
Derangements and Reversibility of Energy Metabolism in Failing Hearts Resulting from Volume Overload: Transcriptomics and Metabolomics Analyses


 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
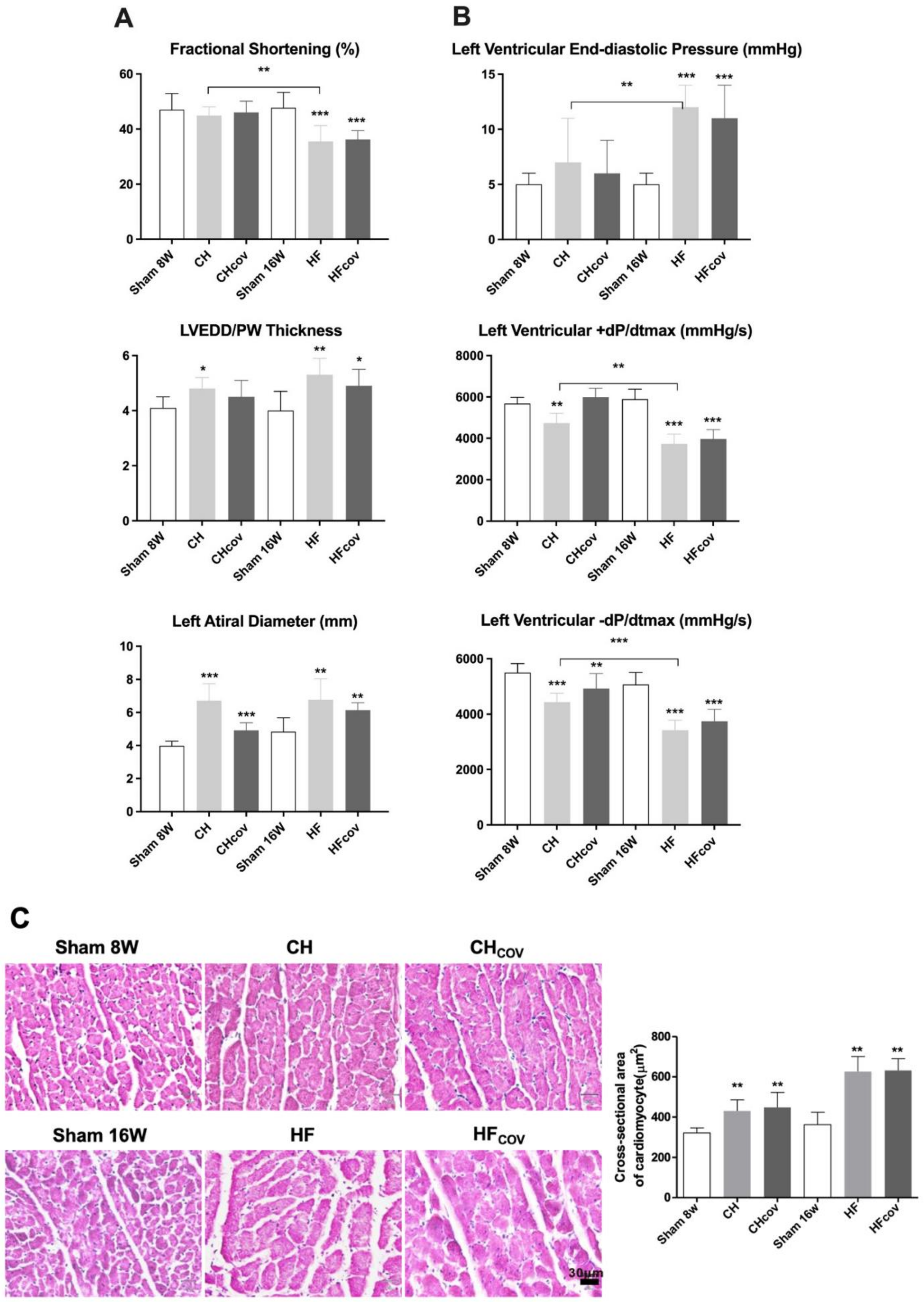
2.1. Cardiac Remodeling after the Creation and Subsequent Correction of VO
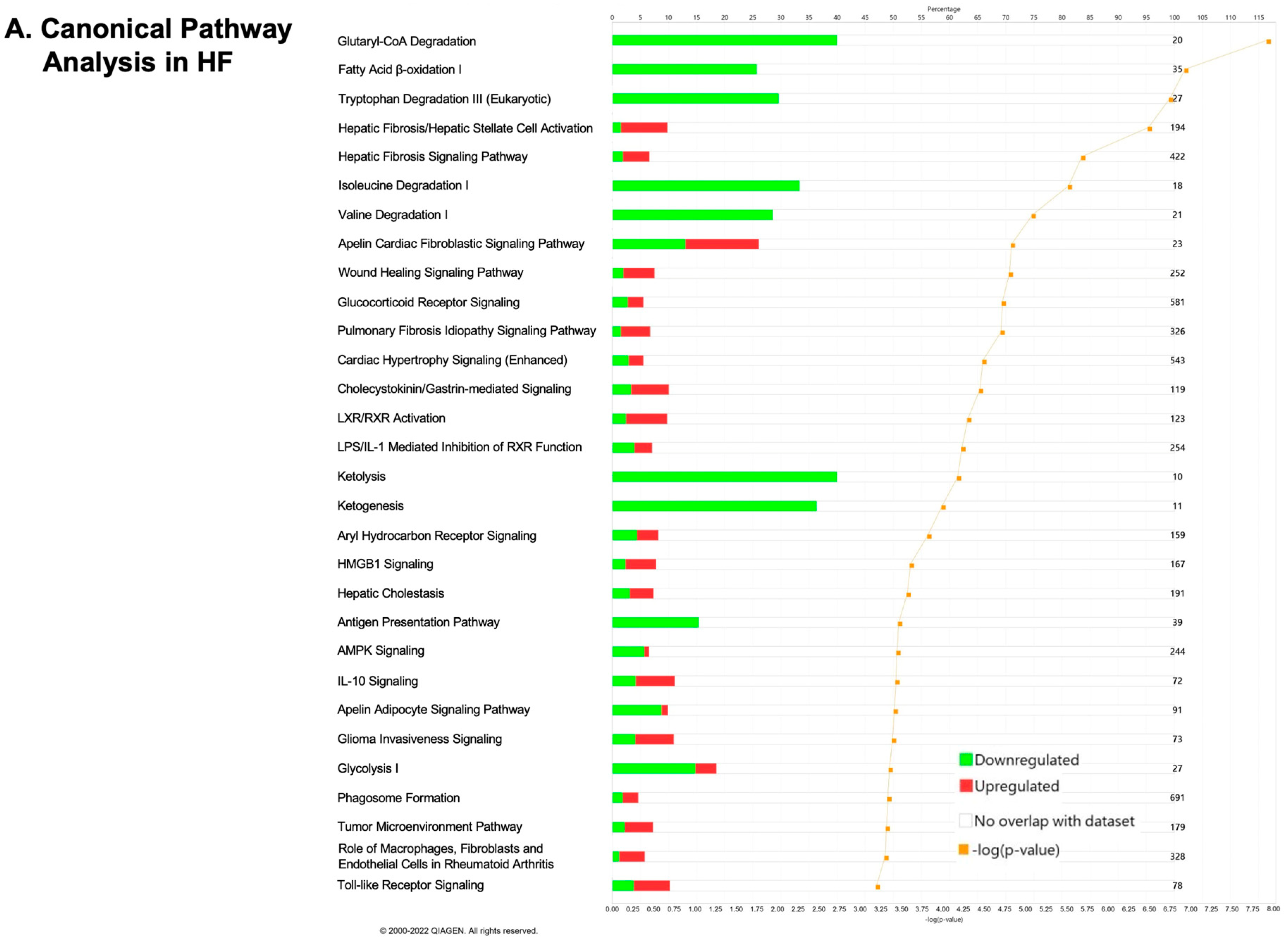
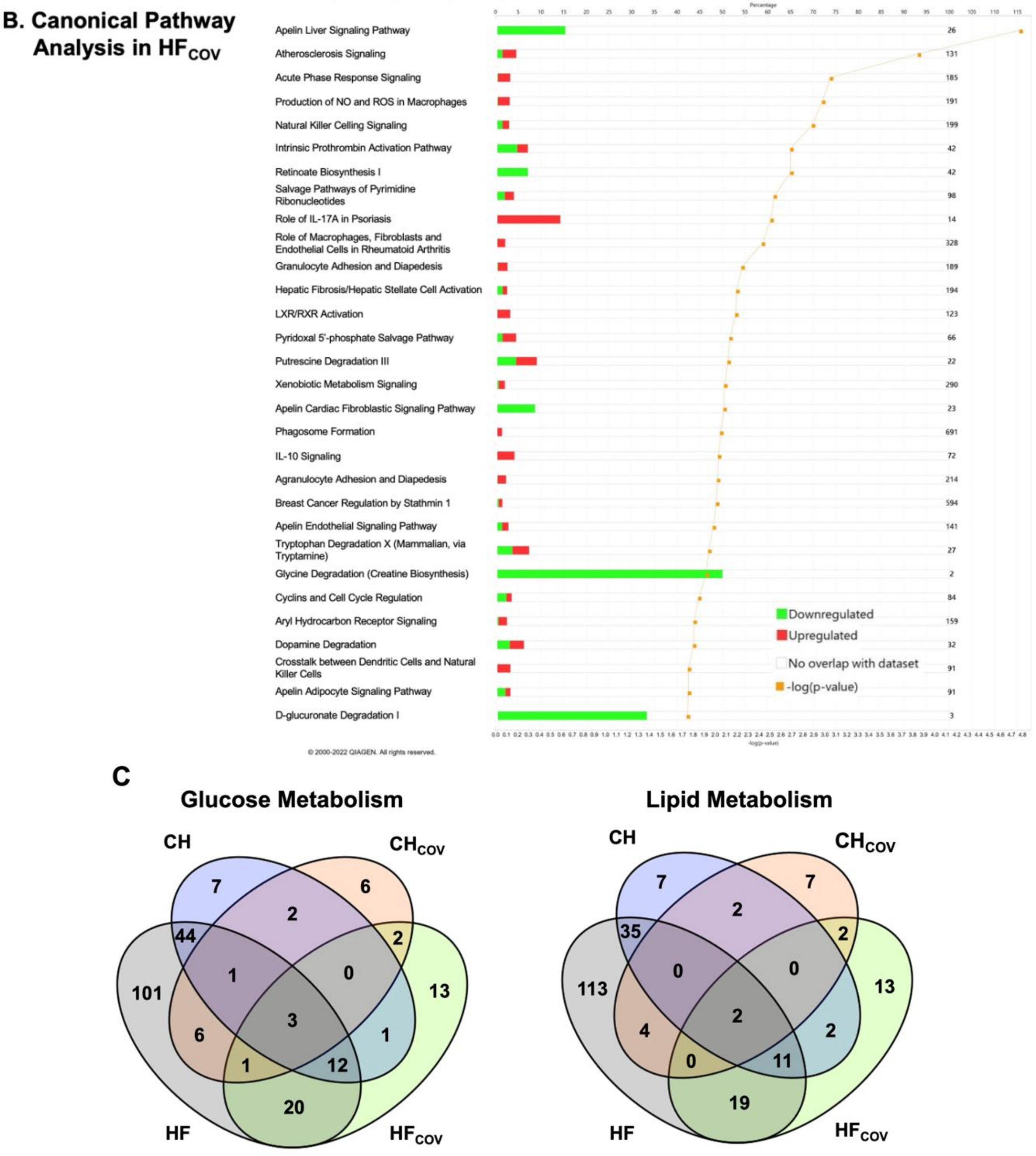
2.2. Transcriptomics Profiling of Cardiac Energy Metabolism in VO-Induced CH and HF
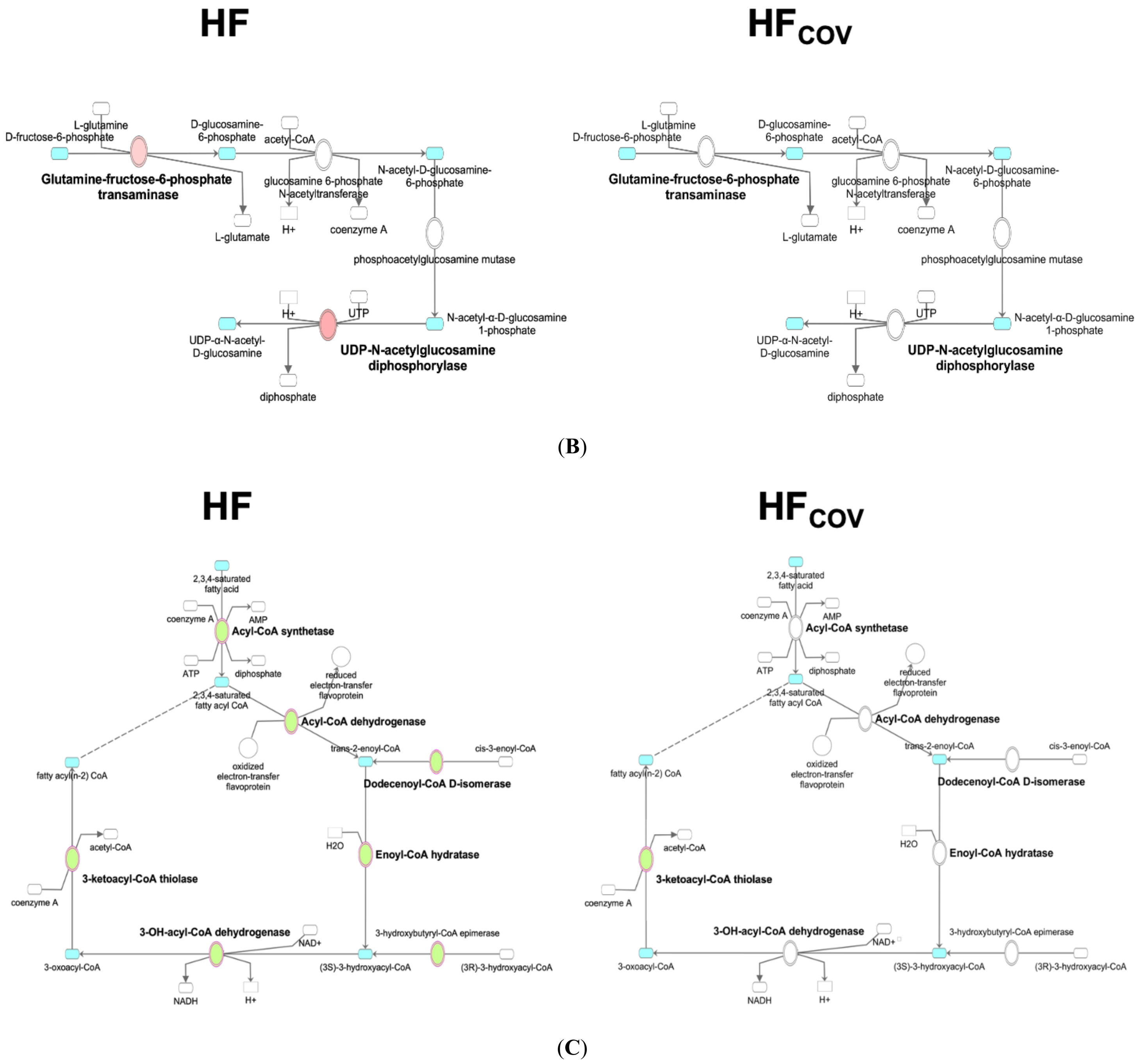
2.3. Transcriptomics Profiling of Cardiac Energy Metabolism after Correction of VO
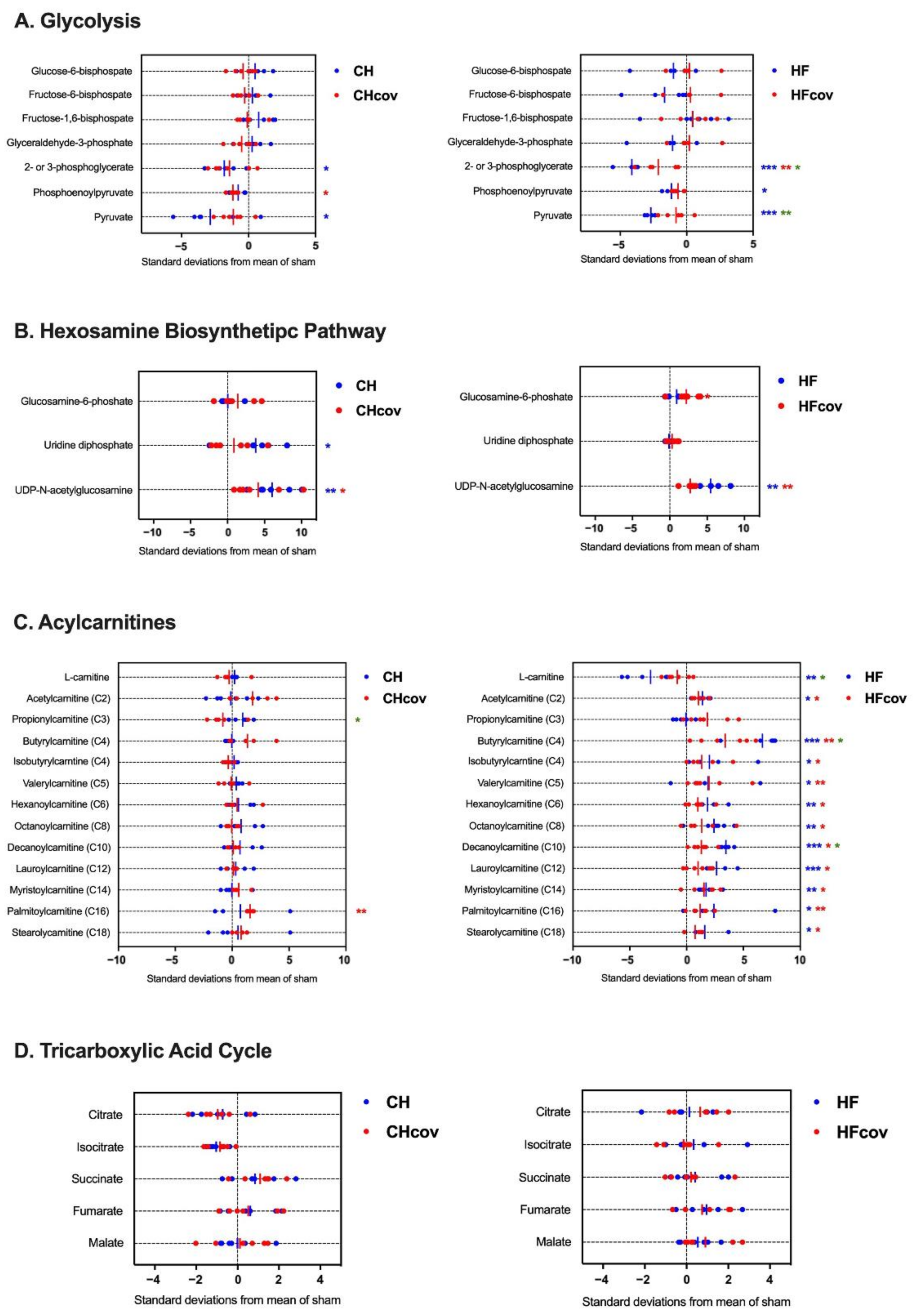
2.4. Metabolomics Profiling of Cardiac Energy Metabolism in VO-induced CH and HF
2.5. Metabolomics Profiling of Cardiac Energy Metabolism after Correction of VO
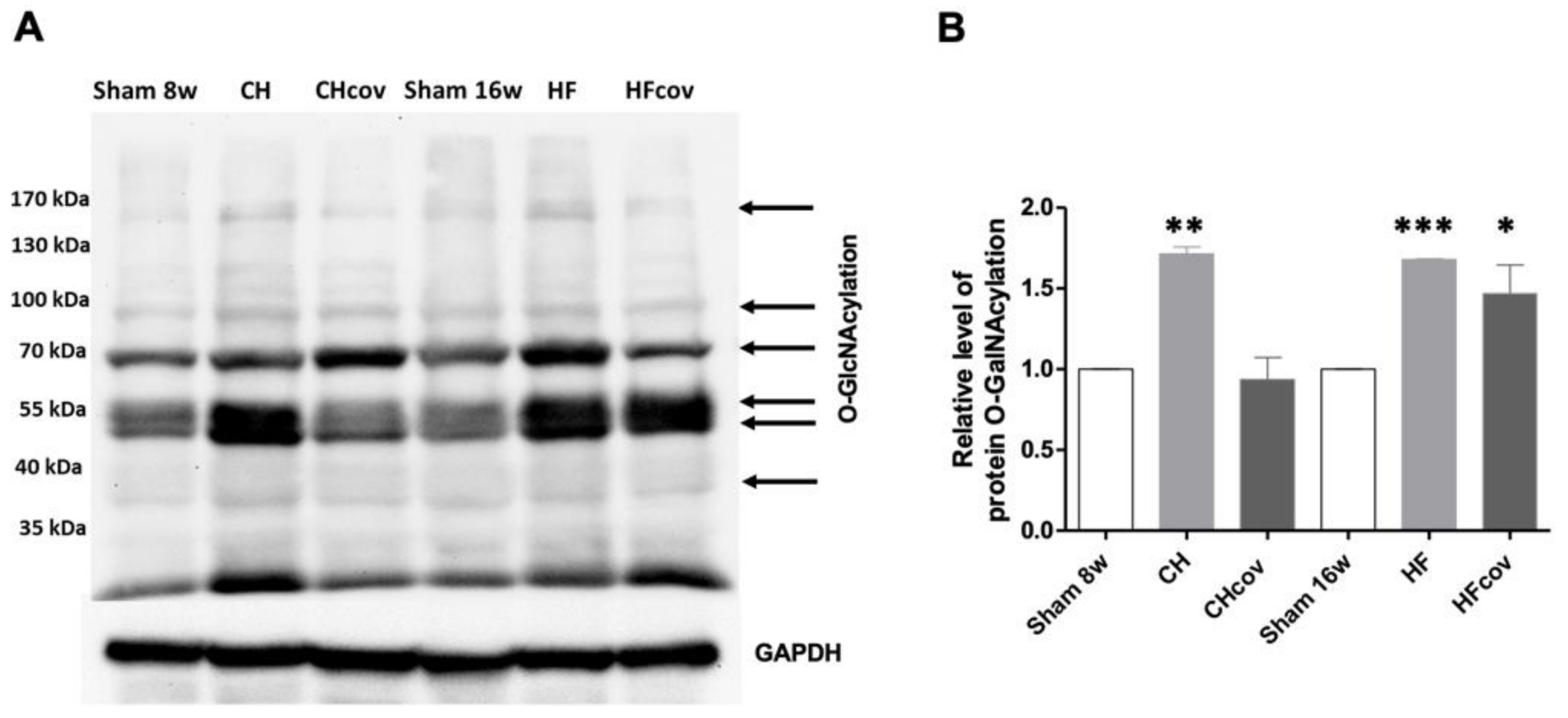
2.6. Elevation of Protein O-linked-N-acetylglucosaminylation (O-GlcNAcylation)
3. Discussion
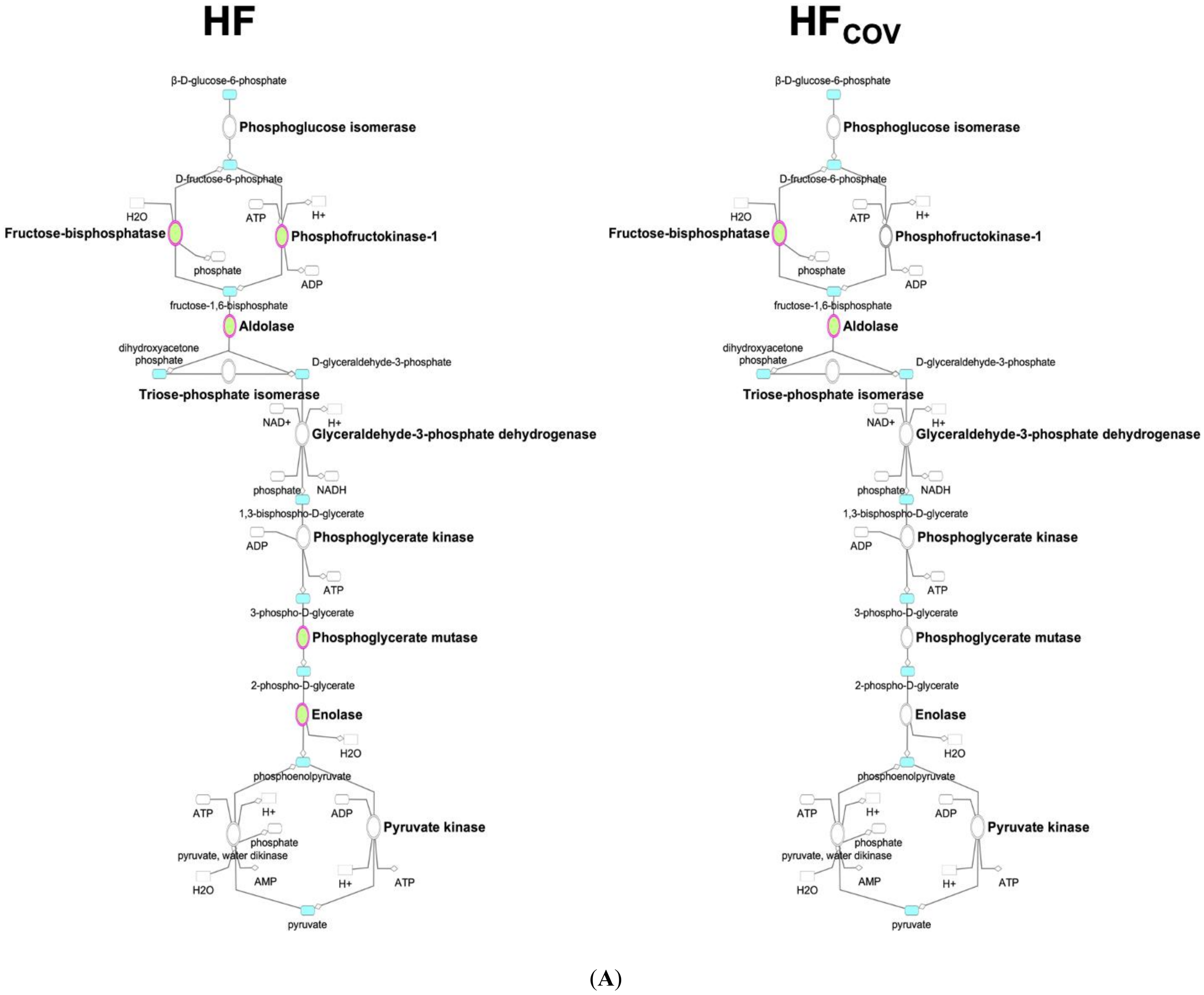
3.1. Downregulation of Glycolysis in Chronic Volume-Overloaded Hearts in CH and HF Phases
3.2. Upregulation of the HBP and Protein O-GlcNAcylation in VO
3.3. Dysregulation of Fatty Acid Metabolism in VO-Induced HF
3.4. No Significant Change in the Gene Expression and Intermediates of the TCA Cycle
3.5. Reversibility of Dysregulated Energy Metabolism after the Correction of VO
3.6. Study Limitations
4. Methods and Materials
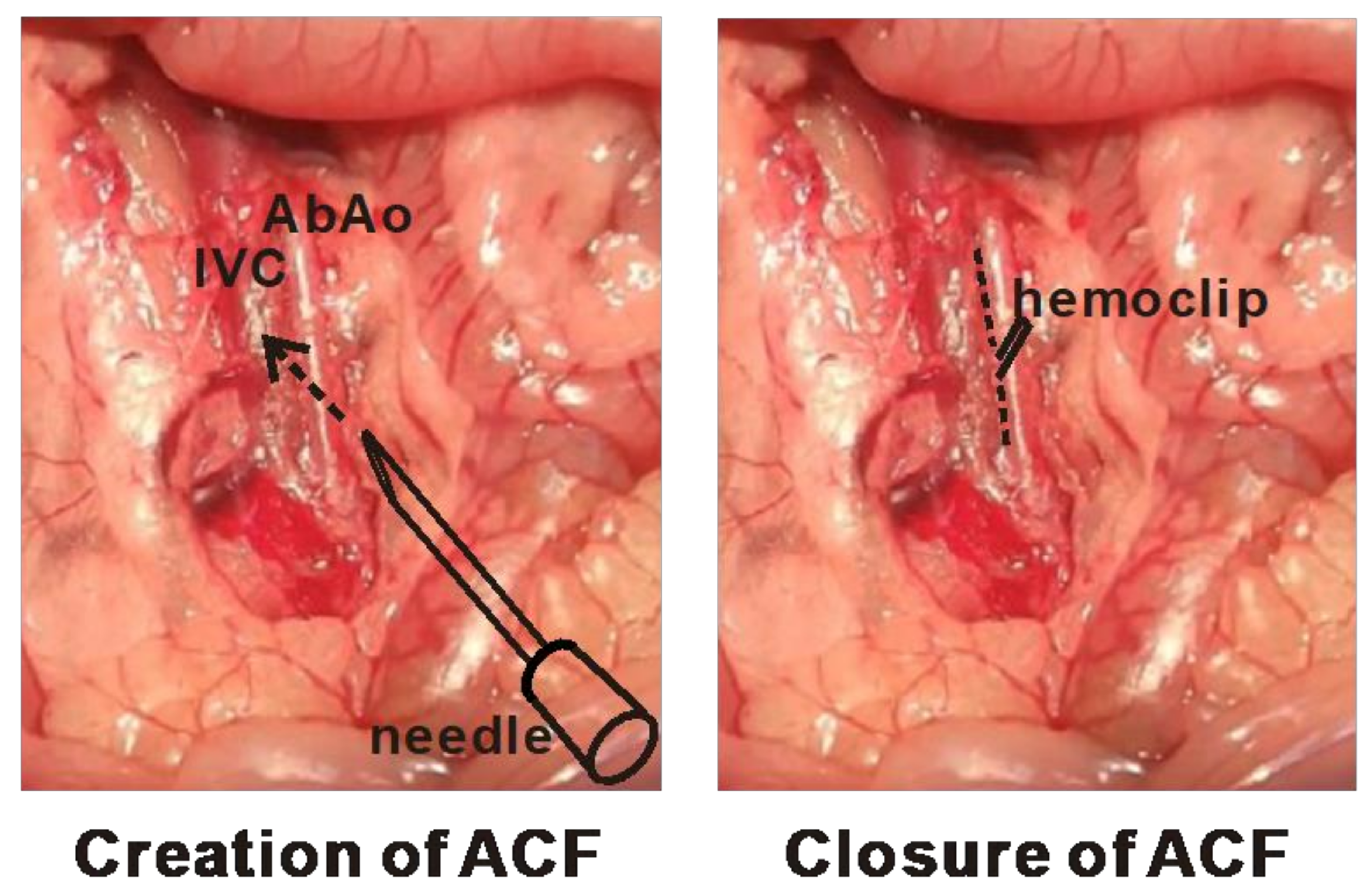
4.1. Animal Surgeries for the Creation and Correction of Cardiac VO
4.2. EchocarDiography and Invasive Hemodynamic Assessment
4.3. Transcriptomics Analysis of Energy Metabolic Pathways
4.4. Targeted Metabolomics Analysis
4.5. Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (qPCR)
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaasch, W.H.; Meyer, T.E. Left ventricular response to mitral regurgitation: Implications for management. Circulation 2008, 118, 2298–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Salvo, T.G.; Acker, M.A.; Dec, G.W.; Byrne, J.G. Mitral valve surgery in advanced heart failure. J. Am. Coll. Cardiol. 2010, 55, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Members, W.C.; Otto, C.M.; Nishimura, R.A.; Bonow, R.O.; Carabello, B.A.; Erwin, J.P., III; Gentile, F.; Jneid, H.; Krieger, E.V.; Mack, M. 2020 ACC/AHA guideline for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2021, 77, e25–e197. [Google Scholar]
- Ingwall, J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009, 81, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Recchia, F.A.; McConnell, P.I.; Bernstein, R.D.; Vogel, T.R.; Xu, X.; Hintze, T.H. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ. Res. 1998, 83, 969–979. [Google Scholar] [CrossRef] [Green Version]
- Vedala, G.; Herrero, P.; De las Fuentes, L.; Rogers, J.G.; Kelly, D.P.; Gropler, R.J. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 271–277. [Google Scholar]
- De las Fuentes, L.; Herrero, P.; Peterson, L.R.; Kelly, D.P.; Gropler, R.J.; Dávila-Román, V.G. Myocardial Fatty Acid Metabolism Independent Predictor of Left Ventricular Mass in Hypertensive Heart Disease. Hypertension 2003, 41, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Brower, G.L.; Henegar, J.R.; Janicki, J.S. Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. Am. J. Physiol. Heart Circ. Physiol 1996, 271, H2071–H2078. [Google Scholar] [CrossRef]
- Huang, M.; Hester, R.L.; Guyton, A.C. Hemodynamic changes in rats after opening an arteriovenous fistula. Am. J. Physiol. 1992, 262, H846–H851. [Google Scholar] [CrossRef]
- Wang, X.; Ren, B.; Liu, S.; Sentex, E.; Tappia, P.S.; Dhalla, N.S. Characterization of cardiac hypertrophy and heart failure due to volume overload in the rat. J. Appl. Physiol. 2003, 94, 752–763. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Pat, B.; Gladden, J.D.; Zheng, J.; Powell, P.; Wei, C.-C.; Cui, X.; Husain, A.; Dell’Italia, L.J. Dynamic molecular and histopathological changes in the extracellular matrix and inflammation in the transition to heart failure in isolated volume overload. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2251–H2260. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-J.; Huang, C.-C.; Chen, P.-R.; Lai, Y.-J. Remodeling matrix synthesis in a rat model of aortocaval fistula and the cyclic stretch: Impaction in pulmonary arterial hypertension-congenital heart disease. Int. J. Mol. Sci. 2020, 21, 4676. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Chen, A.; Lin, S.-Y.; Cheng, M.-L.; Shiao, M.-S.; Mao, T.-Y. A metabolomics approach to investigate the proceedings of mitochondrial dysfunction in rats from prediabetes to diabetes. Saudi J. Biol. Sci. 2021, 28, 4762–4769. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-J.; Tang, H.-Y.; Huang, C.-Y.; Lin, C.-M.; Ho, H.-Y.; Shiao, M.-S.; Cheng, M.-L. Metabolic signature differentiated diabetes mellitus from lipid disorder in elderly Taiwanese. J. Clin. Med. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, D.H.; Wang, Z.V. Glucose metabolism in cardiac hypertrophy and heart failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef]
- Liao, R.; Jain, M.; Cui, L.; D’Agostino, J.; Aiello, F.; Luptak, I.; Ngoy, S.; Mortensen, R.M.; Tian, R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002, 106, 2125–2131. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Musi, N.; D’Agostino, J.; Hirshman, M.F.; Goodyear, L.J. Increased adenosine monophosphate–activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation 2001, 104, 1664–1669. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.D.; Kaulbach, H.C.; Tian, R.; Hopkins, J.C.; Duffy, J.; Doetschman, T.; Minnemann, T.; Boers, M.-E.; Hadro, E.; Oberste-Berghaus, C. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Investig. 1999, 104, 1703–1714. [Google Scholar] [CrossRef] [Green Version]
- Wende, A.R.; Kim, J.; Holland, W.L.; Wayment, B.E.; O’Neill, B.T.; Tuinei, J.; Brahma, M.K.; Pepin, M.E.; McCrory, M.A.; Luptak, I. Glucose transporter 4-deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1098–H1108. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Donthi, R.V.; Wang, J.; Lange, A.J.; Watson, L.J.; Jones, S.P.; Epstein, P.N. Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2889–H2897. [Google Scholar] [CrossRef] [Green Version]
- Donthi, R.V.; Ye, G.; Wu, C.; McClain, D.A.; Lange, A.J.; Epstein, P.N. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J. Biol. Chem. 2004, 279, 48085–48090. [Google Scholar] [CrossRef] [Green Version]
- Marsin, A.; Bertrand, L.; Rider, M.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Nascimben, L.; Ingwall, J.S.; Lorell, B.H.; Pinz, I.; Schultz, V.; Tornheim, K.; Tian, R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004, 44, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Young, M.E.; Yan, J.; Razeghi, P.; Cooksey, R.C.; Guthrie, P.H.; Stepkowski, S.M.; McClain, D.A.; Tian, R.; Taegtmeyer, H. Proposed regulation of gene expression by glucose in rodent heart. Gene Regul. Syst. Biol. 2007, 1, GRSB–S222. [Google Scholar] [CrossRef] [Green Version]
- Lunde, I.G.; Aronsen, J.M.; Kvaløy, H.; Qvigstad, E.; Sjaastad, I.; Tønnessen, T.; Christensen, G.; Grønning-Wang, L.M.; Carlson, C.R. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genom. 2012, 44, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.P.; Zachara, N.E.; Ngoh, G.A.; Hill, B.G.; Teshima, Y.; Bhatnagar, A.; Hart, G.W.; Marbán, E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 2008, 117, 1172–1182. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Marchase, R.B.; Chatham, J.C. Increased O-GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1391–H1399. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.J.; McDonough, P.M.; Swanson, E.; Trost, S.U.; Suzuki, M.; Fukuda, M.; Dillmann, W.H. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J. Biol. Chem. 2003, 278, 44230–44237. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Belke, D.; Suarez, J.; Swanson, E.; Clark, R.; Hoshijima, M.; Dillmann, W.H. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ. Res. 2005, 96, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umapathi, P.; Mesubi, O.O.; Banerjee, P.S.; Abrol, N.; Wang, Q.; Luczak, E.D.; Wu, Y.; Granger, J.M.; Wei, A.-C.; Reyes Gaido, O.E. Excessive O-GlcNAcylation causes heart failure and sudden death. Circulation 2021, 143, 1687–1703. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Rutkowsky, J.M.; Knotts, T.A.; Ono-Moore, K.D.; McCoin, C.S.; Huang, S.; Schneider, D.; Singh, S.; Adams, S.H.; Hwang, D.H. Acylcarnitines activate proinflammatory signaling pathways. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, E1378–E1387. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Knotts, T.A.; Chavez, J.A.; Wang, L.-P.; Hoehn, K.L.; Summers, S.A. Lipid mediators of insulin resistance. Nutr. Rev. 2007, 65, S39–S46. [Google Scholar] [CrossRef]
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006, 55, S9–S15. [Google Scholar] [CrossRef] [Green Version]
- Schooneman, M.G.; Vaz, F.M.; Houten, S.M.; Soeters, M.R. Acylcarnitines Reflecting or Inflicting Insulin Resistance? Diabetes 2013, 62, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Masumura, Y.; Kobayashi, A.; Yamazaki, N. Myocardial free carnitine and fatty acylcarnitine levels in patients with chronic heart failure. Jpn. Circ. J. 1990, 54, 1471–1476. [Google Scholar] [CrossRef]
- Fujino, T.; Kinugawa, K.; Hatano, M.; Nitta, D.; Imamura, T.; Maki, H.; Amiya, E.; Komuro, I. Serum Acylcarnitine Concentration is Elevated in Patients with Acute Decompensated Heart Failure. J. Card. Fail. 2014, 10, S164. [Google Scholar] [CrossRef]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Thompson Legault, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A. Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Kelly, J.P.; McGarrah, R.W.; Hellkamp, A.S.; Fiuzat, M.; Testani, J.M.; Wang, T.S.; Verma, A.; Samsky, M.D.; Donahue, M.P. Prognostic implications of long-chain acylcarnitines in heart failure and reversibility with mechanical circulatory support. J. Am. Coll. Cardiol. 2016, 67, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.V.; Joseph, J.W.; Ilkayeva, O.; Burgess, S.; Lu, D.; Ronnebaum, S.M.; Odegaard, M.; Becker, T.C.; Sherry, A.D.; Newgard, C.B. Compensatory Responses to Pyruvate Carboxylase Suppression in Islet β-Cells Preservation of Glucose-Stimulated Insulin Secretion. J. Biol. Chem. 2006, 281, 22342–22351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, M.A.; Gómez, M.A.; Guillén, F.; Börnstein, B.; Campos, Y.; Rubio, J.; Carlos, S.; Arenas, J.n. Myocardial carnitine and carnitine palmitoyltransferase deficiencies in patients with severe heart failure. Biochim. Biophys. Acta 2000, 1502, 330–336. [Google Scholar] [CrossRef] [Green Version]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ. Heart Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M.; Noland, R.C.; Kovalik, J.-P.; Seiler, S.E.; Davies, M.N.; DeBalsi, K.L.; Ilkayeva, O.R.; Stevens, R.D.; Kheterpal, I.; Zhang, J. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012, 15, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Ebeling, P.; Tuominen, J.A.; Arenas, J.; Garcia-Benayas, C.; Koivisto, V.A. The association of acetyl-L-carnitine with glucose and lipid metabolism in human muscle in vivo: The effect of hyperinsulinemia. Metabolism 1997, 46, 1454–1457. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L. The failing heart relies on ketone bodies as a fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Russell, R., 3rd; Taegtmeyer, H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J. Clin. Investig. 1991, 87, 384. [Google Scholar] [CrossRef] [Green Version]
- Turer, A.T. Using metabolomics to assess myocardial metabolism and energetics in heart failure. J. Mol. Cell. Cardiol. 2013, 55, 12–18. [Google Scholar] [CrossRef]
- Gupte, A.A.; Hamilton, D.J.; Cordero-Reyes, A.M.; Youker, K.A.; Yin, Z.; Estep, J.D.; Stevens, R.D.; Wenner, B.; Ilkayeva, O.; Loebe, M. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ. Cardiovasc. Genet. 2014, 7, 266–276. [Google Scholar] [CrossRef]
- Birner, C.; Dietl, A.; Deutzmann, R.; Schröder, J.; Schmid, P.; Jungbauer, C.; Resch, M.; Endemann, D.; Stark, K.; Riegger, G. Proteomic profiling implies mitochondrial dysfunction in tachycardia-induced heart failure. J. Card. Fail. 2012, 18, 660–673. [Google Scholar] [CrossRef]
- Cieniewski-Bernard, C.; Mulder, P.; Henry, J.-P.; Drobecq, H.; Dubois, E.; Pottiez, G.; Thuillez, C.; Amouyel, P.; Richard, V.; Pinet, F. Proteomic analysis of left ventricular remodeling in an experimental model of heart failure. J. Proteome Res. 2008, 7, 5004–5016. [Google Scholar] [CrossRef]
- Hammer, E.; Goritzka, M.; Ameling, S.; Darm, K.; Steil, L.; Klingel, K.; Trimpert, C.; Herda, L.R.; Dörr, M.; Kroemer, H.K. Characterization of the human myocardial proteome in inflammatory dilated cardiomyopathy by label-free quantitative shotgun proteomics of heart biopsies. J. Proteome Res. 2011, 10, 2161–2171. [Google Scholar] [CrossRef]
- Bugger, H.; Schwarzer, M.; Chen, D.; Schrepper, A.; Amorim, P.A.; Schoepe, M.; Nguyen, T.D.; Mohr, F.W.; Khalimonchuk, O.; Weimer, B.C. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc. Res. 2010, 85, 376–384. [Google Scholar] [CrossRef]
- Shibayama, J.; Yuzyuk, T.N.; Cox, J.; Makaju, A.; Miller, M.; Lichter, J.; Li, H.; Leavy, J.D.; Franklin, S.; Zaitsev, A.V. Metabolic remodeling in moderate synchronous versus dyssynchronous pacing-induced heart failure: Integrated metabolomics and proteomics study. PLoS ONE 2015, 10, e0118974. [Google Scholar] [CrossRef]
- Des Rosiers, C.; Labarthe, F.; Lloyd, S.G.; Chatham, J.C. Cardiac anaplerosis in health and disease: Food for thought. Cardiovasc. Res. 2011, 90, 210–219. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tung, Y.-C.; Cheng, M.-L.; Wu, L.-S.; Tang, H.-Y.; Huang, C.-Y.; Chang, G.-J.; Chang, C.-J. Derangements and Reversibility of Energy Metabolism in Failing Hearts Resulting from Volume Overload: Transcriptomics and Metabolomics Analyses. Int. J. Mol. Sci. 2022, 23, 6809. https://doi.org/10.3390/ijms23126809
Tung Y-C, Cheng M-L, Wu L-S, Tang H-Y, Huang C-Y, Chang G-J, Chang C-J. Derangements and Reversibility of Energy Metabolism in Failing Hearts Resulting from Volume Overload: Transcriptomics and Metabolomics Analyses. International Journal of Molecular Sciences. 2022; 23(12):6809. https://doi.org/10.3390/ijms23126809
Chicago/Turabian StyleTung, Ying-Chang, Mei-Ling Cheng, Lung-Sheng Wu, Hsiang-Yu Tang, Cheng-Yu Huang, Gwo-Jyh Chang, and Chi-Jen Chang. 2022. "Derangements and Reversibility of Energy Metabolism in Failing Hearts Resulting from Volume Overload: Transcriptomics and Metabolomics Analyses" International Journal of Molecular Sciences 23, no. 12: 6809. https://doi.org/10.3390/ijms23126809
APA StyleTung, Y.-C., Cheng, M.-L., Wu, L.-S., Tang, H.-Y., Huang, C.-Y., Chang, G.-J., & Chang, C.-J. (2022). Derangements and Reversibility of Energy Metabolism in Failing Hearts Resulting from Volume Overload: Transcriptomics and Metabolomics Analyses. International Journal of Molecular Sciences, 23(12), 6809. https://doi.org/10.3390/ijms23126809