
SIRT2 Deficiency Exacerbates Hepatic Steatosis via a Putative Role of the ER Stress Pathway

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
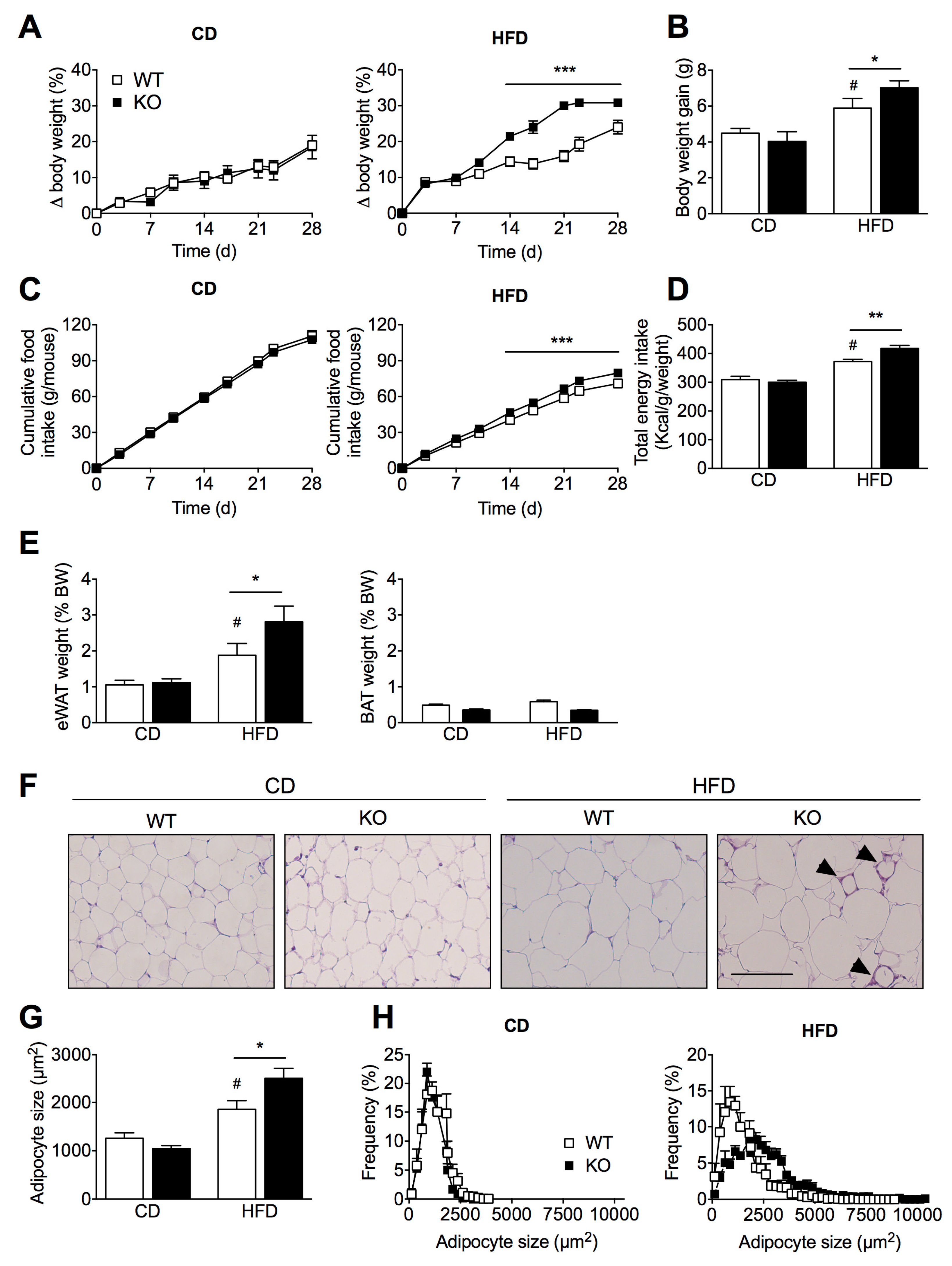
2.1. SIRT2 Deletion in Mice Aggravates HFD-Induced Obesity
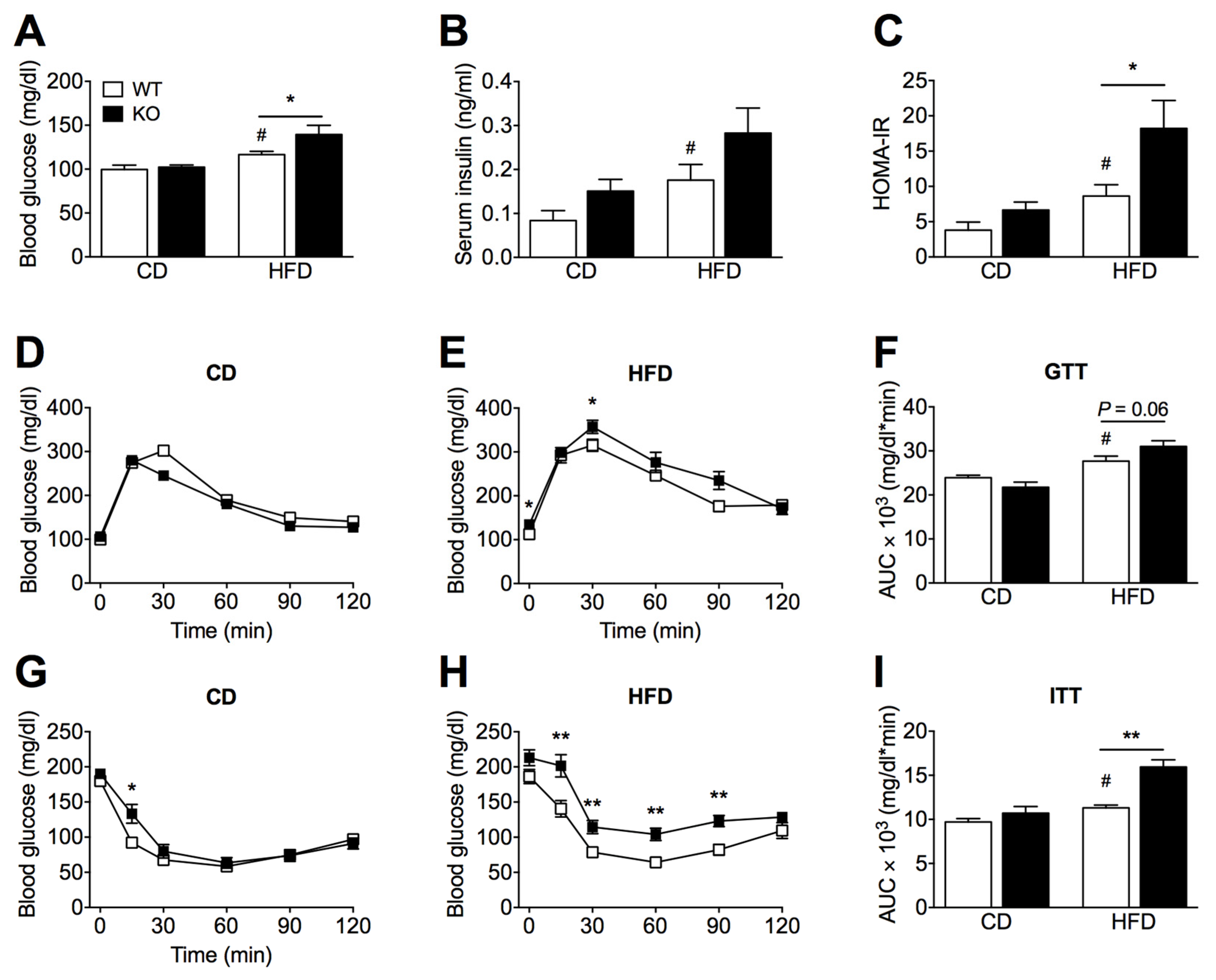
2.2. SIRT2 Deletion Promotes Glucose Intolerance and Insulin Resistance
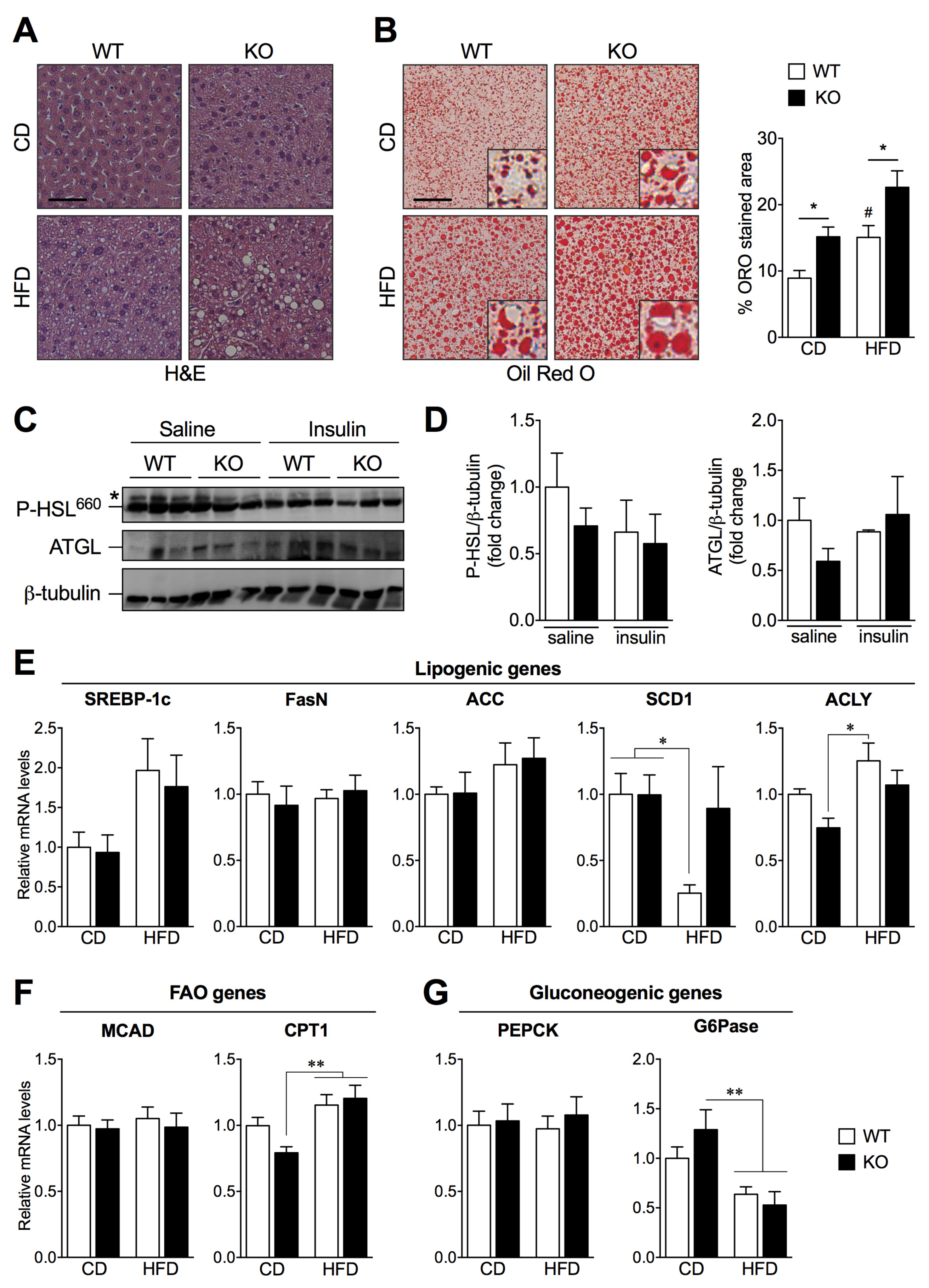
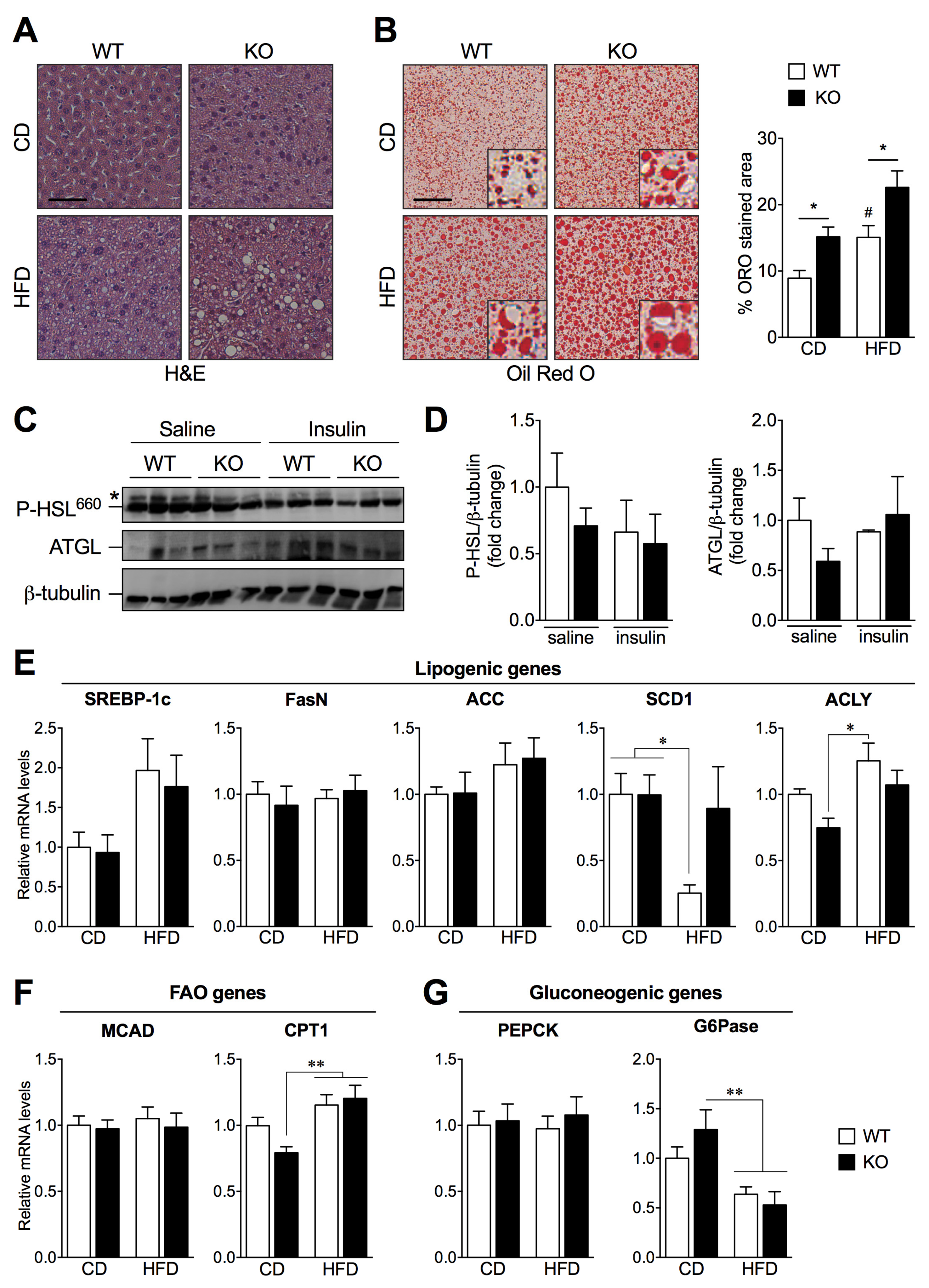
2.3. SIRT2 Deletion Promotes Hepatic Steatosis and Alters the Expression of Key Lipogenic Genes
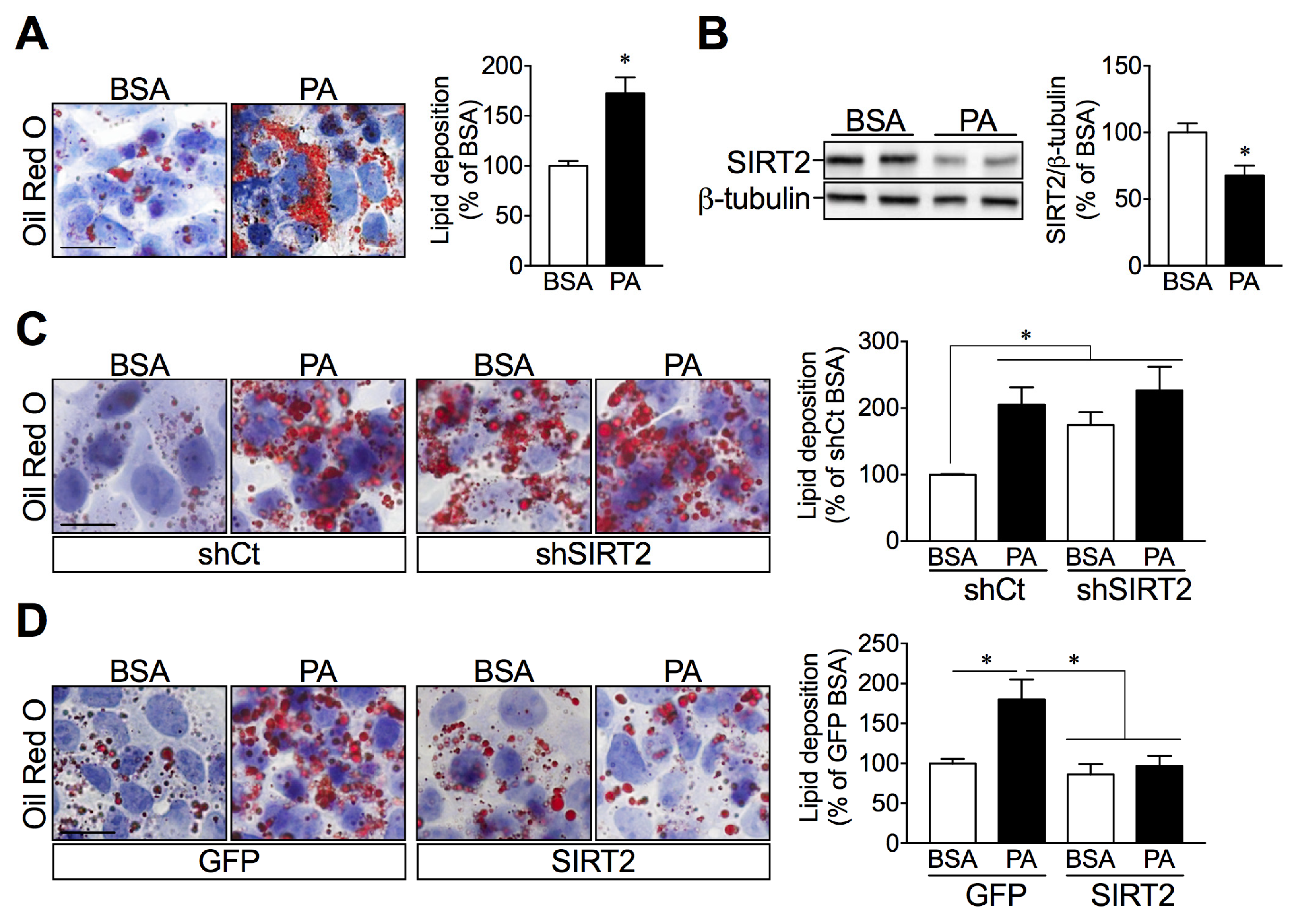
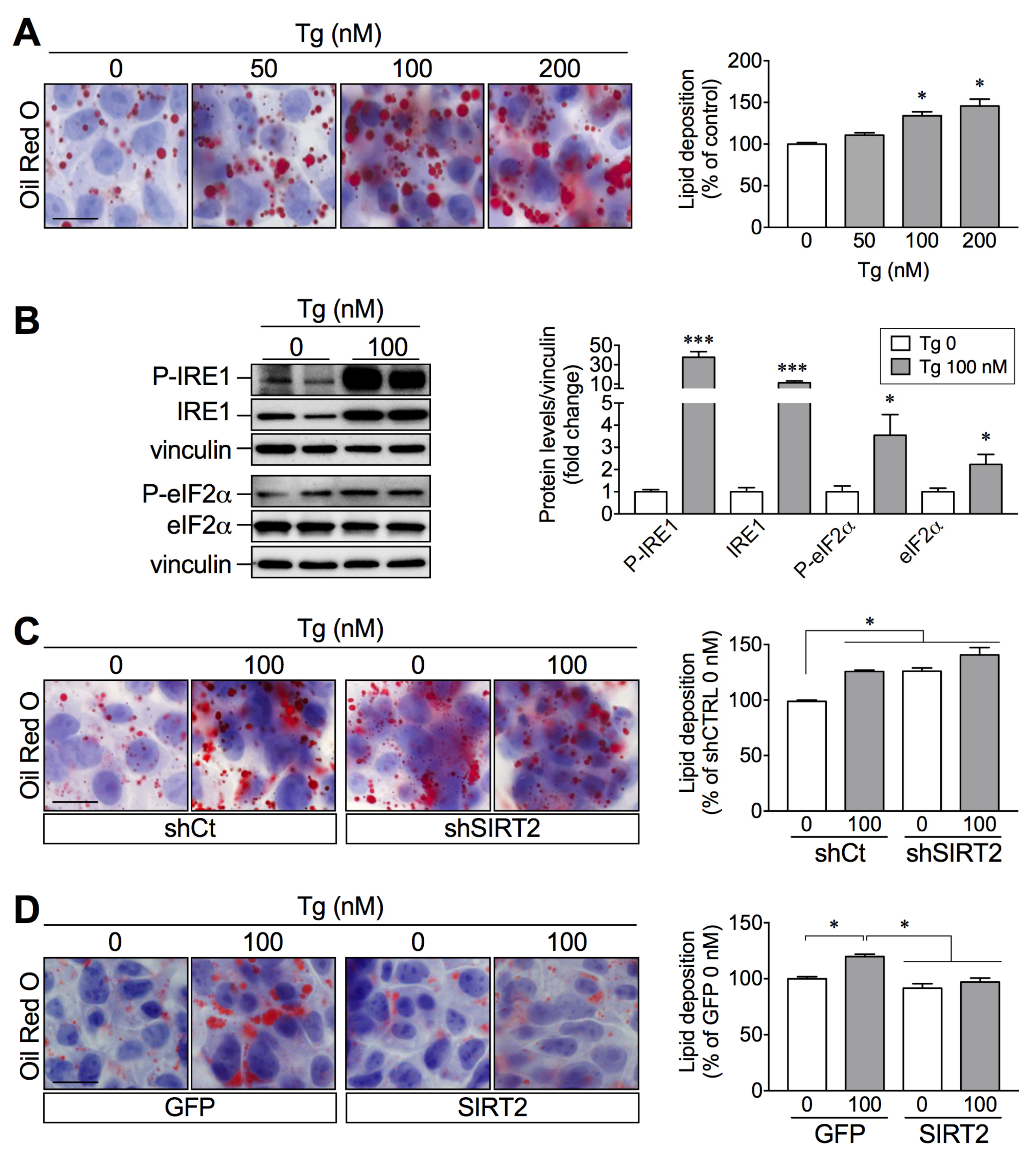
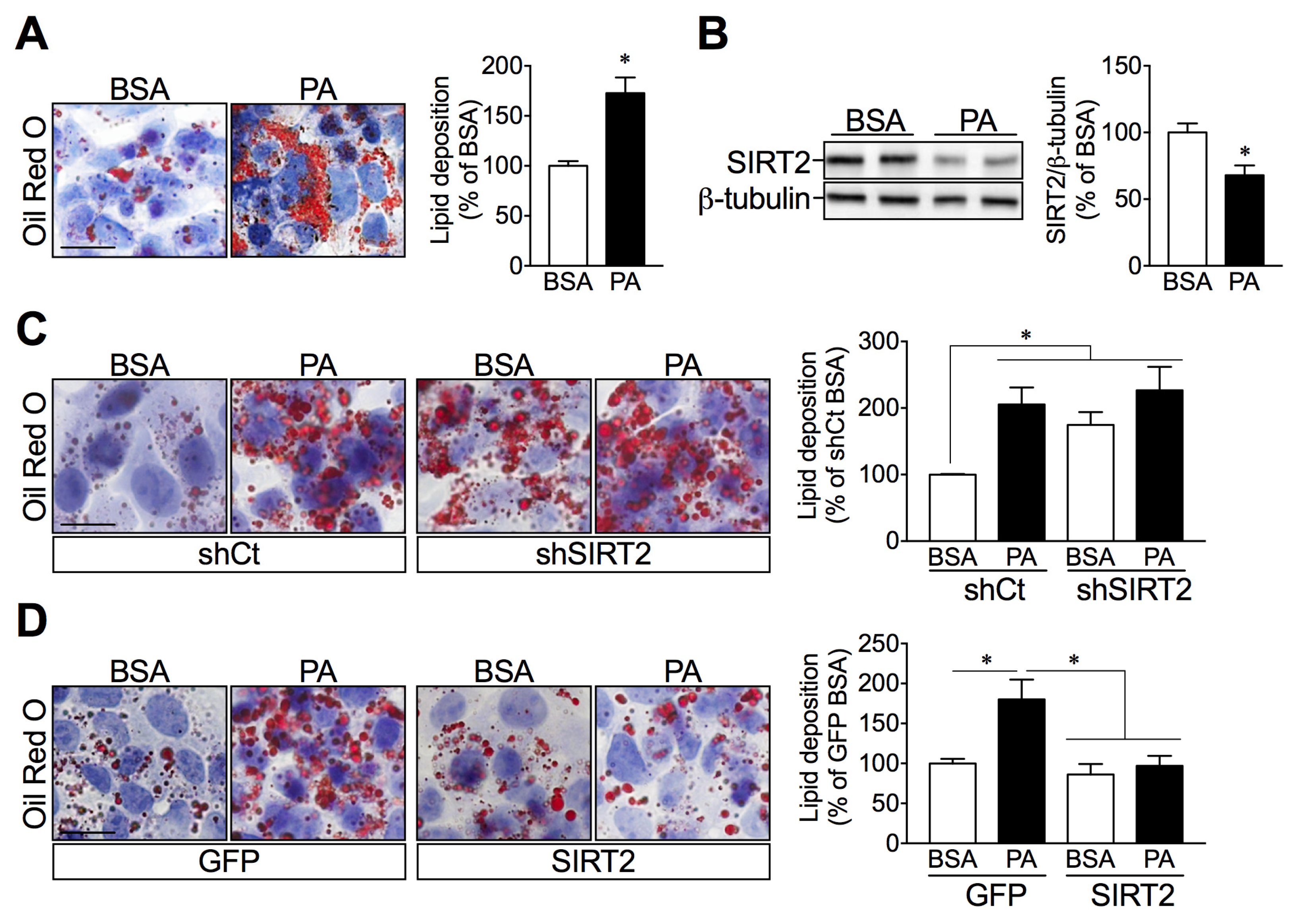
2.4. SIRT2 Silencing in HepG2 Cells Increases Lipid Deposition while SIRT2 Overexpression Attenuates Palmitate-Induced Lipid Overload
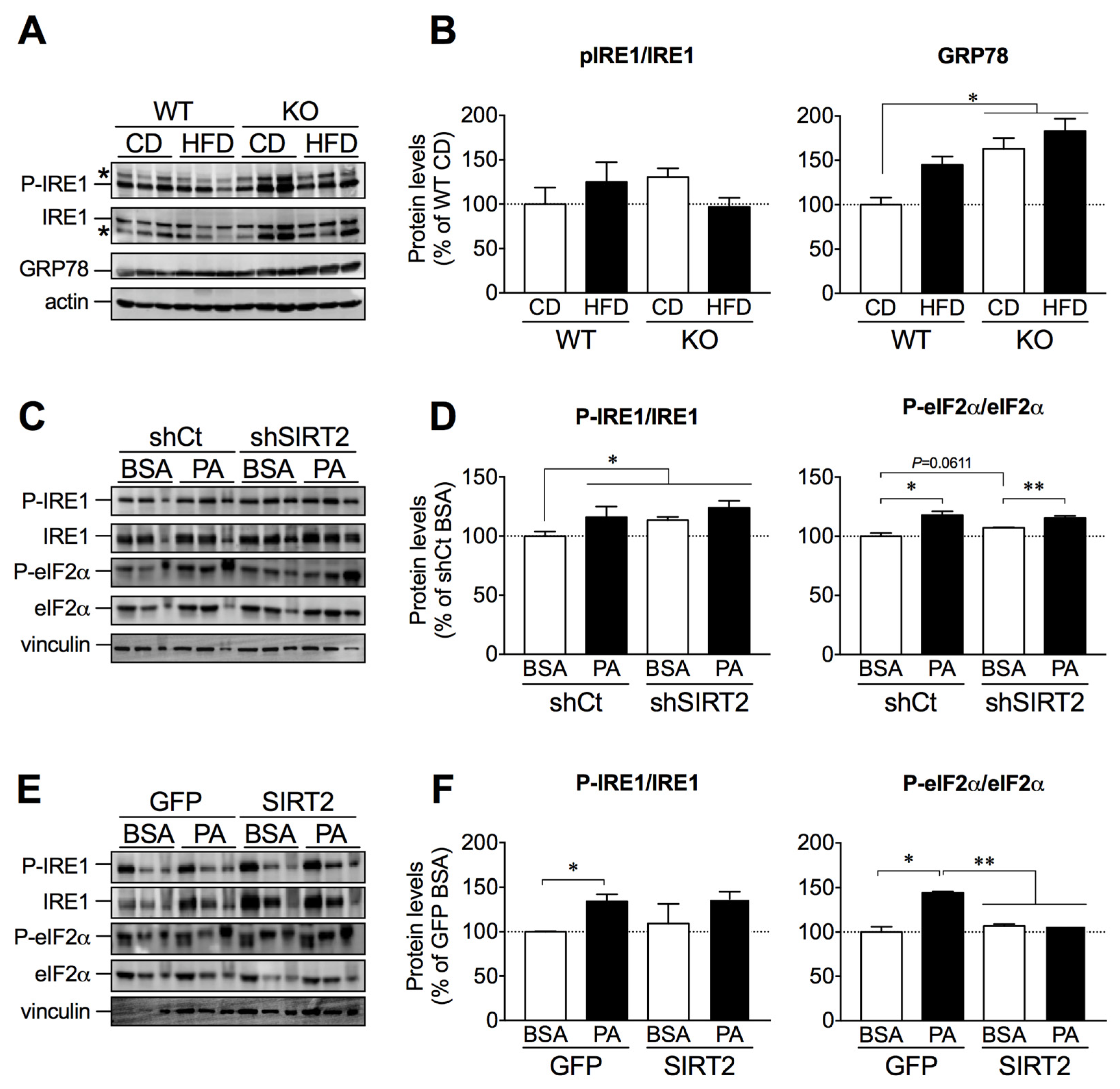
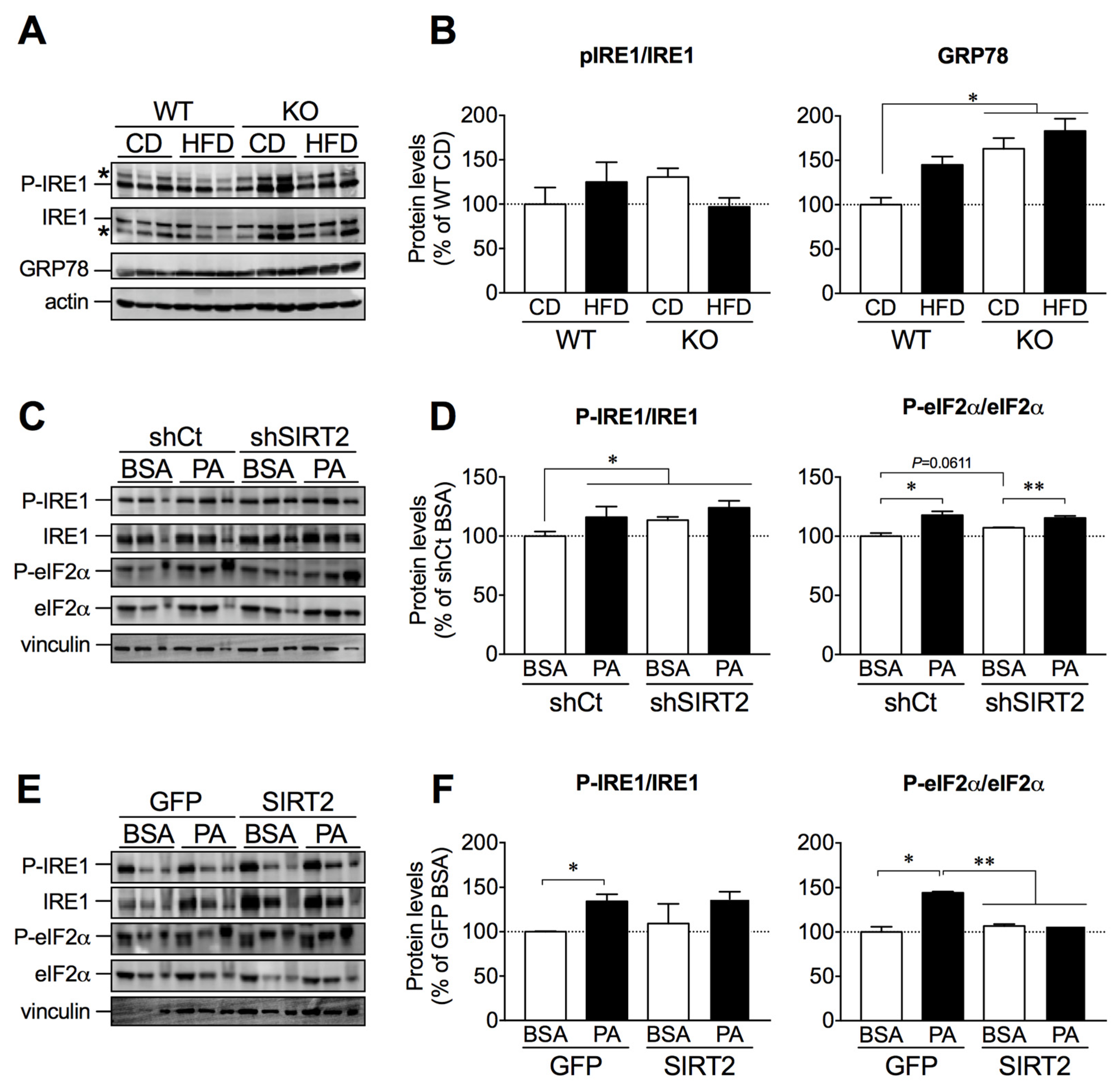
2.5. SIRT2 Links ER Stress and Hepatic Lipid Accumulation
2.6. SIRT2 Overexpression in HepG2 Cells Attenuates Palmitate-Induced ER Stress Activation
3. Discussion
4. Materials and Methods
4.1. Animals and Diets
4.2. Glucose and Insulin Tolerance Tests
4.3. Plasma Biochemistry
4.4. Tissue Histology
4.5. Cell Culture and Treatments
4.6. ORO Staining of HepG2 Cells
4.7. Production of Lentiviral Vectors
4.8. Generation of SIRT2-Silenced and SIRT2-Overexpressing Stable Transfectants
4.9. Western Blotting Analysis
4.10. Quantitative Real-Time PCR
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 299–310. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinology 2012, 8, 287–296. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Ungvari, Z.; Minor, R.K.; Le Couteur, D.G.; de Cabo, R. Are sirtuins viable targets for improving healthspan and lifespan? Nature reviews. Drug Discov. 2012, 11, 443–461. [Google Scholar] [CrossRef] [Green Version]
- Dryden, S.C.; Nahhas, F.A.; Nowak, J.E.; Goustin, A.S.; Tainsky, M.A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 2003, 23, 3173–3185. [Google Scholar] [CrossRef] [Green Version]
- Serrano, L.; Martínez-Redondo, P.; Marazuela-Duque, A.; Vazquez, B.N.; Dooley, S.J.; Voigt, P.; Beck, D.B.; Kane-Goldsmith, N.; Tong, Q.; Rabanal, R.M.; et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013, 27, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Gomes, P.; Outeiro, T.F.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef]
- Krishnan, J.; Danzer, C.; Simka, T.; Ukropec, J.; Walter, K.M.; Kumpf, S.; Mirtschink, P.; Ukropcova, B.; Gasperikova, D.; Pedrazzini, T.; et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012, 26, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Lemos, V.; de Oliveira, R.M.; Naia, L.; Szegö, É.; Ramos, E.; Pinho, S.; Magro, F.; Cavadas, C.; Rego, A.C.; Costa, V.; et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum. Mol. Genet. 2017, 26, 4105–4117. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Hu, F.; Wang, D.; Kang, X.; Feng, X.; Zhang, L.; Zhou, B.; Liu, S.; Yuan, G. SIRT2 prevents liver steatosis and metabolic disorders via deacetylation of HNF4α. Hepatology 2021, 74, 723–740. [Google Scholar] [CrossRef]
- Lantier, L.; Williams, A.S.; Hughey, C.C.; Bracy, D.P.; James, F.D.; Ansari, M.A.; Gius, D.; Wasserman, D.H. SIRT2 knockout exacerbates insulin resistance in high fat-fed mice. PLoS ONE 2018, 13, e0208634. [Google Scholar] [CrossRef]
- Kantartzis, K.; Fritsche, L.; Bombrich, M.; Machann, J.; Schick, F.; Staiger, H.; Kunz, I.; Schoop, R.; Lehn-Stefan, A.; Heni, M.; et al. Effects of resveratrol supplementation on liver fat content in overweight and insulin-resistant subjects: A randomized, double-blind, placebo-controlled clinical trial. Diabetes Obes. Metab. 2018, 20, 1793–1797. [Google Scholar] [CrossRef]
- Theodotou, M.; Fokianos, K.; Moniatis, D.; Kadlenic, R.; Chrysikou, A.; Aristotelous, A.; Mouzouridou, A.; Diakides, J.; Stavrou, E. Effect of resveratrol on non-alcoholic fatty liver disease. Exp. Ther. Med. 2019, 18, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J. Inflammation is necessary for long-term but not short-term high-fat diet–induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Tong, Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1’s repressive interaction with PPARγ. Mol. Biol. Cell 2009, 20, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D.G. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, G.; Davaakhuu, G.; Kaplun, L.; Chung, W.-C.; Rana, A.; Atfi, A.; Miele, L.; Tzivion, G. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J. Biol. Chem. 2014, 289, 6054–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Liu, Y.-H.; Fu, Y.-C.; Liu, X.-M.; Zhou, X.-H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- Wang, R.-H.; Li, C.; Deng, C.-X. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int. J. Biol. Sci. 2010, 6, 682. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 2013, 5, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Bashmakov, Y.; Shimomura, I.; Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. USA 1998, 95, 5987–5992. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Powell, C.A.; Kay, M.K.; Sonkar, R.; Meruvu, S.; Choudhury, M. Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation. Int. J. Mol. Sci. 2021, 22, 1822. [Google Scholar] [CrossRef]
- Achard, C.S.; Laybutt, D.R. Lipid-induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: Adaptation with enhanced insulin signaling or insulin resistance. Endocrinology 2012, 153, 2164–2177. [Google Scholar] [CrossRef]
- DeZwaan-McCabe, D.; Sheldon, R.D.; Gorecki, M.C.; Guo, D.-F.; Gansemer, E.R.; Kaufman, R.J.; Rahmouni, K.; Gillum, M.P.; Taylor, E.B.; Teesch, L.M. ER stress inhibits liver fatty acid oxidation while unmitigated stress leads to anorexia-induced lipolysis and both liver and kidney steatosis. Cell Rep. 2017, 19, 1794–1806. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Zheng, Z.; Mendez, R.; Ha, S.-W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 2012, 211, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Xu, D.; She, L.; Wang, Z.; Yang, N.; Sun, R.; Zhang, Y.; Yan, C.; Wei, Q.; Aa, J.; et al. Silybin inhibits NLRP3 inflammasome assembly through the NAD(+)/SIRT2 pathway in mice with nonalcoholic fatty liver disease. FASEB J. 2018, 32, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Bang, I.H.; Kwon, O.K.; Hao, L.; Park, D.; Chung, M.-J.; Oh, B.-C.; Lee, S.; Bae, E.J.; Park, B.-H. Deacetylation of XBP1s by sirtuin 6 confers resistance to ER stress-induced hepatic steatosis. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Jiang, J.; Zhang, G.; Bu, Y.; Zhang, G.; Zhao, X. Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS ONE 2017, 12, e0183541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, P.; Leal, H.; Mendes, A.F.; Reis, F.; Cavadas, C. Dichotomous sirtuins: Implications for drug discovery in neurodegenerative and cardiometabolic diseases. Trends Pharmacol. Sci. 2019, 40, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Liu, H.; Agostini, M.; Yousefi, S.; Perren, A.; Tschan, M.; Mak, T.; Melino, G.; Simon, H.-U. p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell Death Differ. 2013, 20, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- de Almeida, L.P.; Zala, D.; Aebischer, P.; Deglon, N. Neuroprotective effect of a CNTF-expressing lentiviral vector in the quinolinic acid rat model of Huntington’s disease. Neurobiol. Dis. 2001, 8, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Cunha-Santos, J.; Duarte-Neves, J.; Carmona, V.; Guarente, L.; De Almeida, L.P.; Cavadas, C. Caloric restriction blocks neuropathology and motor deficits in Machado–Joseph disease mouse models through SIRT1 pathway. Nat. Commun. 2016, 7, 11445. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, H.; Cardoso, J.; Valério, P.; Quatorze, M.; Carmona, V.; Cunha-Santos, J.; Pereira de Almeida, L.; Pereira, C.; Cavadas, C.; Gomes, P. SIRT2 Deficiency Exacerbates Hepatic Steatosis via a Putative Role of the ER Stress Pathway. Int. J. Mol. Sci. 2022, 23, 6790. https://doi.org/10.3390/ijms23126790
Leal H, Cardoso J, Valério P, Quatorze M, Carmona V, Cunha-Santos J, Pereira de Almeida L, Pereira C, Cavadas C, Gomes P. SIRT2 Deficiency Exacerbates Hepatic Steatosis via a Putative Role of the ER Stress Pathway. International Journal of Molecular Sciences. 2022; 23(12):6790. https://doi.org/10.3390/ijms23126790
Chicago/Turabian StyleLeal, Helena, João Cardoso, Patrícia Valério, Marta Quatorze, Vítor Carmona, Janete Cunha-Santos, Luís Pereira de Almeida, Cláudia Pereira, Cláudia Cavadas, and Pedro Gomes. 2022. "SIRT2 Deficiency Exacerbates Hepatic Steatosis via a Putative Role of the ER Stress Pathway" International Journal of Molecular Sciences 23, no. 12: 6790. https://doi.org/10.3390/ijms23126790
APA StyleLeal, H., Cardoso, J., Valério, P., Quatorze, M., Carmona, V., Cunha-Santos, J., Pereira de Almeida, L., Pereira, C., Cavadas, C., & Gomes, P. (2022). SIRT2 Deficiency Exacerbates Hepatic Steatosis via a Putative Role of the ER Stress Pathway. International Journal of Molecular Sciences, 23(12), 6790. https://doi.org/10.3390/ijms23126790