
Pharmacotherapy of Itch—Antihistamines and Histamine Receptors as G Protein-Coupled Receptors

 ,
, 
{kind=link}
{kind=link}
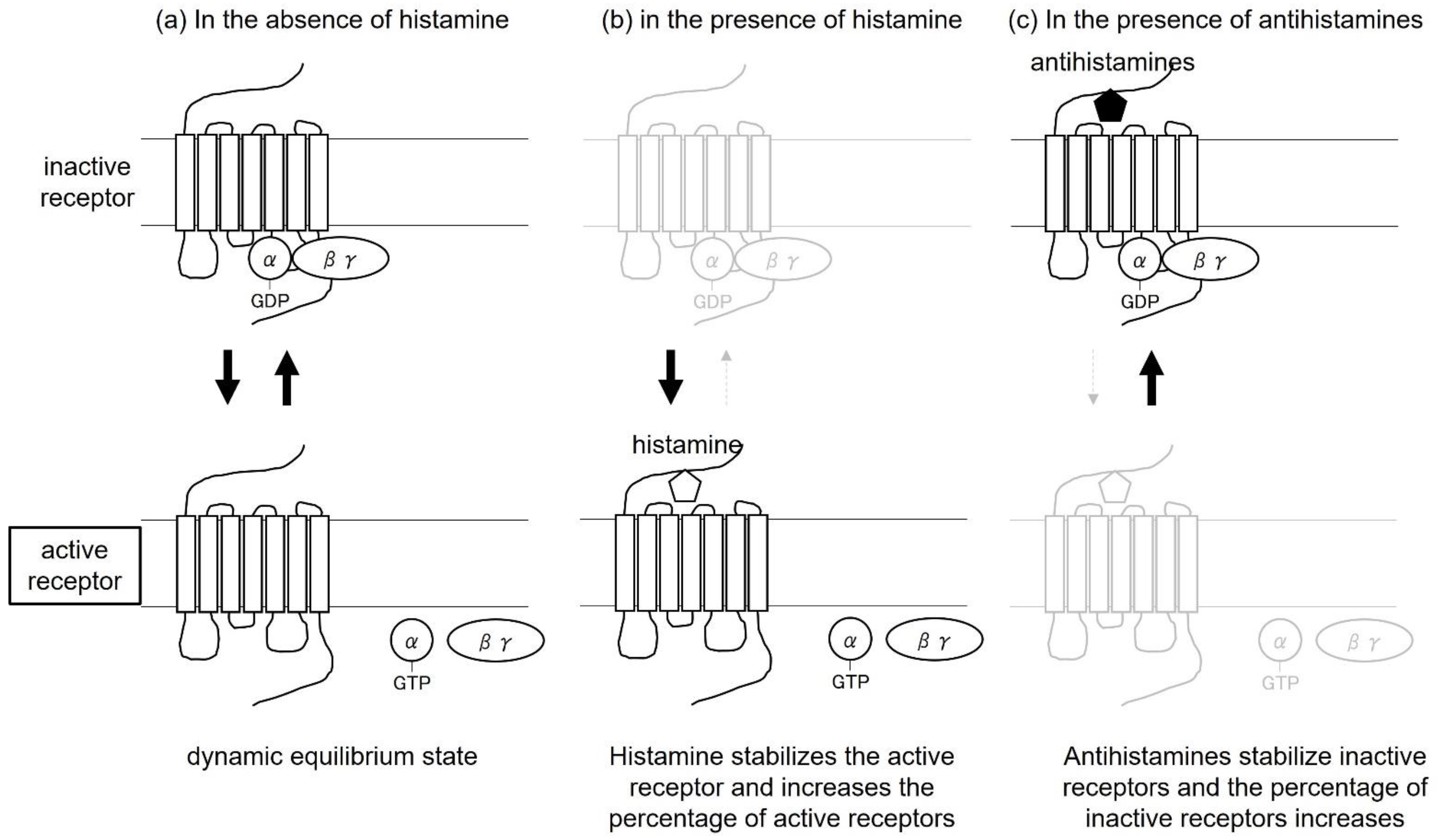{kind=link}
{kind=link}
{kind=link}
Abstract
1. Histamine as an Itch-Inducing Substance
2. Actions of Histamine on Skin and Central Nervous System
3. Histamine-Mediated Skin Diseases
4. Regulation of the Function and Distribution of Histamine Receptors as G Protein-Coupled Receptors—Desensitization Therapy—
5. Intracellular Signaling Mechanism via GPCRs
6. Antihistamines as Biased Agonists
7. Antihistamines as Inverse Agonists
8. Relationship between Biased Agonists and Inverse Agonists
9. Significance in Therapy—Inverse Agonists, Desensitization Therapy, and Biased Agonists—
10. Drug Selection Based on Impaired Performance
11. Drugs with a New Mechanism of Action; Anti-PAF Action
12. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Oetjen, L.K.; Mack, M.R.; Feng, J.; Whelan, T.M.; Niu, H.; Guo, C.J.; Chen, S.; Trier, A.M.; Xu, A.Z.; Tripathi, S.V.; et al. Sensory Neurons Co-Opt Classical Immune Signaling Pathways to Mediate Chronic Itch. Cell 2017, 171, 217–228.e13. [Google Scholar] [CrossRef]
- Dillon, S.R.; Sprecher, C.; Hammond, A.; Bilsborough, J.; Rosenfeld-Franklin, M.; Presnell, S.R.; Haugen, H.S.; Maurer, M.; Harder, B.; Johnston, J.; et al. Interleukin 31, a Cytokine Produced by Activated T Cells, Induces Dermatitis in Mice. Nat. Immunol. 2004, 5, 752–760. [Google Scholar] [CrossRef]
- Nattkemper, L.A.; Tey, H.L.; Valdes-Rodriguez, R.; Lee, H.; Mollanazar, N.K.; Albornoz, C.; Sanders, K.M.; Yosipovitch, G. The Genetics of Chronic Itch: Gene Expression in the Skin of Patients with Atopic Dermatitis and Psoriasis with Severe Itch. J. Investig. Dermatol. 2018, 138, 1311–1317. [Google Scholar] [CrossRef]
- Wilson, S.R.; Thé, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The Epithelial Cell-Derived Atopic Dermatitis Cytokine TSLP Activates Neurons to Induce Itch. Cell 2013, 155, 285–295. [Google Scholar] [CrossRef]
- Meixiong, J.; Anderson, M.; Limjunyawong, N.; Sabbagh, M.F.; Hu, E.; Mack, M.R.; Oetjen, L.K.; Wang, F.; Kim, B.S.; Dong, X. Activation of Mast-Cell-Expressed Mas-Related G-Protein-Coupled Receptors Drives Non-Histaminergic Itch. Immunity 2019, 50, 1163–1171.e5. [Google Scholar] [CrossRef]
- Glatzer, F.; Gschwandtner, M.; Ehling, S.; Rossbach, K.; Janik, K.; Klos, A.; Bäumer, W.; Kietzmann, M.; Werfel, T.; Gutzmer, R. Histamine Induces Proliferation in Keratinocytes from Patients with Atopic Dermatitis through the Histamine 4 Receptor. J. Allergy Clin. Immunol. 2013, 132, 1358–1367. [Google Scholar] [CrossRef]
- Premont, R.T.; Gainetdinov, R.R. Physiological Roles of G Protein-Coupled Receptor Kinases and Arrestins. Annu. Rev. Physiol. 2007, 69, 511–534. [Google Scholar] [CrossRef]
- Moore, C.A.C.; Milano, S.K.; Benovic, J.L. Regulation of Receptor Trafficking by GRKs and Arrestins. Annu. Rev. Physiol. 2007, 69, 451–482. [Google Scholar] [CrossRef]
- Cannavo, A.; Liccardo, D.; Koch, W.J. Targeting Cardiac β-Adrenergic Signaling via GRK2 Inhibition for Heart Failure Therapy. Front. Physiol. 2013, 4, 264. [Google Scholar] [CrossRef]
- Koch, W.J.; Rockman, H.A.; Samama, P.; Hamilton, R.; Bond, R.A.; Milano, C.A.; Lefkowitz, R.J. Cardiac Function in Mice Overexpressing the Beta-Adrenergic Receptor Kinase or a Beta ARK Inhibitor. Science 1995, 268, 1350–1353. [Google Scholar] [CrossRef]
- Chen, M.; Sato, P.Y.; Chuprun, J.K.; Peroutka, R.J.; Otis, N.J.; Ibetti, J.; Pan, S.; Sheu, S.S.; Gao, E.; Koch, W.J. Prodeath Signaling of G Protein-Coupled Receptor Kinase 2 in Cardiac Myocytes after Ischemic Stress Occurs via Extracellular Signal-Regulated Kinase-Dependent Heat Shock Protein 90-Mediated Mitochondrial Targeting. Circ. Res. 2013, 112, 1121–1134. [Google Scholar] [CrossRef]
- Nakaya, M.; Tajima, M.; Kosako, H.; Nakaya, T.; Hashimoto, A.; Watari, K.; Nishihara, H.; Ohba, M.; Komiya, S.; Tani, N.; et al. GRK6 Deficiency in Mice Causes Autoimmune Disease Due to Impaired Apoptotic Cell Clearance. Nat. Commun. 2013, 4, 1532. [Google Scholar] [CrossRef]
- Watari, K.; Nakaya, M.; Kurose, H. Multiple Functions of G Protein-Coupled Receptor Kinases. J. Mol. Signal. 2014, 9, 1. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Ferguson, S.S.G.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.T.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-Arrestin-Dependent Formation of Beta2 Adrenergic Receptor-Src Protein Kinase Complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Burghi, V.; Echeverría, E.B.; Zappia, C.D.; Díaz Nebreda, A.; Ripoll, S.; Gómez, N.; Shayo, C.; Davio, C.A.; Monczor, F.; Fernández, N.C. Biased Agonism at Histamine H 1 Receptor: Desensitization, Internalization and MAPK Activation Triggered by Antihistamines. Eur. J. Pharmacol. 2021, 896, 173913. [Google Scholar] [CrossRef]
- Leurs, R.; Church, M.K.; Taglialatela, M. H1-Antihistamines: Inverse Agonism, Anti-Inflammatory Actions and Cardiac Effects. Clin. Exp. Allergy 2002, 32, 489–498. [Google Scholar] [CrossRef]
- Kawashima, M.; Kouno, K. Investigation of the Effect of Different Duration of Prophylactic Oral Administration of Antihistamines on the Prognosis of Chronic Urticaria. Jpn. J. Clin. Dermatol. 2010, 64, 523–531. [Google Scholar]
- Hide, M.; Morioke, S.; Fukunaga, A.; Hiragun, T.; Chinuki, Y.; Inomata, N.; Masuda, K.; Tanizaki, H.; Yukinobu, N.; Yagami, A.; et al. Urticaria Clinical Practice Guidelines 2018. Jpn. J. Dermatol. 2018, 128, 2503–2624. [Google Scholar]
- Furukawa, F.; Kawada, A.; Hide, M. Comparison of Efficacy between Symptomatic and Prophyalctic Use of Anti-Histamine for Patients with Chronic Urticaria. Jpn. J. Clin. Dermatol. 2009, 63, 691–699. [Google Scholar] [CrossRef]
- Fujieda, S.; Yamada, T.; Kojima, A.; Kudo, M.; Suzaki, H.; Kadokura, Y.; Sanbe, T.; Yoshihashi, H.; Makiyama, K.; Ohki, M.; et al. Clinical Efficacy of Prophylactic Administration of a Second-Generation Antihistamine to Patients with Japanese Cedar Pollinosis a Three-Year, Multicenter, Retrospective Study. Nihon Bika Gakkai Kaishi 2007, 46, 18–28. [Google Scholar] [CrossRef][Green Version]
- Allocco, F.T.; Votypka, V.; DeTineo, M.; Naclerio, R.M.; Baroody, F.M. Effects of Fexofenadine on the Early Response to Nasal Allergen Challenge. Ann. Allergy Asthma Immunol. 2002, 89, 578–584. [Google Scholar] [CrossRef]
- Kawashima, M.; Harada, S. A Study of the Effect on Itch and Quality of Life of Atopic Dermatitis Partients Treated by Standard Therapy with Anti-Allergic Drug. Jpn. J. Clin. Dermatol. 2006, 60, 661–667. [Google Scholar]
- Yanai, K. Ideal Antihistamine Therapy from the Viewpoint of Pharmacological Action. J. Otolaryngol. Jpn. 2019, 123, 196–204. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kawachi, H.; Kurono, Y.; Masuyama, K.; Asako, M.; Okubo, K.; Ota, N.; Okano, M.; Kamijo, A.; Goto, Y.; et al. Nasal Allergy Clinical Practice Guidelines 2020. Allergy 2021, 70, 166–170. [Google Scholar] [CrossRef]
- Yanai, K. Consideration of Antihistamine Selection in Children. Padiatr. Otorhinolaryngol. Jpn. 2018, 39, 275–282. [Google Scholar] [CrossRef]
- Muñoz-Cano, R.; Ainsua-Enrich, E.; Torres-Atencio, I.; Martin, M.; Sánchez-Lopez, J.; Bartra, J.; Picado, C.; Mullol, J.; Valero, A. Effects of Rupatadine on Platelet- Activating Factor-Induced Human Mast Cell Degranulation Compared With Desloratadine and Levocetirizine (The MASPAF Study). J. Investig. Allergol. Clin. Immunol. 2017, 27, 160–168. [Google Scholar] [CrossRef][Green Version]
- Muñoz-Cano, R.M.; Casas-Saucedo, R.; Santiago, A.V.; Bobolea, I.; Ribó, P.; Mullol, J. Platelet-Activating Factor (PAF) in Allergic Rhinitis: Clinical and Therapeutic Implications. J. Clin. Med. 2019, 8, 1338. [Google Scholar] [CrossRef]
- Enomoto, A.; Fukasawa, T.; Terunuma, H.; Nakagawa, K.; Yoshizaki, A.; Sato, S.; Miyagawa, K. Decrease in MAP3Ks Expression Enhances the Cell Death Caused by Hyperthermia. Int. J. Hyperth. 2022, 39, 200–208. [Google Scholar] [CrossRef]
- Fukasawa, T.; Enomoto, A.; Miyagawa, K. Serine-Threonine Kinase 38 Regulates CDC25A Stability and the DNA Damage-Induced G2/M Checkpoint. Cell. Signal. 2015, 27, 1569–1575. [Google Scholar] [CrossRef]
- Enomoto, A.; Fukasawa, T.; Tsumoto, H.; Karube, M.; Nakagawa, K.; Yoshizaki, A.; Sato, S.; Miura, Y.; Miyagawa, K. Prevention of Calpain-Dependent Degradation of STK38 by MEKK2-Mediated Phosphorylation. Sci. Rep. 2019, 9, 16010. [Google Scholar] [CrossRef]
- Enomoto, A.; Fukasawa, T.; Takamatsu, N.; Ito, M.; Morita, A.; Hosoi, Y.; Miyagawa, K. The HSP90 Inhibitor 17-Allylamino-17-Demethoxygeldanamycin Modulates Radiosensitivity by Downregulating Serine/Threonine Kinase 38 via Sp1 Inhibition. Eur. J. Cancer 2013, 49, 3547–3558. [Google Scholar] [CrossRef]
- Fukasawa, T.; Yoshizaki, A.; Ebata, S.; Nakamura, K.; Saigusa, R.; Miura, S.; Yamashita, T.; Hirabayashi, M.; Ichimura, Y.; Taniguchi, T.; et al. Contribution of Soluble Forms of Programmed Death 1 and Programmed Death Ligand 2 to Disease Severity and Progression in Systemic Sclerosis. Arthritis Rheumatol. 2017, 69, 1879–1890. [Google Scholar] [CrossRef]
- Fukasawa, T.; Yoshizaki, A.; Ebata, S.; Yoshizaki-Ogawa, A.; Asano, Y.; Enomoto, A.; Miyagawa, K.; Kazoe, Y.; Mawatari, K.; Kitamori, T.; et al. Single-Cell-Level Protein Analysis Revealing the Roles of Autoantigen-Reactive B Lymphocytes in Autoimmune Disease and the Murine Model. eLife 2021, 10, e67209. [Google Scholar] [CrossRef]
- Nakao, T.; Kazoe, Y.; Mori, E.; Morikawa, K.; Fukasawa, T.; Yoshizaki, A.; Kitamori, T. Cytokine Analysis on a Countable Number of Molecules from Living Single Cells on Nanofluidic Devices. Analyst 2019, 144, 7200–7208. [Google Scholar] [CrossRef]
- Burboa, P.C.; Nepali, P.R.; Lillo, M.A.; Alves, N.G.; Duran, W.N. ENOS Location Is a Fundamental Mechanism for Inactivation of Inflammation-Induced Hyperpermeability. FASEB J. 2022, 36 (Suppl. 1). [Google Scholar] [CrossRef]
- Jiang, T.; Samapati, R.; Klassen, S.; Lei, D.; Erfinanda, L.; Jankowski, V.; Simmons, S.; Yin, J.; Arenz, C.; Dietrich, A.; et al. Stimulation of the EP 3 Receptor Causes Lung Edema by Activation of TRPC6 in Pulmonary Endothelial Cells. Eur. Respir. J. 2022, 59, 2102635. [Google Scholar] [CrossRef]
- Poto, R.; Cristinziano, L.; Modestino, L.; de Paulis, A.; Marone, G.; Loffredo, S.; Galdiero, M.R.; Varricchi, G. Neutrophil Extracellular Traps, Angiogenesis and Cancer. Biomedicines 2022, 10, 431. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Abdelmonaem, A.A.; Abdel-Gaber, S.A.; Hafez, H.M.; Abdel Hafez, S.M.N.; Yehia Abdelzaher, W. Rupatadine Ameliorated Ulcerative Colitis in Rats via Modulation of Platelet-Activatiweng Factor/Interleukin-6/Vascular Endothelial Growth Factor Signalling Pathway. J. Pharm. Pharmacol. 2022, 74, 537–546. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukasawa, T.; Yoshizaki-Ogawa, A.; Enomoto, A.; Miyagawa, K.; Sato, S.; Yoshizaki, A. Pharmacotherapy of Itch—Antihistamines and Histamine Receptors as G Protein-Coupled Receptors. Int. J. Mol. Sci. 2022, 23, 6579. https://doi.org/10.3390/ijms23126579
Fukasawa T, Yoshizaki-Ogawa A, Enomoto A, Miyagawa K, Sato S, Yoshizaki A. Pharmacotherapy of Itch—Antihistamines and Histamine Receptors as G Protein-Coupled Receptors. International Journal of Molecular Sciences. 2022; 23(12):6579. https://doi.org/10.3390/ijms23126579
Chicago/Turabian StyleFukasawa, Takemichi, Asako Yoshizaki-Ogawa, Atsushi Enomoto, Kiyoshi Miyagawa, Shinichi Sato, and Ayumi Yoshizaki. 2022. "Pharmacotherapy of Itch—Antihistamines and Histamine Receptors as G Protein-Coupled Receptors" International Journal of Molecular Sciences 23, no. 12: 6579. https://doi.org/10.3390/ijms23126579
APA StyleFukasawa, T., Yoshizaki-Ogawa, A., Enomoto, A., Miyagawa, K., Sato, S., & Yoshizaki, A. (2022). Pharmacotherapy of Itch—Antihistamines and Histamine Receptors as G Protein-Coupled Receptors. International Journal of Molecular Sciences, 23(12), 6579. https://doi.org/10.3390/ijms23126579