
Chl1, an ATP-Dependent DNA Helicase, Inhibits DNA:RNA Hybrids Formation at DSB Sites to Maintain Genome Stability in S. pombe

Abstract
:1. Introduction
2. Results
2.1. Localization of Chl1 at DSB
2.2. Rpc37 Recruits Chl1 to DSB Sites
2.3. Chl1 Suppresses DNA:RNA Hybrid Accumulation at DSB Sites
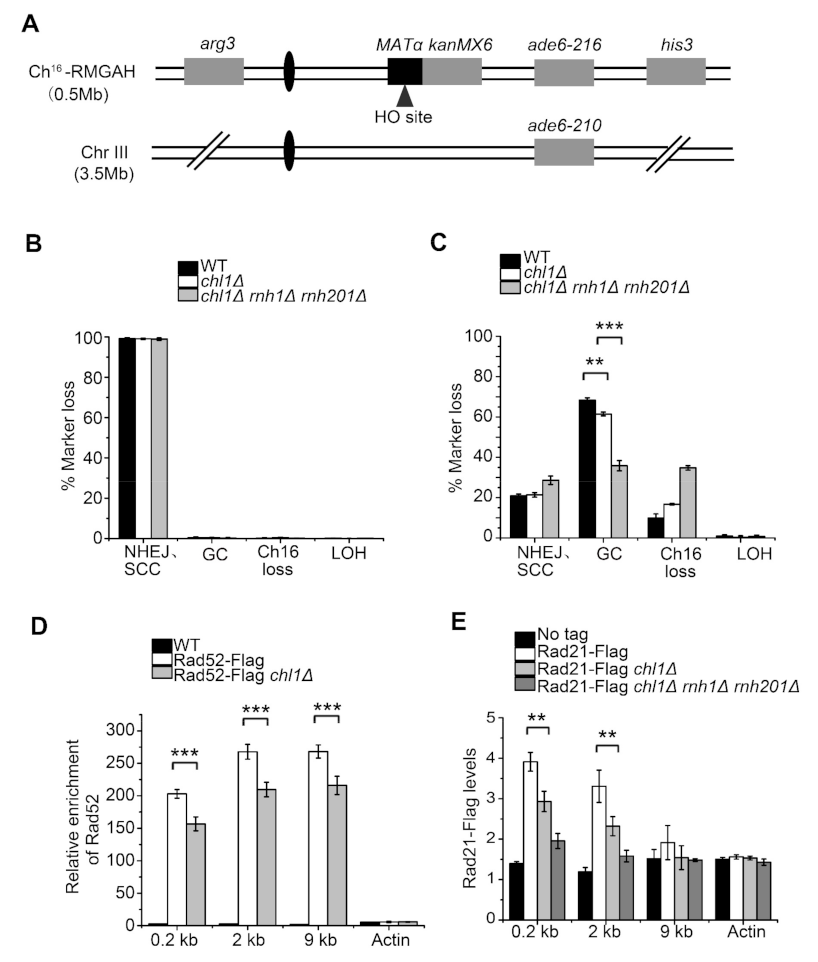
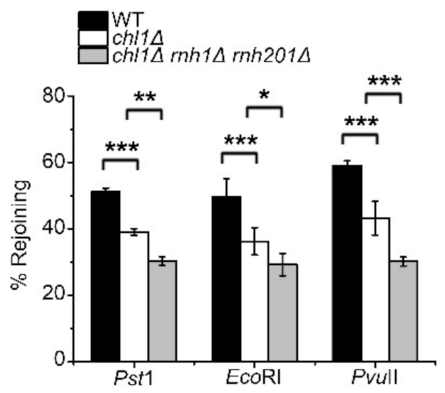
2.4. Chl1 Mutants Display Reduced DNA Repair Efficiency by HR and NHEJ Pathways
3. Discussion
4. Materials and Methods
4.1. Yeast Strains and Growth Condition
4.2. HO-Induced DSB
4.3. Spot Assay
4.4. Yeast Two-Hybrid Analysis
4.5. ChIP Assays
4.6. DNA:RNA Hybrid Immunoprecipitation (DRIP)
4.7. Coimmunoprecipitation (Co-IP)
4.8. Mass Spectrometry
4.9. Western Blot
4.10. Site-Specific DSB Assay
4.11. Plasmid Rejoining Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yasuhira, S.; Saito, T.; Maesawa, C.; Masuda, T. Sensor and effector kinases in DNA damage checkpoint regulate capacity for homologous recombination repair of fission yeast in G2 phase. DNA Repair 2012, 11, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef]
- Lin, Z.; Kong, H.; Nei, M.; Ma, H. Origins and evolution of the recA/RAD51 gene family. evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 10328–10333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persky, N.S.; Lovett, S.T. Mechanisms of recombination. lessons from E. coli. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hua, Y.; Li, R.; Kong, D. Cdc24 Is Essential for Long-range End Resection in the Repair of Double-stranded DNA Breaks. J. Biol. Chem. 2016, 291, 24961–24973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Chung, W.-H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 Helicase and Two Nucleases Dna2 and Exo1 Resect DNA Double-Strand Break Ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S.; Rothstein, R.; Lisby, M. Mechanisms and Regulation of Mitotic Recombination in Saccharomyces cerevisiae. Genetics 2014, 198, 795–835. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [Green Version]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar]
- West, S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003, 4, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E.; Ira, G.; Malkova, A.; Sugawara, N. Repairing a double-strand chromosome break by homologous recombination. revisiting Robin Holliday’s model. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2004, 359, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Palmbos, P.L.; Wu, D.; Wilson, T.E. Nonhomologous End Joining in Yeast. Annu. Rev. Genet. 2005, 39, 431–451. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Germain, D.R.; Poon, H.-Y.; Hildebrandt, M.R.; Monckton, E.A.; McDonald, D.; Hendzel, M.; Godbout, R. DEAD Box 1 Facilitates Removal of RNA and Homologous Recombination at DNA Double-Strand Breaks. Mol. Cell. Biol. 2016, 36, 2794–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 2016, 167, 1001–1013.e7. [Google Scholar] [CrossRef] [Green Version]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Ba, Z.; Gao, M.; Wu, Y.; Ma, Y.; Amiard, S.; White, C.I.; Danielsen, J.M.R.; Yang, Y.-G.; Qi, Y. A Role for Small RNAs in DNA Double-Strand Break Repair. Cell 2012, 149, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Hua, Y.; Wang, J.; Li, L.; Yuan, J.; Zhang, B.; Wang, Z.; Ji, J.; Kong, D. RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell 2021, 184, 1314–1329.e1310. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Puget, N.; Lin, Y.-L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Crouch, R.J.; Ribonuclease, H. The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.-T.; Hawley, B.R.; Skalka, G.L.; Baldock, R.A.; Smith, E.M.; Bader, A.S.; Malewicz, M.; Watts, F.Z.; Wilczynska, A.; Bushell, M. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018, 9, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuhide, H.; Lahti, J.M. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000, 28, 917–924. [Google Scholar]
- Yu, Y.; Ren, J.-Y.; Zhang, J.-M.; Suo, F.; Fang, X.-F.; Wu, F.; Du, L.-L. A proteome-wide visual screen identifies fission yeast proteins localizing to DNA double-strand breaks. DNA Repair 2013, 12, 433–443. [Google Scholar] [CrossRef]
- Shah, N.; Inoue, A.; Lee, S.W.; Beishline, K.; Lahti, J.M.; Noguchi, E. Roles of ChlR1 DNA helicase in replication recovery from DNA damage. Exp. Cell Res. 2013, 319, 2244–2253. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Ooka, M.; Kawasumi, R.; Miyata, K.; Takata, M.; Hirota, K.; Branzei, D. Warsaw breakage syndrome DDX11 helicase acts jointly with RAD17 in the repair of bulky lesions and replication through abasic sites. Proc. Natl. Acad. Sci. USA 2018, 115, 8412–8417. [Google Scholar] [CrossRef] [Green Version]
- Gerring, S.L.; Spencer, F.; Hieter, P. The CHL 1 (CTF 1) gene product of Saccharomyces cerevisiae is important for chromosome transmission and normal cell cycle progression in G2/M. EMBO J. 1990, 9, 4347–4358. [Google Scholar]
- Skibbens, R.V. Chl1p, a DNA Helicase-Like Protein in Budding Yeast, Functions in Sister-Chromatid Cohesion. Genetics 2004, 166, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.S.; Kroll, E.S.; Lundblad, V.; Spencer, F.A. Sacharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol. Cell. Biol. 2001, 21, 3144–3158. [Google Scholar]
- Samora Catarina, P.; Saksouk, J.; Goswami, P.; Wade Ben, O.; Singleton Martin, R.; Bates Paul, A.; Lengronne, A.; Costa, A.; Uhlmann, F. Ctf4 Links DNA Replication with Sister Chromatid Cohesion Establishment by Recruiting the Chl1 Helicase to the Replisome. Mol. Cell 2016, 63, 371–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinline-Purvis, H.; Savory, A.P.; Cullen, J.K.; Davé, A.; Moss, J.; Bridge, W.L.; Marguerat, S.; Bähler, J.; Ragoussis, J.; Mott, R.; et al. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 2009, 28, 3400–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, C.-C.; Deegan, R.S.; Subramanian, L.; Gal, C.; Sarkar, S.; Blaikley, E.J.; Walker, C.; Hulme, L.; Bernhard, E.; Codlin, S.; et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat. Commun. 2014, 5, 4091. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, C.; Nasmyth, K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr. Biol. 2001, 11, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Ström, L.; Lindroos, H.B.; Shirahige, K.; Sjögren, C. Postreplicative Recruitment of Cohesin to Double-Strand Breaks Is Required for DNA Repair. Mol. Cell 2004, 16, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zhang, N.; Pati, D. Cohesin subunit RAD21: From biology to disease. Gene 2020, 758, 144966. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P.; Armache, K.J.; Baumli, S.; Benkert, S.; Brueckner, F.; Buchen, C.; Damsma, G.E.; Dengl, S.; Geiger, S.R.; Jasiak, A.J.; et al. Structure of eukaryotic RNA polymerases. Annu. Rev. Biophys. 2008, 37, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Amann, J.; Valentine, M.; Kidd, V.J.; Lahti, J.M. Localization of chi1-related helicase genes to human chromosome regions 12p11 and 12p13. similarity between parts of these genes and conserved human telomeric-associated DNA. Genomics 1996, 32, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Amann, J.; Kidd, V.J.; Lahti, J.M. Characterization of Putative Human Homologues of the Yeast Chromosome Transmission Fidelity Gene, CHL1. J. Biol. Chem. 1997, 272, 3823–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Sommers, J.A.; Khan, I.; de Winter, J.P.; Brosh, R.M., Jr. Biochemical characterization of Warsaw breakage syndrome helicase. J. Biol. Chem. 2012, 287, 1007–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, A.; Shin, J.-H.; Kim, D.-H.; Bermudez, V.P.; Kelman, Z.; Seo, Y.-S.; Hurwitz, J. Studies with the Human Cohesin Establishment Factor, ChlR1. J. Biol. Chem. 2008, 283, 20925–20936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharti, S.K.; Khan, I.; Banerjee, T.; Sommers, J.A.; Wu, Y.; Brosh, R.M., Jr. Molecular functions and cellular roles of the ChlR1 (DDX11) helicase defective in the rare cohesinopathy Warsaw breakage syndrome. Cell. Mol. Life Sci. 2014, 71, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; George, F.; Kuper, J.; Hamon, F.; Shin-Ya, K.; Teulade-Fichou, M.-P.; Kisker, C.; Brosh, R.M., Jr. Specialization among Iron-Sulfur Cluster Helicases to Resolve G-quadruplex DNA Structures That Threaten Genomic Stability. J. Biol. Chem. 2013, 288, 28217–28229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Hundseth, K.; Ding, H.; Vidhyasagar, V.; Inoue, A.; Nguyen, C.-H.; Zain, R.; Lee, J.S.; Wu, Y. A Distinct Triplex DNA Unwinding Activity of ChlR1 Helicase. J. Biol. Chem. 2015, 290, 5174–5189. [Google Scholar] [CrossRef] [Green Version]
- Parish, J.L.; Rosa, J.; Wang, X.; Lahti, J.M.; Doxsey, S.J.; Androphy, E. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J. Cell Sci. 2006, 119, 4857–4865. [Google Scholar] [CrossRef]
- Laha, S.; Das, S.P.; Hajra, S.; Sau, S.; Sinha, P. The budding yeast protein Chl1p is required to preserve genome integrity upon DNA damage in S-phase. Nucleic Acids Res. 2006, 34, 5880–5891. [Google Scholar] [CrossRef] [Green Version]
- Forsburg, S.L.; Rhind, N. Basic methods for fission yeast. Yeast 2006, 23, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Hentges, P.; Van Driessche, B.; Tafforeau, L.; Vandenhaute, J.; Carr, A.M. Three novel antibiotic marker cassettes for gene disruption and marker switching inSchizosaccharomyces pombe. Yeast 2005, 22, 1013–1019. [Google Scholar] [CrossRef]
- Tang, Y.; Yin, Z.; Zeng, Y.; Zhang, Q.; Chen, L.; He, Y.; Lu, P.; Ye, D.; Zhang, X. MTOPVIB interacts with AtPRD1 and plays important roles in formation of meiotic DNA double-strand breaks in Arabidopsis. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.-L.; Nakamura, T.M.; Russell, P. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 2006, 20, 1583–1596. [Google Scholar] [CrossRef] [Green Version]
- Zaratiegui, M.; Castel, S.E.; Irvine, D.V.; Kloc, A.; Ren, J.; Li, F.; De Castro, E.; Marín, L.; Chang, A.-Y.; Goto, D.; et al. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature 2011, 479, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Taneja, N.; Zofall, M.; Balachandran, V.; Thillainadesan, G.; Sugiyama, T.; Wheeler, D.; Zhou, M.; Grewal, S.I. SNF2 Family Protein Fft3 Suppresses Nucleosome Turnover to Promote Epigenetic Inheritance and Proper Replication. Mol. Cell 2017, 66, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Reddy, B.; Thompson, J.; Wang, H.; Noma, K.-I.; Yates, J.R., III; Jia, S. Regulation of Set9-mediated H4K20 methylation by a PWWP domain protein. Mol. Cell 2009, 33, 428–437. [Google Scholar]
- Manolis, K.G.; Nimmo, E.R.; Hartsuiker, E.; Carr, A.M.; Jeggo, P.A.; Allshire, R.C. Novel functional requirements for non-homologous DNA end joining in Schizosaccharomyces pombe. EMBO J. 2001, 20, 210–221. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Genotype |
|---|---|
| LLD3650 | h- his3D1 ura4-D18 crb2Δ::ura4 rad22-CFP::KanMX6 arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 nmt41-HO-his3 |
| ywp746 | h- his3D1 ura4-D18 crb2Δ::ura4 rad22-CFP::KanMX6 arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 nmt41-HO-his3 chl1-flag::HphMX6 |
| SPJ1577 | h+ Leu1-32 ura4DS/E |
| ywp168 ywp1545 | h+ Leu1-32 ura4DS/E chl1Δ::HphMX6 h+ Leu1-32 ura4DS/E rnh1∆::NatMX4 rnh201∆::KanMX4 chl1∆::HphMX6 |
| ywp702 | h+ leu1-32 ura4 DS/E Chl1-5xflag::KanMX6 |
| ywp1108 | h+ leu1-32 ura4 DS/E rpc37-13xmyc::NatMX6 Chl1-5xflag::HphMX6 |
| ywp1106 | h+ leu1-32 ura4 DS/E rpc37-13xmyc::NatMX6 |
| ywp1005 | h- his3D1 ura4-D18 crb2Δ::ura4 rad22-CFP::KanMX6 nmt41-HO-his3arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 rpc37-5xflag::HphMX6 chl11Δ::NatMX6 |
| ywp1328 | h- his3D1 ura4-D18 crb2Δ::ura4 rad22-CFP::KanMX6 nmt41-HO-his3 arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 rpc37-5xflag::HphMX6 |
| ywp1330 | h- his3D1 ura4-D18 crb2Δ::ura4 rad22-CFP::KanMX6 nmt41-HO-his3 arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 chl1Δ::NatMX6 |
| ywp635 ywp637 | h+ arg3-D4 Ch16-RMGAH leu1::nmt41-Leu2 h+ arg3-D4 Ch16-RMGAH leu1::nmt41-HO-Leu2 |
| ywp659 ywp661 | h+ arg3-D4 Ch16-RMGAH leu1::nmt41-Leu2 chl1Δ::HphMX6 h+ arg3-D4 Ch16-RMGAH leu1::nmt41-HO-Leu2 chl1Δ::HphMX6 |
| ywp1705 | h+ arg3-D4 Ch16-RMGAH leu1::nmt41-Leu2 chl1Δ::HphMX6rnh1Δ::NatMX6 rnh201Δ::KanMX6 |
| ywp1707 | h+ arg3-D4 Ch16-RMGAH leu1::nmt41-HO-Leu2 chl1Δ::HphMX6 rnh1Δ::NatMX6 rnh201Δ::KanMX6 |
| ywp852 | h- his3D1 ura4-D18 crb2∆::ura4 nmt41-HO-his3 arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 rad52-5xflag::HphMX6 |
| Ywp992 | h- his3D1 ura4-D18 crb2∆::ura4 nmt41-HO-his3 arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 rad52-5xflag::HphMX6 chl11∆::NatMX6 |
| ywp1709 | h- his3D1 ura4-D18 crb2∆::ura4 rad22-CFP::KanMX6 nmt41-HO-his3 arg3::HOsite-KanMX4 leu1-32::YFP-Crb2::leu1 rad21-5xflag::HphMX6 chl1∆::NatMX6 rnh1Δ::NatMX6 rnh201Δ::KanMX6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, D.; Du, Z.; Xu, H.; Bao, X. Chl1, an ATP-Dependent DNA Helicase, Inhibits DNA:RNA Hybrids Formation at DSB Sites to Maintain Genome Stability in S. pombe. Int. J. Mol. Sci. 2022, 23, 6631. https://doi.org/10.3390/ijms23126631
He D, Du Z, Xu H, Bao X. Chl1, an ATP-Dependent DNA Helicase, Inhibits DNA:RNA Hybrids Formation at DSB Sites to Maintain Genome Stability in S. pombe. International Journal of Molecular Sciences. 2022; 23(12):6631. https://doi.org/10.3390/ijms23126631
Chicago/Turabian StyleHe, Deyun, Zhen Du, Huiling Xu, and Xiaoming Bao. 2022. "Chl1, an ATP-Dependent DNA Helicase, Inhibits DNA:RNA Hybrids Formation at DSB Sites to Maintain Genome Stability in S. pombe" International Journal of Molecular Sciences 23, no. 12: 6631. https://doi.org/10.3390/ijms23126631
APA StyleHe, D., Du, Z., Xu, H., & Bao, X. (2022). Chl1, an ATP-Dependent DNA Helicase, Inhibits DNA:RNA Hybrids Formation at DSB Sites to Maintain Genome Stability in S. pombe. International Journal of Molecular Sciences, 23(12), 6631. https://doi.org/10.3390/ijms23126631