
Alpha-Synuclein Autoimmune Decline in Prodromal Multiple System Atrophy and Parkinson’s Disease

 ,
,
Abstract
:1. Introduction
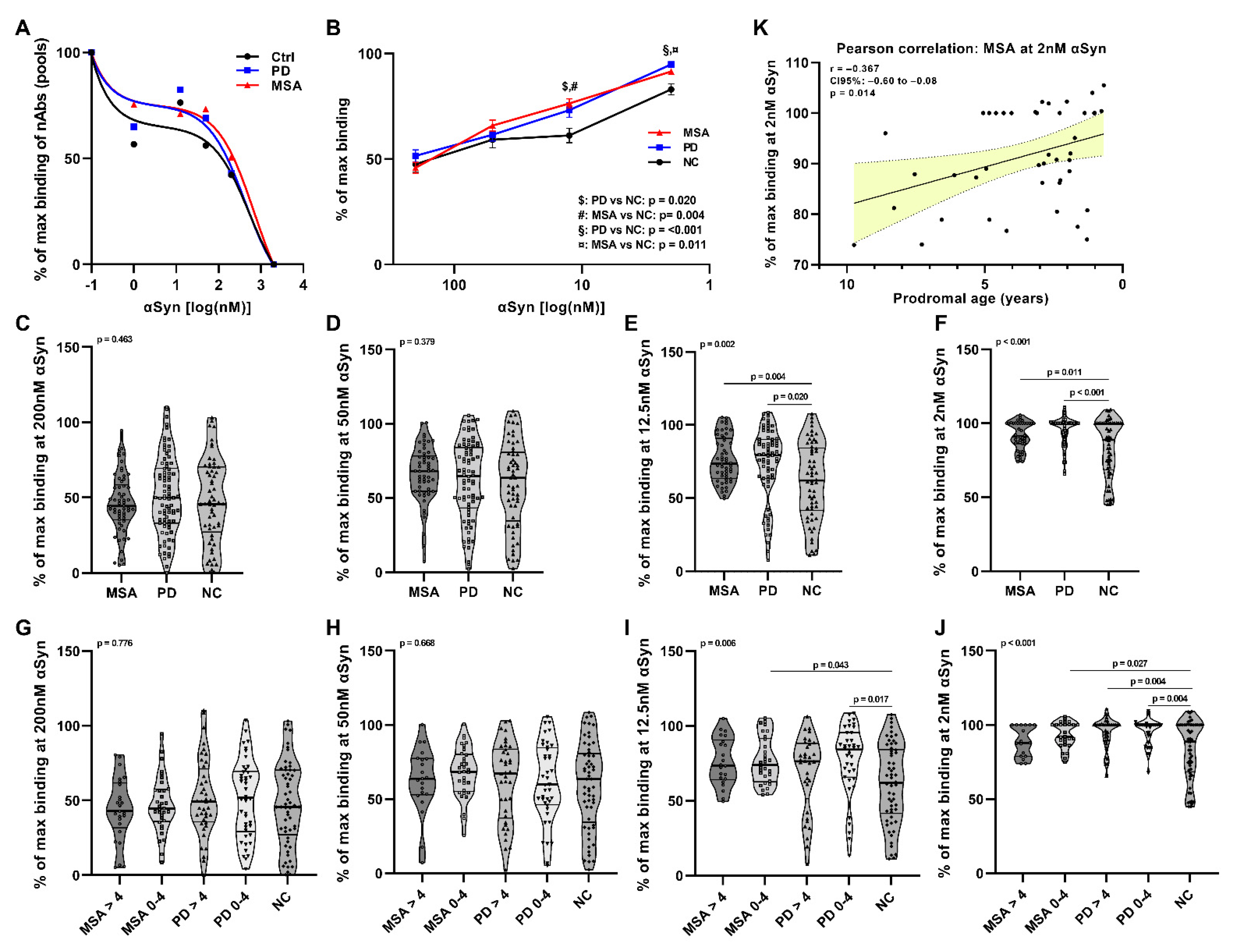
2. Results
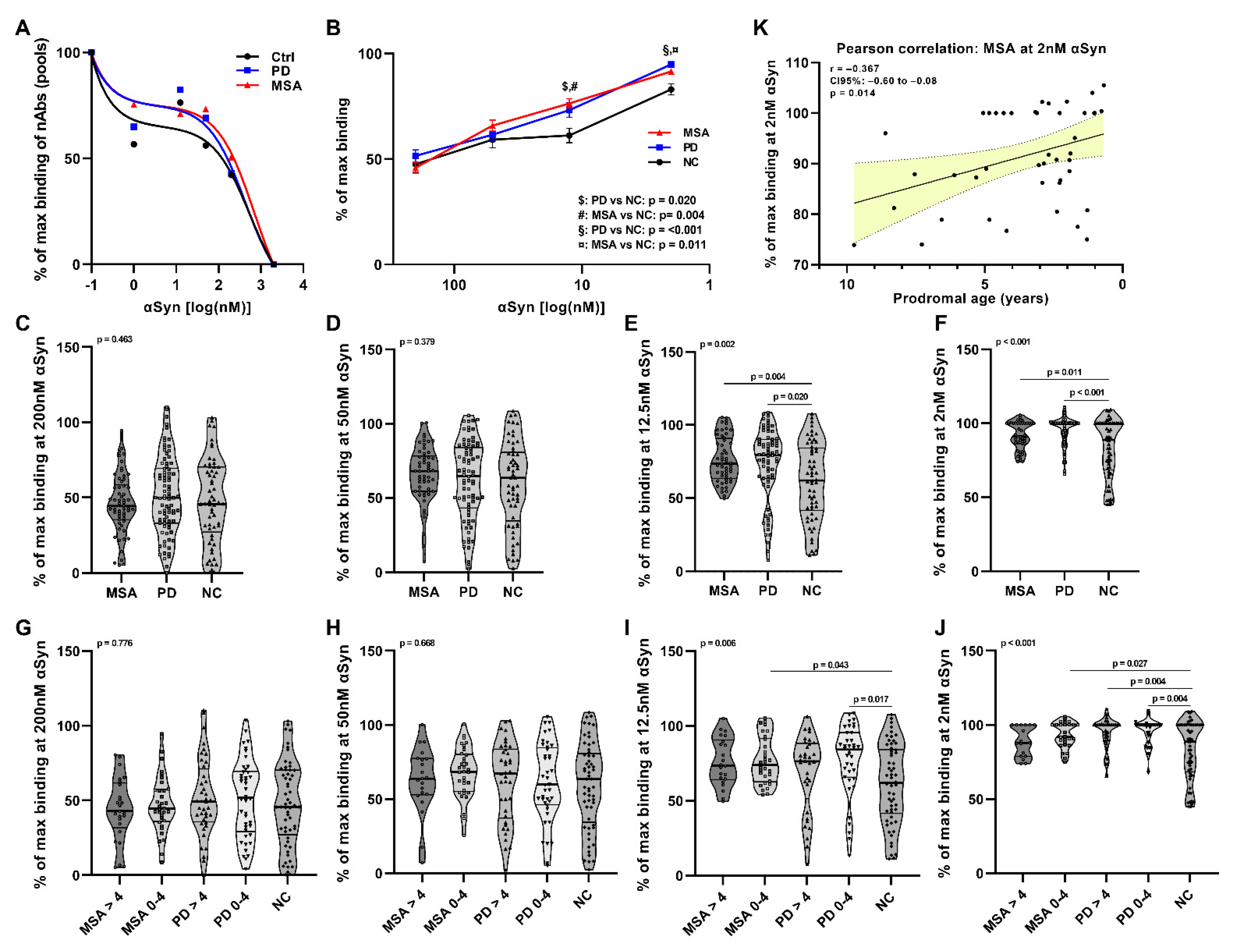
2.1. Affinity Measurement of Anti-αSyn nAbs in pMSA and pPD
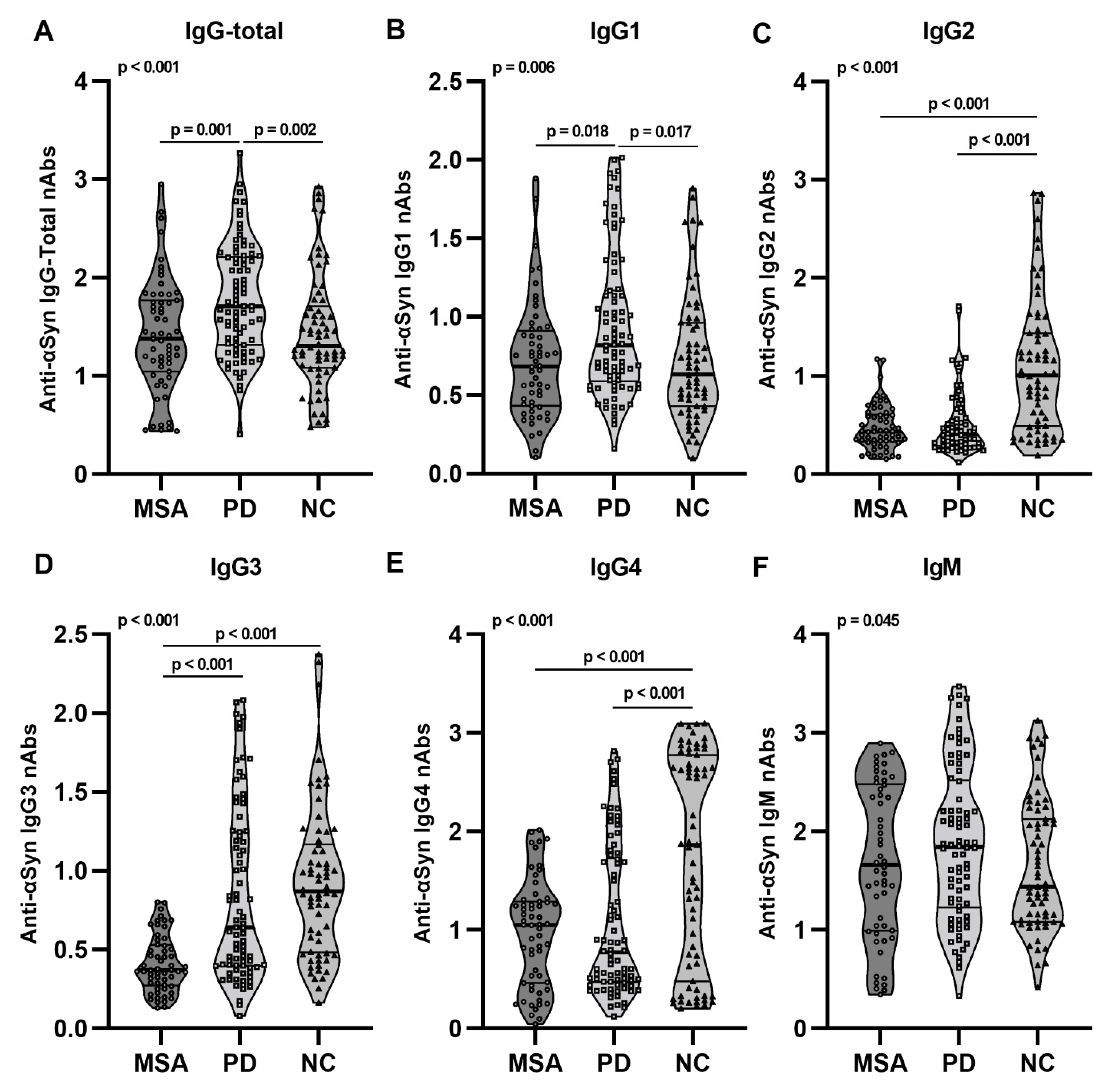
2.2. Anti-αSyn IgG Subclasses and IgM nAbs in pMSA and pPD
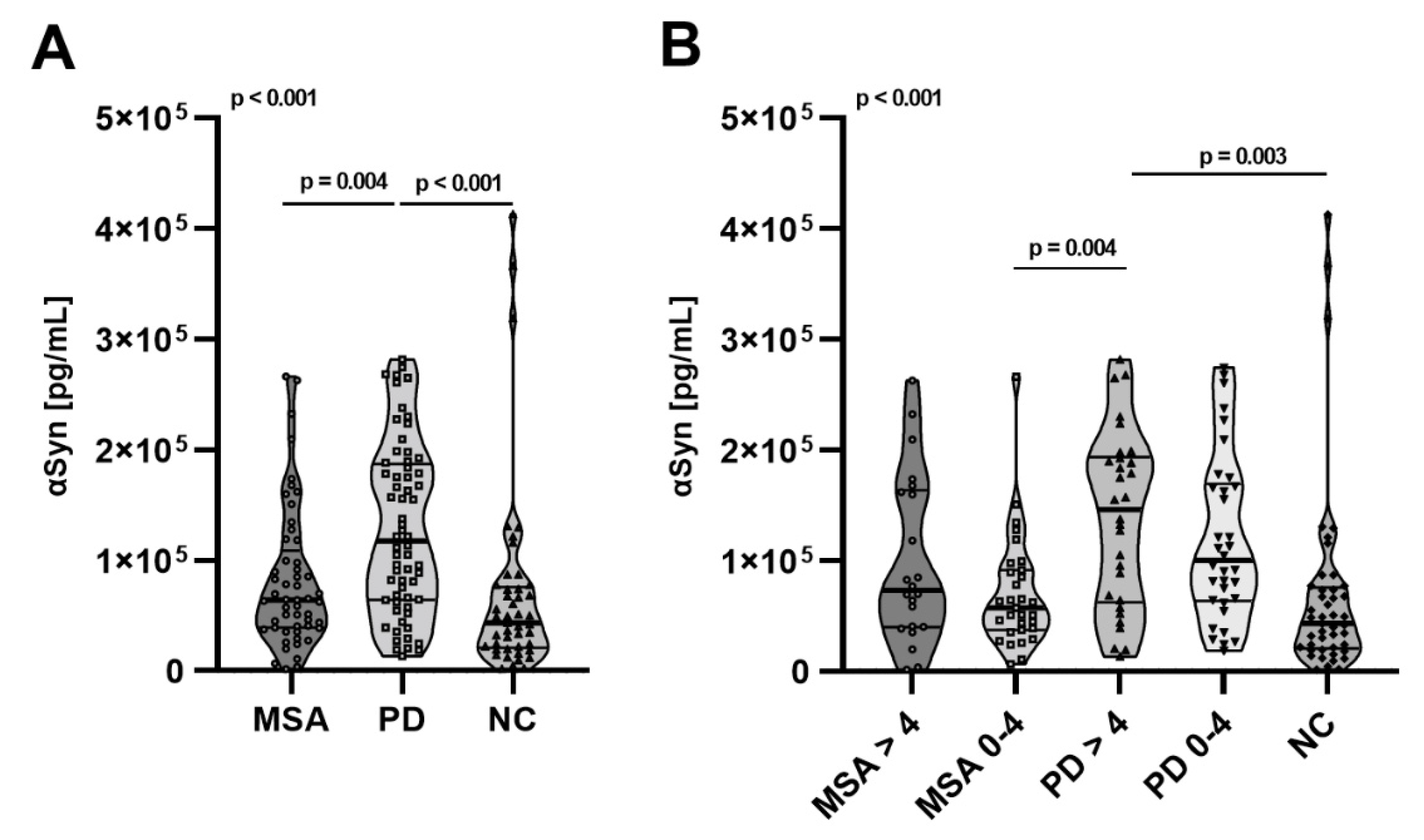
2.3. Total αSyn Levels in pMSA and pPD
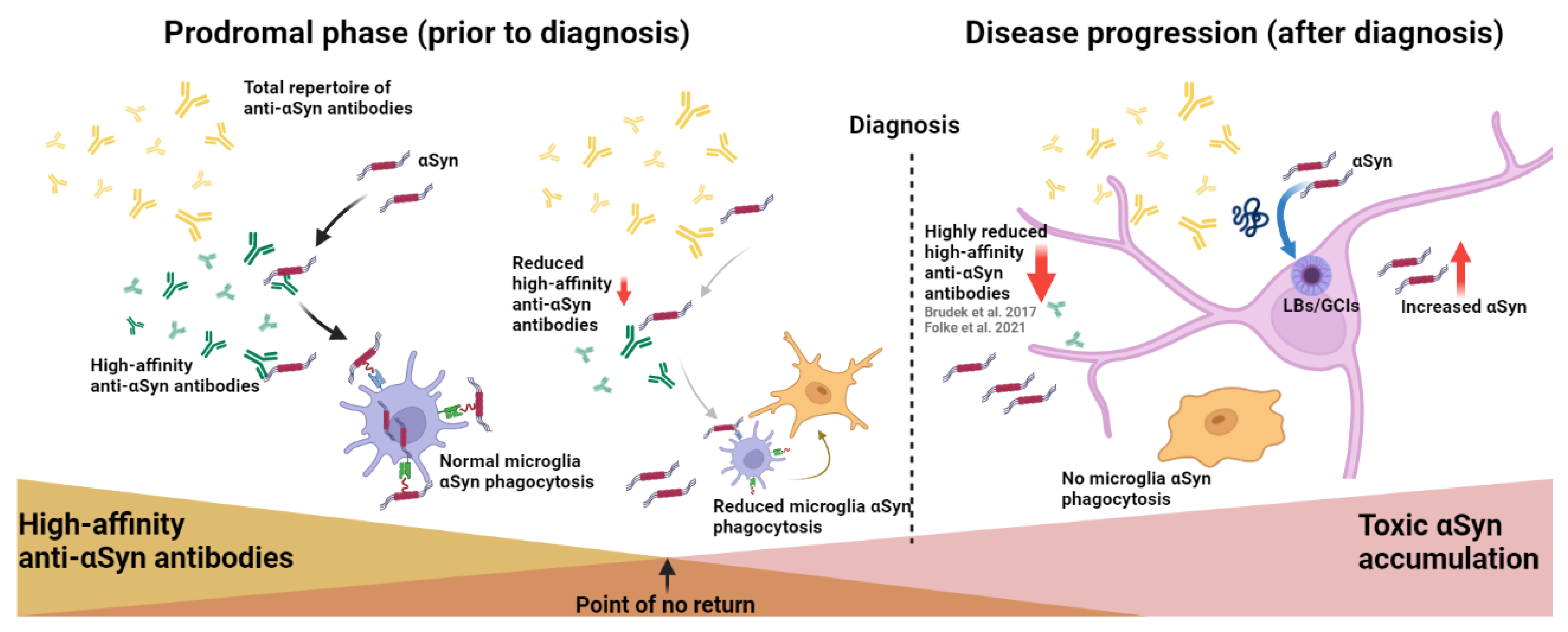
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. αSyn Affinity Measurement Using Electrochemiluminescence Assay
4.3. nAbs-αSyn Subclass Measurements Using ELISA
4.4. αSyn Measurement Using MSD Platform
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years on. J. Parkinson’s Dis. 2017, 7, S53–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doppler, K.; Ebert, S.; Uçeyler, N.; Trenkwalder, C.; Ebentheuer, J.; Volkmann, J.; Sommer, C. Cutaneous neuropathy in Parkinson’s disease: A window into brain pathology. Acta Neuropathol. 2014, 128, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Doppler, K.; Weis, J.; Karl, K.; Ebert, S.; Ebentheuer, J.; Trenkwalder, C.; Klebe, S.; Volkmann, J.; Sommer, C. Distinctive distribution of phospho-alpha-synuclein in dermal nerves in multiple system atrophy. Mov. Disord. 2015, 30, 1688–1692. [Google Scholar] [CrossRef]
- Beach, T.G.; Serrano, G.E.; Kremer, T.; Canamero, M.; Dziadek, S.; Sade, H.; Derkinderen, P.; Corbillé, A.-G.; Letournel, F.; Munoz, D.G.; et al. Immunohistochemical Method and Histopathology Judging for the Systemic Synuclein Sampling Study (S4). J. Neuropathol. Exp. Neurol. 2018, 77, 793–802. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Takahashi, H.; Ohama, E.; Ikuta, F. Parkinson’s disease: An immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 1990, 79, 581–583. [Google Scholar] [CrossRef]
- Qualman, S.J.; Haupt, H.M.; Yang, P.; Hamilton, S.R. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson’s disease. Gastroenterology 1984, 87, 848–856. [Google Scholar] [CrossRef]
- Kuzkina, A.; Schulmeyer, L.; Monoranu, C.-M.; Volkmann, J.; Sommer, C.; Doppler, K. The aggregation state of α-synuclein deposits in dermal nerve fibers of patients with Parkinson’s disease resembles that in the brain. Parkinsonism Relat. Disord. 2019, 64, 66–72. [Google Scholar] [CrossRef]
- Wang, Z.; Becker, K.; Donadio, V.; Siedlak, S.; Yuan, J.; Rezaee, M.; Incensi, A.; Kuzkina, A.; Orrú, C.D.; Tatsuoka, C.; et al. Skin α-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol. 2021, 78, 30–40. [Google Scholar] [CrossRef]
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.A.; Gradinaru, V. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 2020, 23, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Maass, F.; Rikker, S.; Dambeck, V.; Warth, C.; Tatenhorst, L.; Csoti, I.; Schmitz, M.; Zerr, I.; Leha, A.; Bähr, M.; et al. Increased alpha-synuclein tear fluid levels in patients with Parkinson’s disease. Sci. Rep. 2020, 10, 8507. [Google Scholar] [CrossRef] [PubMed]
- Hamm-Alvarez, S.F.; Janga, S.R.; Edman, M.C.; Feigenbaum, D.; Freire, D.; Mack, W.J.; Okamoto, C.T.; Lew, M.F. Levels of oligomeric α-Synuclein in reflex tears distinguish Parkinson’s disease patients from healthy controls. Biomark. Med. 2019, 13, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Abd Elhadi, S.; Grigoletto, J.; Poli, M.; Arosio, P.; Arkadir, D.; Sharon, R. α-Synuclein in blood cells differentiates Parkinson’s disease from healthy controls. Ann. Clin. Transl. Neurol. 2019, 6, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.U. Homeostatic roles of naturally occurring antibodies: An overview. J. Autoimmun. 2007, 29, 287–294. [Google Scholar] [CrossRef]
- Lutz, H.U. Naturally Occurring Antibodies (NAbs); Springer: New York, NY, USA, 2012; Volume 750, ISBN 9788578110796. [Google Scholar]
- Huang, Y.-R.; Xie, X.-X.; Ji, M.; Yu, X.-L.; Zhu, J.; Zhang, L.-X.; Liu, X.-G.; Wei, C.; Li, G.; Liu, R.-T. Naturally occurring autoantibodies against α-synuclein rescues memory and motor deficits and attenuates α-synuclein pathology in mouse model of Parkinson’s disease. Neurobiol. Dis. 2019, 124, 202–217. [Google Scholar] [CrossRef]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.-L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Smith, L.M.; Schiess, M.C.; Coffey, M.P.; Klaver, A.C.; Loeffler, D.A. α-Synuclein and Anti-α-Synuclein Antibodies in Parkinson’s Disease, Atypical Parkinson Syndromes, REM Sleep Behavior Disorder, and Healthy Controls. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Folke, J.; Rydbirk, R.; Løkkegaard, A.; Salvesen, L.; Hejl, A.-M.; Starhof, C.; Bech, S.; Winge, K.; Christensen, S.; Pedersen, L.Ø.; et al. Distinct Autoimmune Anti-α-Synuclein Antibody Patterns in Multiple System Atrophy and Parkinson’s Disease. Front. Immunol. 2019, 10, 2253. [Google Scholar] [CrossRef]
- Folke, J.; Rydbirk, R.; Løkkegaard, A.; Hejl, A.-M.; Winge, K.; Starhof, C.; Salvesen, L.; Pedersen, L.Ø.; Aznar, S.; Pakkenberg, B.; et al. Cerebrospinal fluid and plasma distribution of anti-α-synuclein IgMs and IgGs in multiple system atrophy and Parkinson’s disease. Parkinsonism Relat. Disord. 2021, 87, 98–104. [Google Scholar] [CrossRef]
- Scott, K.M.; Kouli, A.; Yeoh, S.L.; Clatworthy, M.R.; Williams-Gray, C.H. A Systematic Review and Meta-Analysis of Alpha Synuclein Auto-Antibodies in Parkinson’s Disease. Front. Neurol. 2018, 9, 815. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudek, T.; Winge, K.; Folke, J.; Christensen, S.; Fog, K.; Pakkenberg, B.; Pedersen, L.O. Autoimmune antibody decline in Parkinson’s disease and Multiple System Atrophy; a step towards immunotherapeutic strategies. Mol. Neurodegener. 2017, 12, 44. [Google Scholar] [CrossRef]
- de Natale, E.R.; Wilson, H.; Politis, M. Predictors of RBD progression and conversion to synucleinopathies. Curr. Neurol. Neurosci. Rep. 2022, 22, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Iashchishyn, I.A.; Forsgren, L.; Morozova-Roche, L.A. Immunochemical Detection of α-Synuclein Autoantibodies in Parkinson’s Disease: Correlation between Plasma and Cerebrospinal Fluid Levels. ACS Chem. Neurosci. 2017, 8, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Gruden, M.A.; Casaite, V.; Meskys, R.; Forsgren, L.; Morozova-Roche, L.A. Alpha-Synuclein Reactive Antibodies As Diagnostic Biomarkers in Blood Sera of Parkinson’s Disease Patients. PLoS ONE 2011, 6, e18513. [Google Scholar] [CrossRef] [PubMed]
- Shalash, A.; Salama, M.; Makar, M.; Roushdy, T.; Elrassas, H.H.; Mohamed, W.; El-Balkimy, M.; Donia, M.A. Elevated serum α-synuclein autoantibodies in patients with parkinson’s disease relative to Alzheimer’s disease and controls. Front. Neurol. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Evetts, S.; Hu, M.; Talbot, K.; Wade-Martins, R.; Davis, J.J. An impedimetric assay of α-synuclein autoantibodies in early stage Parkinson’s disease. RSC Adv. 2014, 4, 58773–58777. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Hayes, J.M.; Wormald, M.R.; Rudd, P.M.; Davey, G.P. Fc gamma receptors: Glycobiology and therapeutic prospects. J. Inflamm. Res. 2016, 9, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Albus, A.; Gold, M.; Bach, J.-P.; Burg-Roderfeld, M.; Jördens, M.; Kirchhein, Y.; Kronimus, Y.; Mengel, D.; Zerr, I.; Dodel, R. Extending the functional characteristics of naturally occurring autoantibodies against β-Amyloid, Prion Protein and α-Synuclein. PLoS ONE 2018, 13, e0202954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braczynski, A.K.; Sevenich, M.; Gering, I.; Kupreichyk, T.; Agerschou, E.D.; Kronimus, Y.; Habib, P.; Stoldt, M.; Willbold, D.; Schulz, J.B.; et al. Alpha-Synuclein-Specific Naturally Occurring Antibodies Inhibit Aggregation In Vitro and In Vivo. Biomolecules 2022, 12, 469. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Tropea, T.F.; Baratta, L.R.; Zuroff, L.; Diaz-Ortiz, M.E.; Zhang, B.; Shinoda, K.; Rezk, A.; Alcalay, R.N.; Chen-Plotkin, A.; et al. Abnormal B-Cell and Tfh-Cell Profiles in Patients with Parkinson Disease: A Cross-sectional Study. Neurol.-Neuroimmunol. NeuroInflamm. 2022, 9, e1125. [Google Scholar] [CrossRef] [PubMed]
- Bas, J.; Calopa, M.; Mestre, M.; Molleví, D.G.; Cutillas, B.; Ambrosio, S.; Buendia, E. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J. Neuroimmunol. 2001, 113, 146–152. [Google Scholar] [CrossRef]
- Stevens, C.H.; Rowe, D.; Morel-Kopp, M.-C.; Orr, C.; Russell, T.; Ranola, M.; Ward, C.; Halliday, G.M. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 2012, 252, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Luo, M.; Zhou, W.; Jin, X.; Xu, Z.; Yan, S.; Li, Y.; Xu, C.; Cheng, R.; Huang, Y.; et al. Global Characterization of Peripheral B Cells in Parkinson’s Disease by Single-Cell RNA and BCR Sequencing. Front. Immunol. 2022, 13, 814239. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [Green Version]
- Contaldi, E.; Magistrelli, L.; Comi, C.T. Lymphocytes in Parkinson’s Disease. J. Parkinson’s Dis. 2022, 1–10. [Google Scholar] [CrossRef]
- Williams, G.P.; Marmion, D.J.; Schonhoff, A.M.; Jurkuvenaite, A.; Won, W.-J.; Standaert, D.G.; Kordower, J.H.; Harms, A.S.T. cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol. 2020, 139, 855–874. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Chen, X.; Zhang, L.; Wei, Q.; Liu, H.; Feng, W.; Chen, Y.; Shang, H. Elevated Percentage of CD3+ T-Cells and CD4+/CD8+ Ratios in Multiple System Atrophy Patients. Front. Neurol. 2020, 11, 658. [Google Scholar] [CrossRef]
- Folke, J.; Ferreira, N.; Brudek, T.; Borghammer, P.; Van Den Berge, N. Passive Immunization in Alpha-Synuclein Preclinical Animal Models. Biomolecules 2022, 12, 168. [Google Scholar] [CrossRef] [PubMed]
- Allen Reish, H.E.; Standaert, D.G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinson’s Dis. 2015, 5, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordström, E.; Eriksson, F.; Sigvardson, J.; Johannesson, M.; Kasrayan, A.; Jones-Kostalla, M.; Appelkvist, P.; Söderberg, L.; Nygren, P.; Blom, M.; et al. ABBV-0805, a novel antibody selective for soluble aggregated α-synuclein, prolongs lifespan and prevents buildup of α-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol. Dis. 2021, 161, 105543. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, B.; Li, X.; Fu, C.; Wang, C.; Kang, X. α-Synuclein: A Multifunctional Player in Exocytosis, Endocytosis, and Vesicle Recycling. Front. Neurosci. 2019, 13, 28. [Google Scholar] [CrossRef]
- Shameli, A.; Xiao, W.; Zheng, Y.; Shyu, S.; Sumodi, J.; Meyerson, H.J.; Harding, C.V.; Maitta, R.W. A critical role for alpha-synuclein in development and function of T lymphocytes. Immunobiology 2016, 221, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Shameli, A.; Harding, C.V.; Meyerson, H.J.; Maitta, R.W. Late stages of hematopoiesis and B cell lymphopoiesis are regulated by α-synuclein, a key player in Parkinson’s disease. Immunobiology 2014, 219, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Lee, H.-J.; Masliah, E.; Lee, S.-J. Non-cell-autonomous Neurotoxicity of α-synuclein Through Microglial Toll-like Receptor 2. Exp. Neurobiol. 2016, 25, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.M.; Yang, D.; Li, X.-Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Högl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 2019, 142, 744–759. [Google Scholar] [CrossRef]
- Koga, S.; Aoki, N.; Uitti, R.J.; Van Gerpen, J.A.; Cheshire, W.P.; Josephs, K.A.; Wszolek, Z.K.; Langston, J.W.; Dickson, D.W. When DLB, PD, and PSP masquerade as MSA. Neurology 2015, 85, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 2nd ed.; SAGE Publications: Newbury Park, CA, USA, 2011. [Google Scholar]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous inference in general parametric models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pMSA (n = 59) | pPD (n = 82) | NC (n = 67) | p-Value | |
|---|---|---|---|---|
| Sex, Female (%) § | 66.1 | 44.8 | 46.3 | 0.063 |
| Age at sample, years # | 57.9 (16.9) (20–90) * | 69.8 (11.5)(20–93) | 61.7 (11.8) (28–89) * | <0.001 |
| Age at diagnosis, years ¤ | 61.7 (16.5) (25–91) | 74.3 (10.6) (23–93) | - | <0.001 |
| Prodromal age, years ¤ | 3.8 (2.6) (0.6–10.1) | 4.6 (3.1) (0.8–11.8) | - | 0.113 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folke, J.; Bergholt, E.; Pakkenberg, B.; Aznar, S.; Brudek, T. Alpha-Synuclein Autoimmune Decline in Prodromal Multiple System Atrophy and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 6554. https://doi.org/10.3390/ijms23126554
Folke J, Bergholt E, Pakkenberg B, Aznar S, Brudek T. Alpha-Synuclein Autoimmune Decline in Prodromal Multiple System Atrophy and Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(12):6554. https://doi.org/10.3390/ijms23126554
Chicago/Turabian StyleFolke, Jonas, Emil Bergholt, Bente Pakkenberg, Susana Aznar, and Tomasz Brudek. 2022. "Alpha-Synuclein Autoimmune Decline in Prodromal Multiple System Atrophy and Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 12: 6554. https://doi.org/10.3390/ijms23126554
APA StyleFolke, J., Bergholt, E., Pakkenberg, B., Aznar, S., & Brudek, T. (2022). Alpha-Synuclein Autoimmune Decline in Prodromal Multiple System Atrophy and Parkinson’s Disease. International Journal of Molecular Sciences, 23(12), 6554. https://doi.org/10.3390/ijms23126554