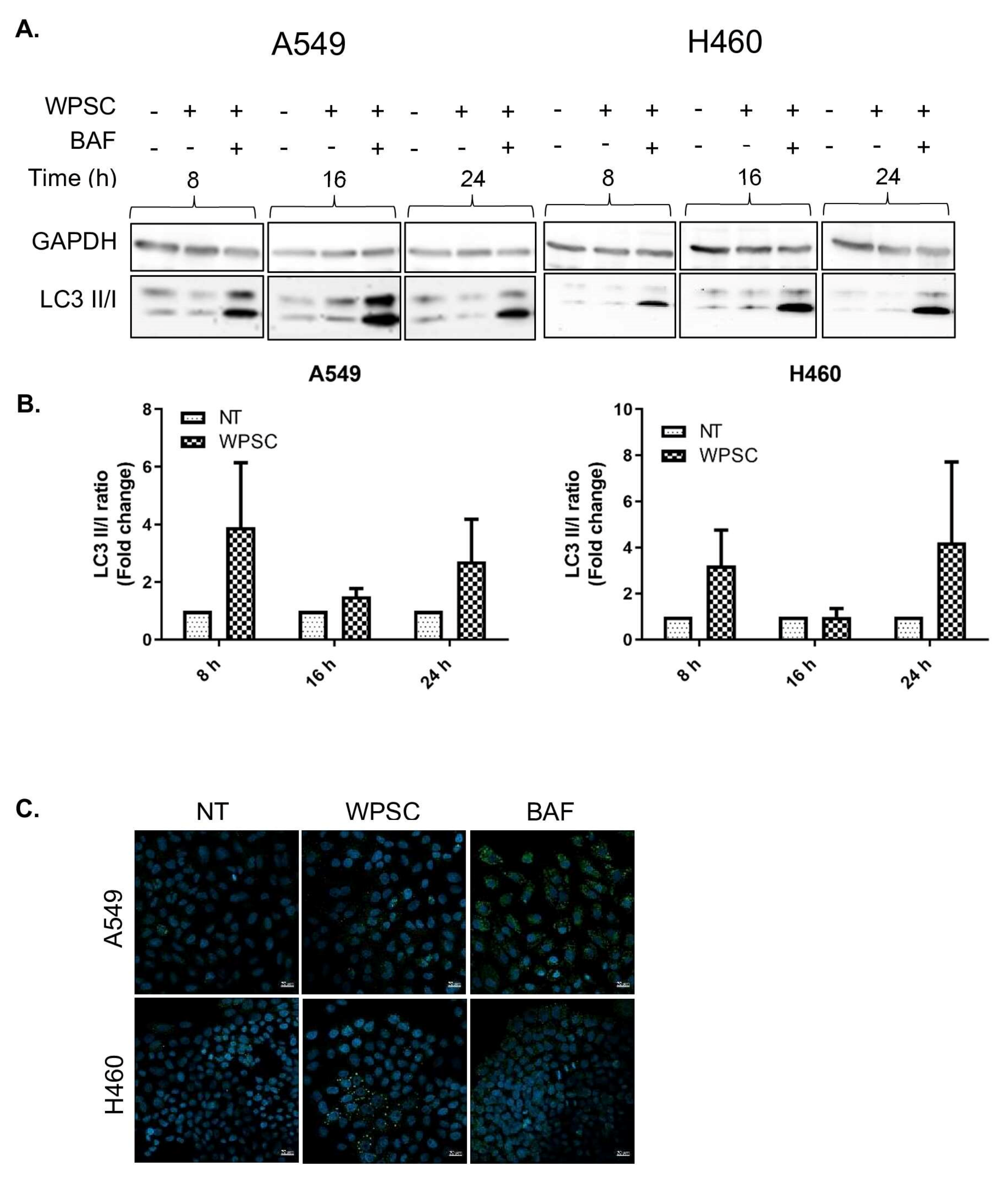
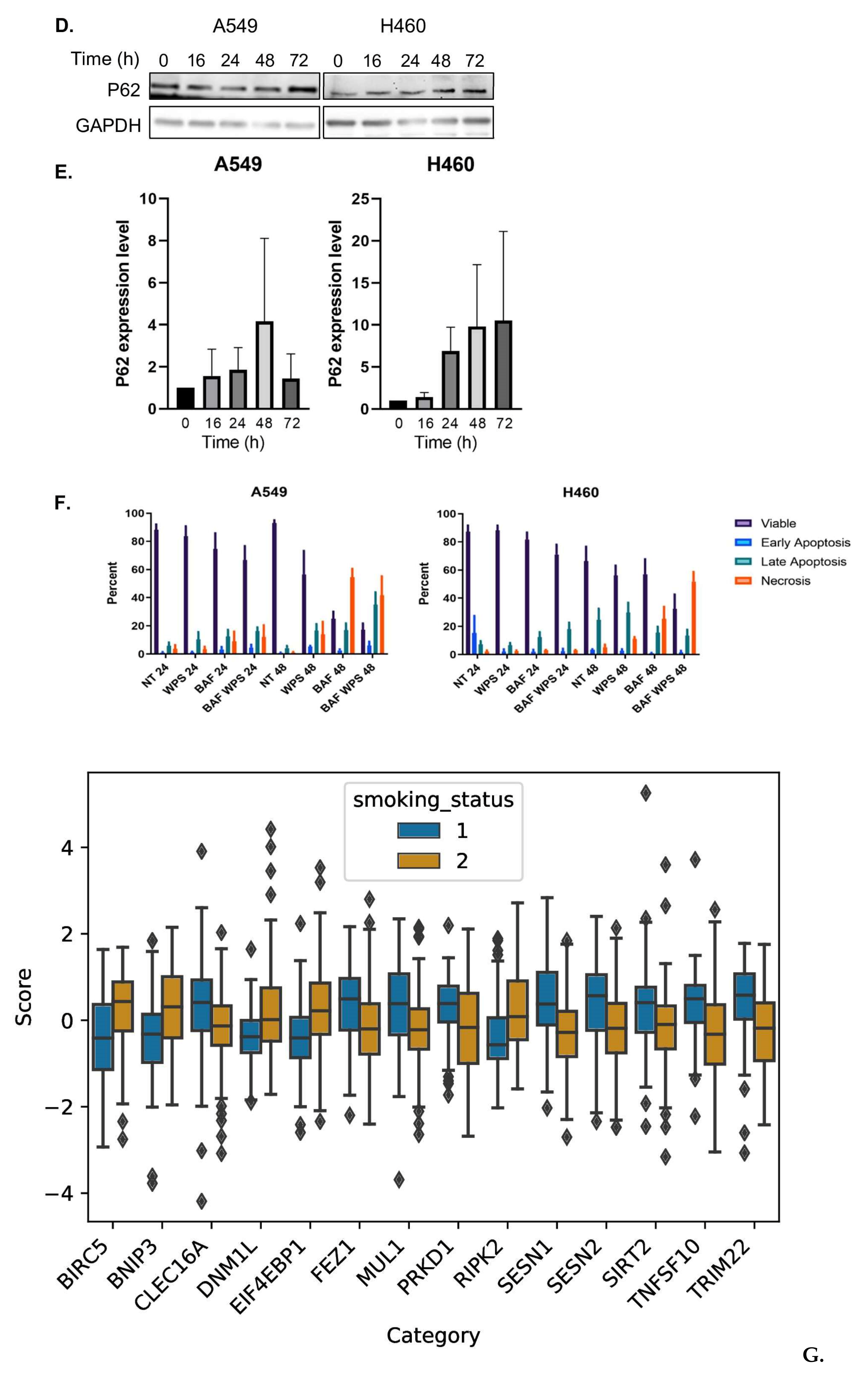
2.1. Waterpipe Smoke Condensate Increases Apoptosis and Activates Autophagy in Lung Cancer Cell Lines
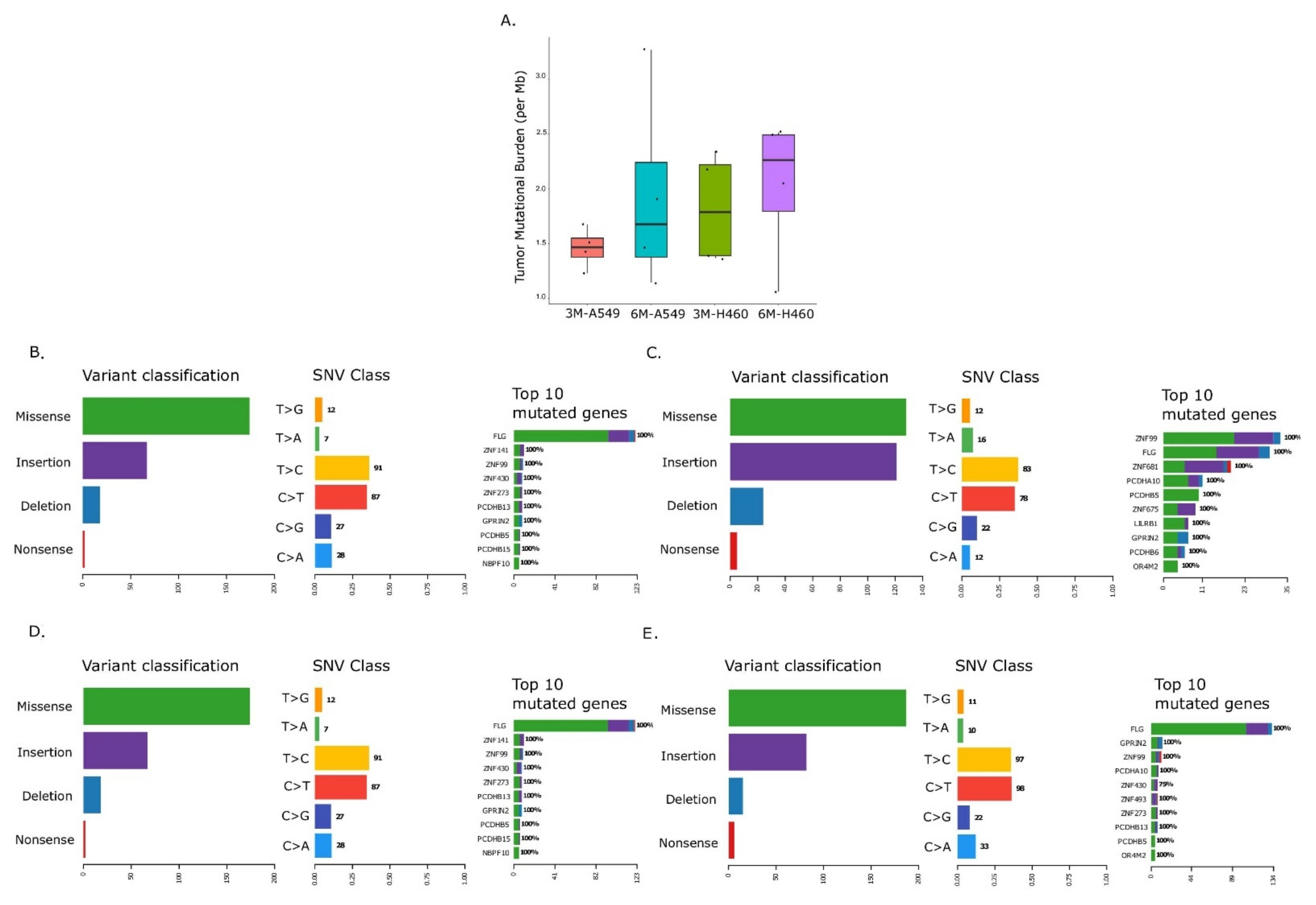
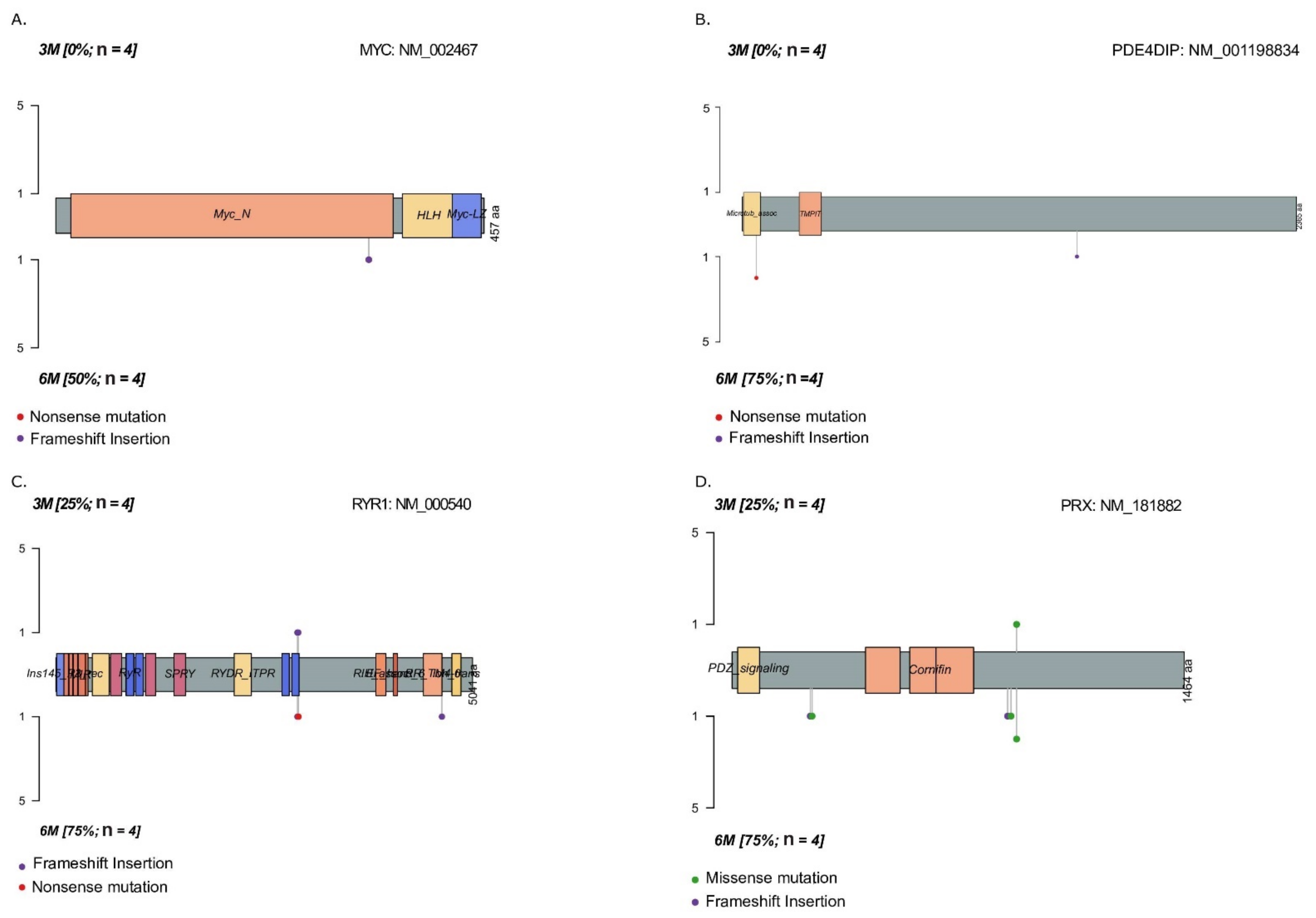
2.2. Temporal Changes in Mutational Landscape of Long-Term Exposure to Waterpipe Smoke in Lung Cancer Cell Lines Genomes
2.3. Smoking Is a Key Determinant of TMB of Lung Adenocarcinoma Patients
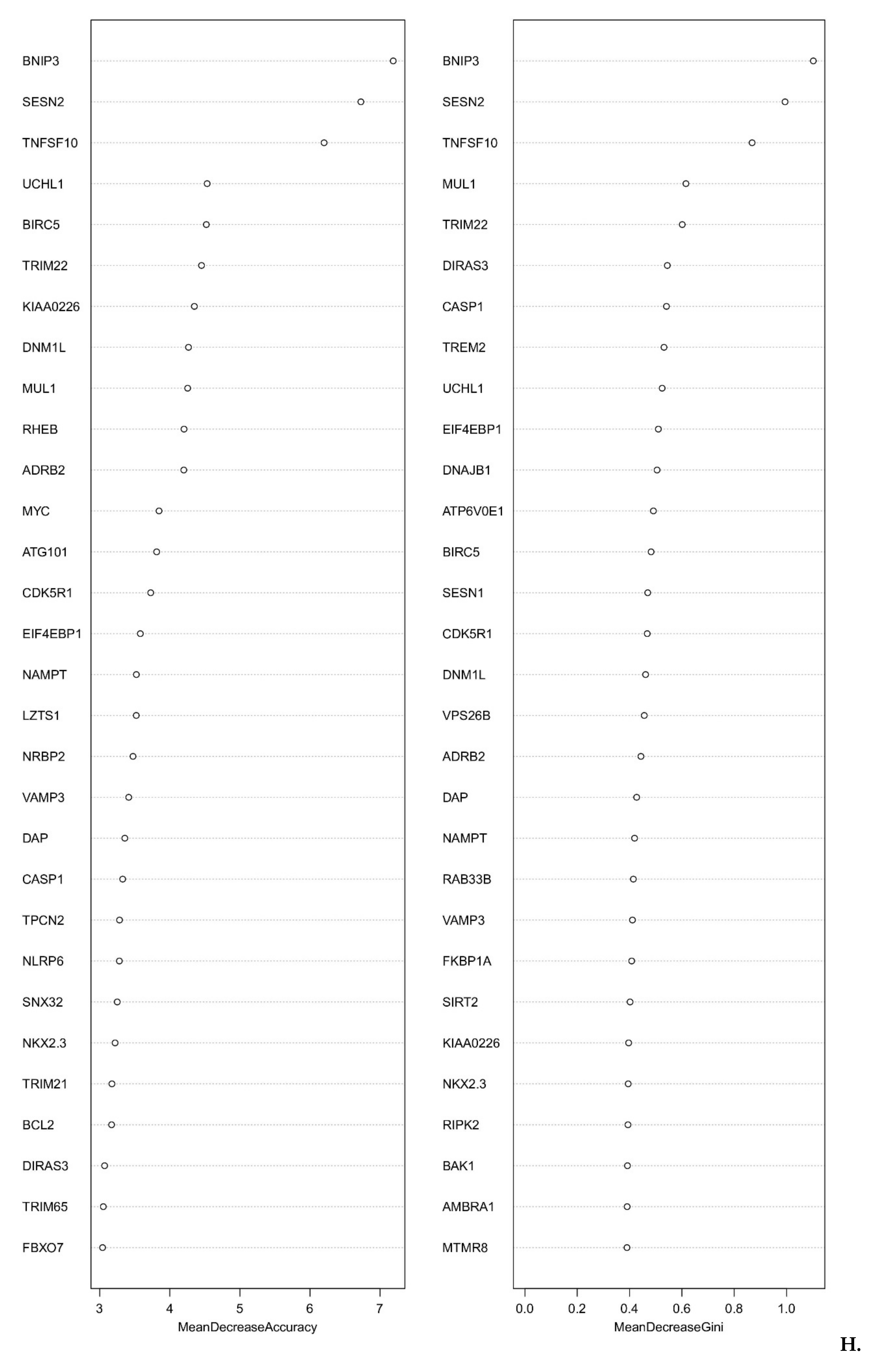
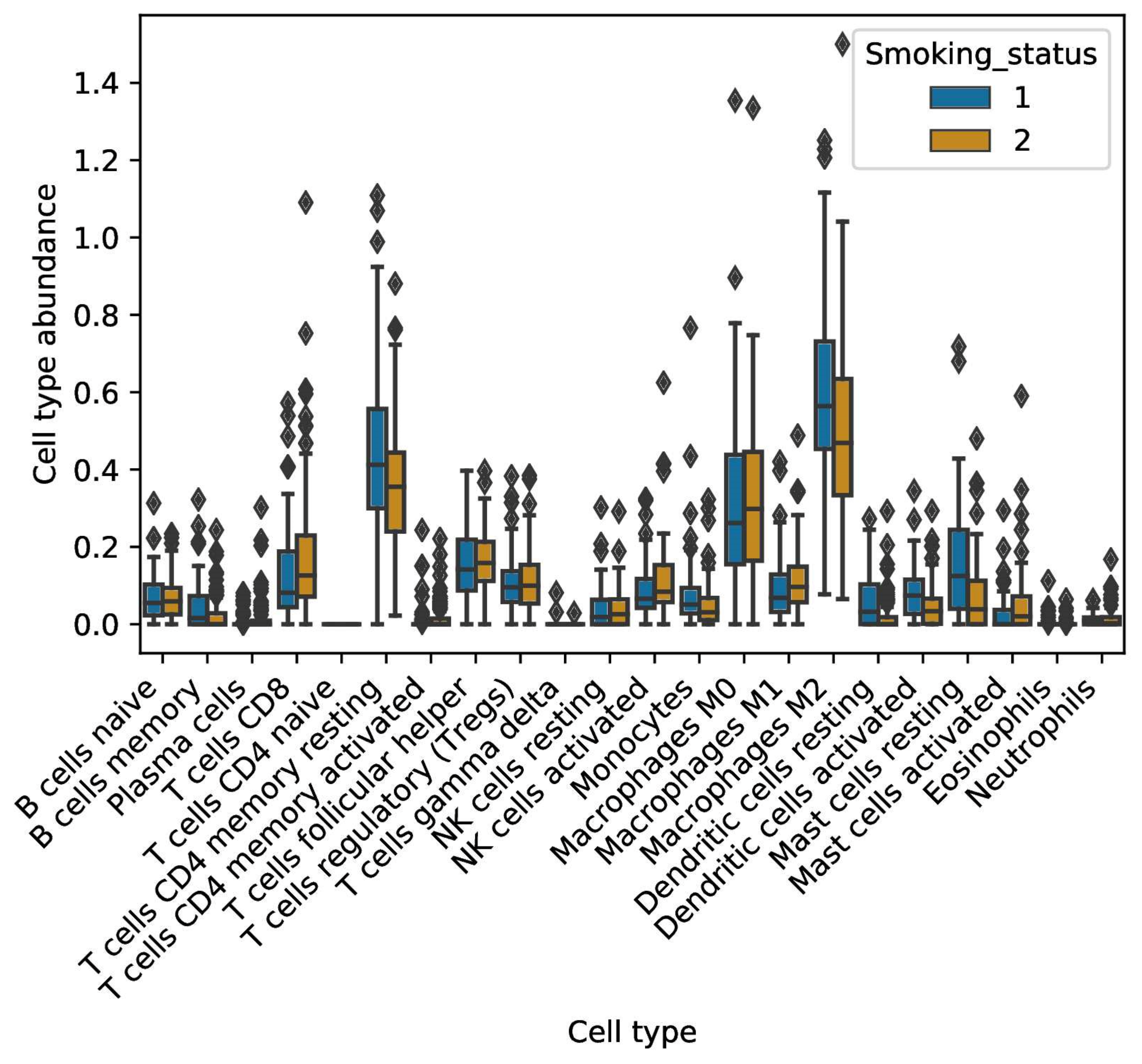
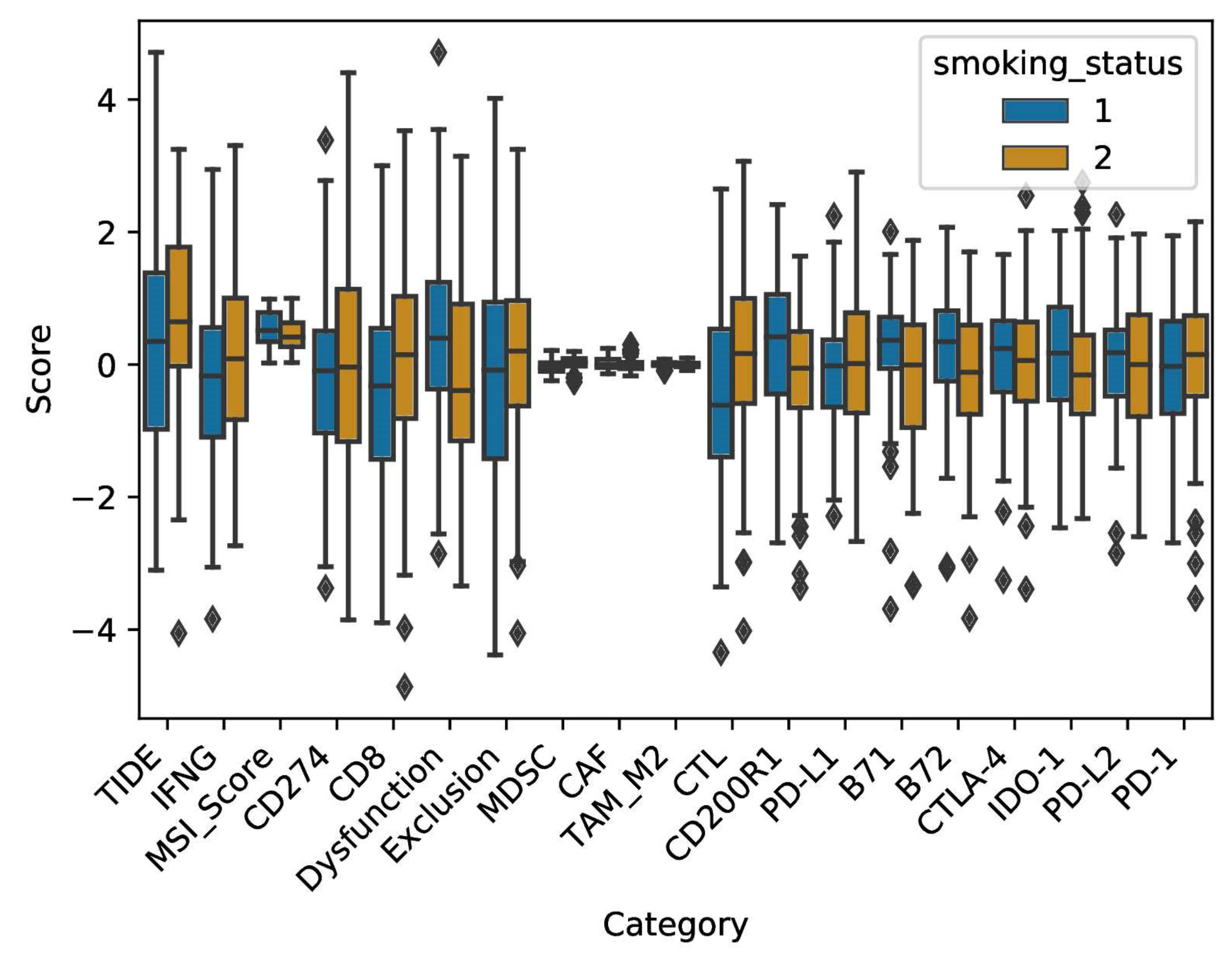
2.4. Smoke Exposure Is Associated with a Reprogramed Tumor Immune Microenvironment
3. Discussion
4. Materials and Methods
4.1. Waterpipe Smoke Sampling and Analysis
4.2. Cell Culture
4.3. MTT Assay
4.4. Flow Cytometry
4.5. Statistical Analysis
4.6. Antibodies Used in This Study
4.7. Immunoblotting
4.8. Immunofluorescence
4.9. Whole Exome Sequencing Variant Analysis
4.10. TCGA Analyses
4.11. Tumor Mutational Burden (TMB) Analysis
4.12. Immune Cell Abundance Analysis
4.13. TIDEPY Analysis
4.14. Immune Checkpoint Analysis
4.15. Autophagy Modeling Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Maziak, W. The global epidemic of waterpipe smoking. Addict. Behav. 2011, 36, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awan, K.H.; Siddiqi, K.; Patil, S.; Hussain, Q.A. Assessing the Effect of Waterpipe Smoking on Cancer Outcome—A Systematic Review of Current Evidence. Asian Pac. J. Cancer Prev. 2017, 18, 495–502. [Google Scholar] [CrossRef]
- Jabra, E.; Al-Omari, A.; Haddadin, F.; Alam, W.; Ammar, K.; Charafeddine, M.; Alrawashdeh, M.; Kasasbeh, N.; Habis, C.; Mukherji, D.; et al. Waterpipe Smoking among Bladder Cancer Patients: A Cross-Sectional Study of Lebanese and Jordanian Populations. J. Smok. Cessat. 2021, 2021, 6615832. [Google Scholar] [CrossRef]
- Elsayed, Y.; Dalibalta, S.; Abu-Farha, N. Chemical analysis and potential health risks of hookah charcoal. Sci. Total Environ. 2016, 569, 262–268. [Google Scholar] [CrossRef]
- Schubert, J.; Muller, F.D.; Schmidt, R.; Luch, A.; Schulz, T.G. Waterpipe smoke: Source of toxic and carcinogenic VOCs, phenols and heavy metals? Arch. Toxicol. 2015, 89, 2129–2139. [Google Scholar] [CrossRef]
- Arazi, H.; Taati, B.; Rafati Sajedi, F.; Suzuki, K. Salivary Antioxidants Status Following Progressive Aerobic Exercise: What Are the Differences between Waterpipe Smokers and Non-Smokers? Antioxidants 2019, 8, 418. [Google Scholar] [CrossRef] [Green Version]
- Khabour, O.F.; Alzoubi, K.H.; Bani-Ahmad, M.; Dodin, A.; Eissenberg, T.; Shihadeh, A. Acute exposure to waterpipe tobacco smoke induces changes in the oxidative and inflammatory markers in mouse lung. Inhal. Toxicol. 2012, 24, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Golbidi, S.; Li, H.; Laher, I. Oxidative Stress: A Unifying Mechanism for Cell Damage Induced by Noise, (Water-Pipe) Smoking, and Emotional Stress-Therapeutic Strategies Targeting Redox Imbalance. Antioxid. Redox Signal. 2018, 28, 741–759. [Google Scholar] [CrossRef]
- Zaarour, R.F.; Prasad, P.; Venkatesh, G.H.; Khouzam, R.A.; Amirtharaj, F.; Zeinelabdin, N.; Rifath, A.; Terry, S.; Nawafleh, H.; El Sayed, Y.; et al. Waterpipe smoke condensate influences epithelial to mesenchymal transition and interferes with the cytotoxic immune response in non-small cell lung cancer cell lines. Oncol. Rep. 2021, 45, 879–890. [Google Scholar] [CrossRef]
- Alsaad, A.M.; Al-Arifi, M.N.; Maayah, Z.H.; Attafi, I.M.; Alanazi, F.E.; Belali, O.M.; Alhoshani, A.; Asiri, Y.A.; Korashy, H.M. Genotoxic impact of long-term cigarette and waterpipe smoking on DNA damage and oxidative stress in healthy subjects. Toxicol. Mech. Methods 2019, 29, 119–127. [Google Scholar] [CrossRef]
- Su, M.; Mei, Y.; Sinha, S. Role of the Crosstalk between Autophagy and Apoptosis in Cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Van Westphal, C.; Carpenter-Thompson, R.; Mohanty, D.K.; Vij, N. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Radic. Biol. Med. 2016, 97, 441–453. [Google Scholar] [CrossRef]
- Shivalingappa, P.C.; Hole, R.; Westphal, C.V.; Vij, N. Airway Exposure to E-Cigarette Vapors Impairs Autophagy and Induces Aggresome Formation. Antioxid. Redox Signal. 2016, 24, 186–204. [Google Scholar] [CrossRef] [Green Version]
- Tran, I.; Ji, C.; Ni, I.; Min, T.; Tang, D.; Vij, N. Role of Cigarette Smoke-Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes with Pembrolizumab Versus Chemotherapy for Metastatic Non-Small-Cell Lung Cancer with PD-L1 Tumor Proportion Score ≥ 50. J. Clin. Oncol. 2021, 39, 2339–2349. [Google Scholar] [CrossRef]
- Mamdani, H.; Matosevic, S.; Khalid, A.B.; Durm, G.; Jalal, S.I. Immunotherapy in Lung Cancer: Current Landscape and Future Directions. Front. Immunol. 2022, 13, 823618. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou Khouzam, R.; Zaarour, R.F.; Brodaczewska, K.; Azakir, B.; Venkatesh, G.H.; Thiery, J.; Terry, S.; Chouaib, S. The Effect of Hypoxia and Hypoxia-Associated Pathways in the Regulation of Antitumor Response: Friends or Foes? Front. Immunol. 2022, 13, 828875. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Q.; Chen, H. Development of a Novel Autophagy-Related Prognostic Signature and Nomogram for Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 591356. [Google Scholar] [CrossRef]
- Titz, B.; Sewer, A.; Schneider, T.; Elamin, A.; Martin, F.; Dijon, S.; Luettich, K.; Guedj, E.; Vuillaume, G.; Ivanov, N.V.; et al. Alterations in the sputum proteome and transcriptome in smokers and early-stage COPD subjects. J. Proteom. 2015, 128, 306–320. [Google Scholar] [CrossRef] [Green Version]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Steiling, K.; Kadar, A.Y.; Bergerat, A.; Flanigon, J.; Sridhar, S.; Shah, V.; Ahmad, Q.R.; Brody, J.S.; Lenburg, M.E.; Steffen, M.; et al. Comparison of proteomic and transcriptomic profiles in the bronchial airway epithelium of current and never smokers. PLoS ONE 2009, 4, e5043. [Google Scholar] [CrossRef]
- Airoldi, L.; Magagnotti, C.; Iannuzzi, A.R.; Marelli, C.; Bagnati, R.; Pastorelli, R.; Colombi, A.; Santaguida, S.; Chiabrando, C.; Schiarea, S.; et al. Effects of cigarette smoking on the human urinary proteome. Biochem. Biophys. Res. Commun. 2009, 381, 397–402. [Google Scholar] [CrossRef]
- Jessie, K.; Pang, W.W.; Haji, Z.; Rahim, A.; Hashim, O.H. Proteomic analysis of whole human saliva detects enhanced expression of interleukin-1 receptor antagonist, thioredoxin and lipocalin-1 in cigarette smokers compared to non-smokers. Int. J. Mol. Sci. 2010, 11, 4488–4505. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.O.; Gumus, Z.H.; Kacker, A.; Choksi, V.L.; Bocker, J.M.; Zhou, X.K.; Yantiss, R.K.; Hughes, D.B.; Du, B.; Judson, B.L.; et al. Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res. 2010, 3, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.T.; Lin, X.; Liu, Y.; Chirieac, L.R.; McGovern, R.; Wain, J.; Heist, R.; Skaug, V.; Zienolddiny, S.; Haugen, A.; et al. Cigarette smoking increases copy number alterations in nonsmall-cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 16345–16350. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Chen, B.; Aran, D.; Charalel, J.; Yau, C.; Wolf, D.M.; van‘t Veer, L.J.; Butte, A.J.; Goldstein, T.; Sirota, M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun. 2019, 10, 3574. [Google Scholar] [CrossRef] [Green Version]
- Adcock, I.M.; Mortaz, E.; Alipoor, S.D.; Garssen, J.; Akbar Velayati, A. In Vitro effects of water-pipe smoke condensate on the endocytic activity of Type II alveolar epithelial cells (A549) with bacillus Calmette-Guerin. Int. J. Mycobacteriol. 2016, 5 (Suppl. S1), S157–S158. [Google Scholar] [CrossRef] [Green Version]
- Shihadeh, A.; Eissenberg, T.; Rammah, M.; Salman, R.; Jaroudi, E.; El-Sabban, M. Comparison of tobacco-containing and tobacco-free waterpipe products: Effects on human alveolar cells. Nicotine Tob. Res. 2014, 16, 496–499. [Google Scholar] [CrossRef] [Green Version]
- Mortaz, E.; Alipoor, S.D.; Movassaghi, M.; Varahram, M.; Ghorbani, J.; Folkerts, G.; Garssen, J.; Adcock, I.M. Water-pipe smoke condensate increases the internalization of Mycobacterium Bovis of type II alveolar epithelial cells (A549). BMC Pulm. Med. 2017, 17, 68. [Google Scholar] [CrossRef] [Green Version]
- Khalil, C.; Chahine, J.B.; Chahla, B.; Hobeika, T.; Khnayzer, R.S. Characterization and cytotoxicity assessment of nargile smoke using dynamic exposure. Inhal. Toxicol. 2019, 31, 343–356. [Google Scholar] [CrossRef]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and senescence: A new insight in selected human diseases. J. Cell Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef]
- Friedberg, E.C. DNA damage and repair. Nature 2003, 421, 436–440. [Google Scholar] [CrossRef]
- Chen, Y.S.; Song, H.X.; Lu, Y.; Li, X.; Chen, T.; Zhang, Y.; Xue, J.X.; Liu, H.; Kan, B.; Yang, G.; et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis. Esophagus 2011, 24, 437–443. [Google Scholar] [CrossRef]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007, 14, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yue, W.; Chen, H. Effect of artesunate on apoptosis and autophagy in tamoxifen resistant breast cancer cells (TAM-R). Transl. Cancer Res. 2019, 8, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Relitti, N.; Brindisi, M.; Magnano, S.; Zisterer, D.; Gemma, S.; Butini, S.; Campiani, G. Autophagy modulators for the treatment of oral and esophageal squamous cell carcinomas. Med. Res. Rev. 2020, 40, 1002–1060. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Sharp, D.W.; Yang, Y.; Laddha, S.V.; Ibrahim, M.; Bommareddy, P.K.; Hu, Z.S.; Vieth, J.; Haas, M.; Bosenberg, M.W.; et al. Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat. Cancer 2020, 1, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Mei, P.; Freitag, C.E.; Wei, L.; Zhang, Y.; Parwani, A.V.; Li, Z. High tumor mutation burden is associated with DNA damage repair gene mutation in breast carcinomas. Diagn. Pathol. 2020, 15, 50. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Raparia, K.; Agte, S.; Pan, A.; Mohindra, N.; Villaflor, V.; Giles, F. Association of Tumor Mutational Burden with DNA Repair Mutations and Response to Anti-PD-1/PD-L1 Therapy in Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2019, 20, 88–96.e86. [Google Scholar] [CrossRef]
- Parikh, A.R.; He, Y.; Hong, T.S.; Corcoran, R.B.; Clark, J.W.; Ryan, D.P.; Zou, L.; Ting, D.T.; Catenacci, D.V.; Chao, J.; et al. Analysis of DNA Damage Response Gene Alterations and Tumor Mutational Burden Across 17,486 Tubular Gastrointestinal Carcinomas: Implications for Therapy. Oncologist 2019, 24, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Chen, Y. Autophagy controls programmed death-ligand 1 expression on cancer cells (Review). Biomed. Rep. 2021, 15, 84. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.R.; Anderson, E.; et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef]
- Qiu, Z.; Lin, A.; Li, K.; Lin, W.; Wang, Q.; Wei, T.; Zhu, W.; Luo, P.; Zhang, J. A novel mutation panel for predicting etoposide resistance in small-cell lung cancer. Drug Des. Devel. Ther. 2019, 13, 2021–2041. [Google Scholar] [CrossRef] [Green Version]
- Skaaby, T.; Husemoen, L.L.; Thyssen, J.P.; Meldgaard, M.; Thuesen, B.H.; Pisinger, C.; Jorgensen, T.; Carlsen, K.; Johansen, J.D.; Menne, T.; et al. Filaggrin loss-of-function mutations and incident cancer: A population-based study. Br. J. Dermatol. 2014, 171, 1407–1414. [Google Scholar] [CrossRef]
- Ting, C.H.; Lee, K.Y.; Wu, S.M.; Feng, P.H.; Chan, Y.F.; Chen, Y.C.; Chen, J.Y. FOSB(-)PCDHB13 Axis Disrupts the Microtubule Network in Non-Small Cell Lung Cancer. Cancers 2019, 11, 107. [Google Scholar] [CrossRef] [Green Version]
- Al-Sawalha, N.A.; Migdadi, A.M.; Alzoubi, K.H.; Khabour, O.F.; Qinna, N.A. Effect of waterpipe tobacco smoking on airway inflammation in murine model of asthma. Inhal. Toxicol. 2017, 29, 46–52. [Google Scholar] [CrossRef]
- Nemmar, A.; Yuvaraju, P.; Beegam, S.; Ali, B.H. Short-term nose-only water-pipe (shisha) smoking exposure accelerates coagulation and causes cardiac inflammation and oxidative stress in mice. Cell Physiol. Biochem. 2015, 35, 829–840. [Google Scholar] [CrossRef]
- Nemmar, A.; Al-Salam, S.; Yuvaraju, P.; Beegam, S.; Yasin, J.; Ali, B.H. Chronic exposure to water-pipe smoke induces cardiovascular dysfunction in mice. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H329–H339. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Hasan, A.; Rizvi, S.F.; Parveen, S.; Pathak, N.; Nazir, A.; Mir, S.S. Crosstalk between ROS and Autophagy in Tumorigenesis: Understanding the Multifaceted Paradox. Front. Oncol. 2022, 12, 852424. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef] [Green Version]
- Vijayalingam, S.; Pillai, S.G.; Rashmi, R.; Subramanian, T.; Sagartz, J.E.; Chinnadurai, G. Overexpression of BH3-Only Protein BNIP3 Leads to Enhanced Tumor Growth. Genes Cancer 2010, 1, 964–971. [Google Scholar] [CrossRef]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Renal. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef]
- Pan, C.; Chen, Z.; Li, C.; Han, T.; Liu, H.; Wang, X. Sestrin2 as a gatekeeper of cellular homeostasis: Physiological effects for the regulation of hypoxia-related diseases. J. Cell Mol. Med. 2021, 25, 5341–5350. [Google Scholar] [CrossRef]
- Chen, K.B.; Xuan, Y.; Shi, W.J.; Chi, F.; Xing, R.; Zeng, Y.C. Sestrin2 expression is a favorable prognostic factor in patients with non-small cell lung cancer. Am. J. Transl. Res. 2016, 8, 1903–1909. [Google Scholar]
- Di Rienzo, M.; Romagnoli, A.; Antonioli, M.; Piacentini, M.; Fimia, G.M. TRIM proteins in autophagy: Selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020, 27, 887–902. [Google Scholar] [CrossRef]
- Ji, J.; Ding, K.; Luo, T.; Zhang, X.; Chen, A.; Zhang, D.; Li, G.; Thorsen, F.; Huang, B.; Li, X.; et al. TRIM22 activates NF-kappaB signaling in glioblastoma by accelerating the degradation of IkappaBalpha. Cell Death Differ. 2021, 28, 367–381. [Google Scholar] [CrossRef]
- He, W.; Wang, Q.; Xu, J.; Xu, X.; Padilla, M.T.; Ren, G.; Gou, X.; Lin, Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy 2012, 8, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, A.; Pagano, M.; Russo, G.; Russo, A. Role of Autophagy in Cancer Cell Response to Nucleolar and Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2020, 21, 7334. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Pagano, M.; Borzacchiello, L.; Mele, L.; Russo, A.; Russo, G.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine Increases the Sensitivity of Human Colorectal Cancer Cells to 5-Fluorouracil by Inhibiting P-Glycoprotein Expression and NF-κB Activation. Int. J. Mol. Sci. 2021, 22, 9286. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Pagano, M.; Pecoraro, A.; Borzacchiello, L.; Mele, L.; Cacciapuoti, G.; Porcelli, M.; Russo, G.; Russo, A. S-Adenosyl-l-Methionine Overcomes uL3-Mediated Drug Resistance in p53 Deleted Colon Cancer Cells. Int. J. Mol. Sci. 2020, 22, 103. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, Q.; Shen, J.; Wei, T.; Shen, W.; Zhang, N.; Luo, P.; Zhang, J. The Effect of Smoking on the Immune Microenvironment and Immunogenicity and Its Relationship with the Prognosis of Immune Checkpoint Inhibitors in Non-small Cell Lung Cancer. Front. Cell Dev. Biol. 2021, 9, 745859. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Bergstrom, E.N.; Ni Huang, M.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A tool for visualizing and exploring patterns of small mutational events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Guo, D.; Wang, M.; Shen, Z.; Zhu, J. A new immune signature for survival prediction and immune checkpoint molecules in lung adenocarcinoma. J. Transl. Med. 2020, 18, 123. [Google Scholar] [CrossRef] [Green Version]
- Ling, B.; Ye, G.; Zhao, Q.; Jiang, Y.; Liang, L.; Tang, Q. Identification of an Immunologic Signature of Lung Adenocarcinomas Based on Genome-Wide Immune Expression Profiles. Front. Mol. Biosci. 2020, 7, 603701. [Google Scholar] [CrossRef]
- Diaz-Uriarte, R. GeneSrF and varSelRF: A web-based tool and R package for gene selection and classification using random forest. BMC Bioinform. 2007, 8, 328. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaarour, R.F.; Sharda, M.; Azakir, B.; Hassan Venkatesh, G.; Abou Khouzam, R.; Rifath, A.; Nizami, Z.N.; Abdullah, F.; Mohammad, F.; Karaali, H.; et al. Genomic Analysis of Waterpipe Smoke-Induced Lung Tumor Autophagy and Plasticity. Int. J. Mol. Sci. 2022, 23, 6848. https://doi.org/10.3390/ijms23126848
Zaarour RF, Sharda M, Azakir B, Hassan Venkatesh G, Abou Khouzam R, Rifath A, Nizami ZN, Abdullah F, Mohammad F, Karaali H, et al. Genomic Analysis of Waterpipe Smoke-Induced Lung Tumor Autophagy and Plasticity. International Journal of Molecular Sciences. 2022; 23(12):6848. https://doi.org/10.3390/ijms23126848
Chicago/Turabian StyleZaarour, Rania Faouzi, Mohak Sharda, Bilal Azakir, Goutham Hassan Venkatesh, Raefa Abou Khouzam, Ayesha Rifath, Zohra Nausheen Nizami, Fatima Abdullah, Fatin Mohammad, Hajar Karaali, and et al. 2022. "Genomic Analysis of Waterpipe Smoke-Induced Lung Tumor Autophagy and Plasticity" International Journal of Molecular Sciences 23, no. 12: 6848. https://doi.org/10.3390/ijms23126848
APA StyleZaarour, R. F., Sharda, M., Azakir, B., Hassan Venkatesh, G., Abou Khouzam, R., Rifath, A., Nizami, Z. N., Abdullah, F., Mohammad, F., Karaali, H., Nawafleh, H., Elsayed, Y., & Chouaib, S. (2022). Genomic Analysis of Waterpipe Smoke-Induced Lung Tumor Autophagy and Plasticity. International Journal of Molecular Sciences, 23(12), 6848. https://doi.org/10.3390/ijms23126848