
Immunity, Ion Channels and Epilepsy

 , and
, and
Abstract
1. Introduction
2. Scope of Review
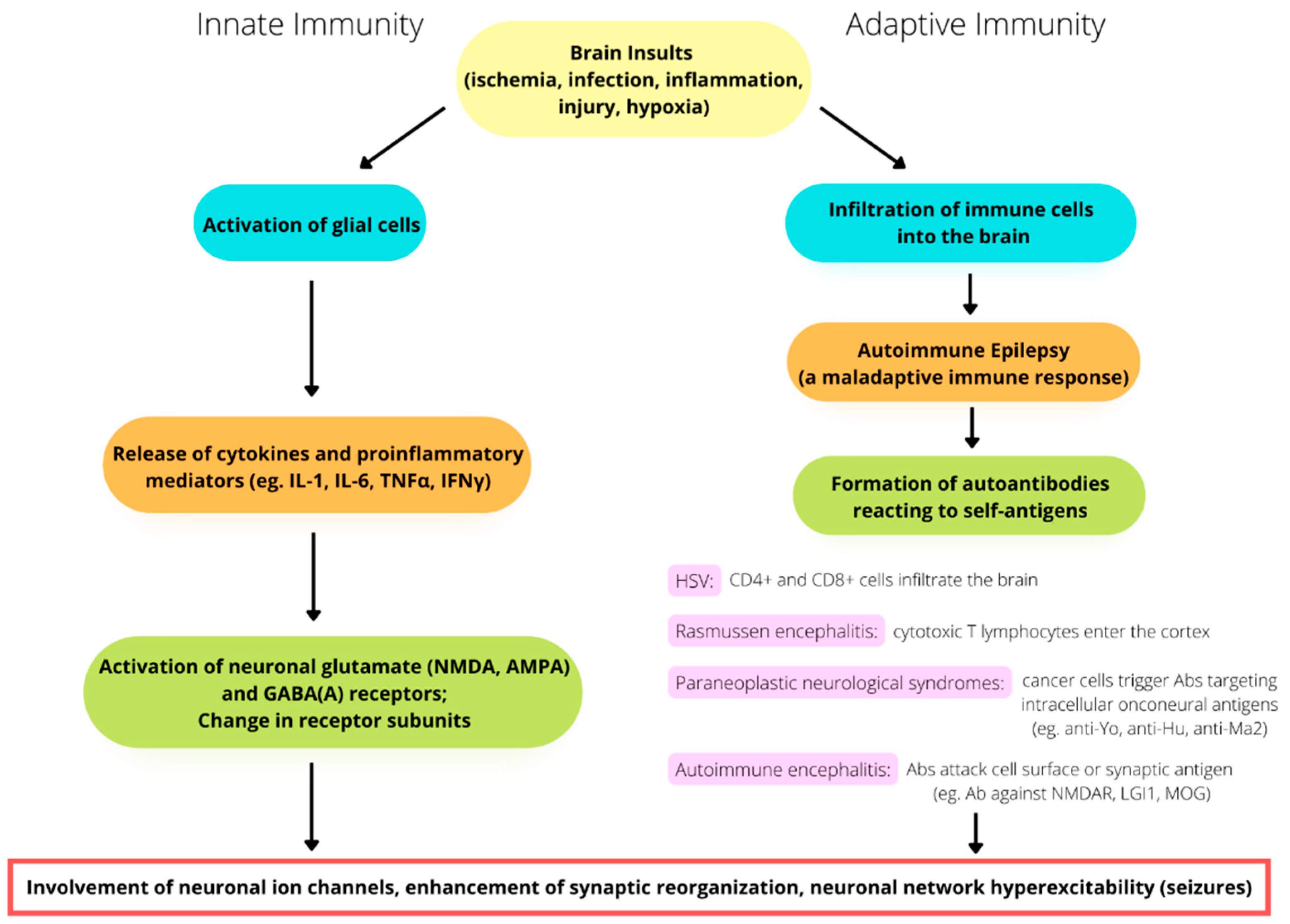
3. Activation of Innate Immunity and Neuroinflammation
4. Adaptive Immunity in Seizures and Epilepsy
5. Involvement of Ion Channels and the Immune System in Epileptogenesis and Epilepsy: Some Examples
6. Clinical Aspects of AE
7. Antibodies against Surface Epitopes
7.1. NMDA Receptor (NMDAR) Encephalitis
7.2. Leucine-Rich Glioma-Inactivated Protein 1 (LGI1) Encephalitis
7.3. Anti-GABA-B Receptor (Anti-GABABR) Encephalitis
7.4. Anti-GABA-A Receptor (Anti-GABAAR) Encephalitis
7.5. Anti-Caspr2 Encephalitis
7.6. Dipeptidyl-Peptidase-Like Protein 6 (DPPX) Antibodies
7.7. α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) Receptor Antibody
8. Antibodies of Intracellular Epitopes
8.1. Glutamic Acid Decarboxylase (GAD65) Antibody
8.2. Antineuronal Nuclear Antibody Type 1 (ANNA-1, Anti-Hu)
8.3. Antineuronal Antibodies Ma1 and Ma2
8.4. Collapsin Response Mediator Protein-5

{kind=link}
| Intracellular Epitopes | ||||
|---|---|---|---|---|
| Target of Antibodies | Clinical Symptoms/Syndromes | Functions of Targets | Underlying Tumors | Ref. |
| GAD65 | AE, LE, stiff-person syndrome, ataxia, type I diabetes | Synthesis of GABA | Lung, thymoma | [136,137,138,139] |
| ANNA-1 (anti-Hu) | AE, epilepsia partialis continua, sensory and autonomic neuropathy | HuD-specific T cells triggered by cytokines | SCLC | [61,141,142,143,144] |
| Ma1 and Ma2 (Ta) | LE, brainstem Encephalitis, tonic-clonic or focal unawareness seizure | RNA transcription regulation | SCLC, bladder, testicular germ cell, breast cancer | [8,147] |
| CRMP-5 | AE, chorea, ataxia, cranial neuropathy, sensorimotor polyneuropathy | Guides the developing axons in the nervous system | SCLC, thymoma | [105,150,151] |
| Surface Epitopes | ||||
| NMDA receptor | LE, movement disorders, psychosis | Long-term potentiation of synaptic plasticity | Ovarian teratoma | [66,103,104,105] |
| LGI1 protein | LE, faciobrachial dystonic seizure, neuromyotonia | Binds to presynaptic ADAM23 and postsynaptic ADAM22 to modulate AMPA receptors, VGKC currents, and synaptic neurotransmission/plasticity | Thymoma (rare) | [108,109,110,111,112] |
| GABAB receptor | LE, seizures, memory loss | Mediates slow and prolonged inhibitory action | SCLC | [115,116,117,118] |
| GABAA receptor | Seizures, psychosis, cognitive impairment | Ligand-gated chloride channel that mediates fast inhibitory transmission | SCLC, thymoma, NHL | [119,120,121,122] |
| Caspr2 | Seizures, cognitive decline, neuromyotonia, neuropathic pain | Cell adhesion molecule in the juxtaparanodal complex organizes the distribution of potassium channels | Thymoma (rare) | [113,124,125,126,127] |
| DPPX | Seizures, hallucination, PERM, prodromal diarrhea | Cell-surface auxiliary subunit of the Kv4.2 potassium channel | Lymphoma (rare) | [128,129,130,131] |
| AMPA receptor | LE, seizures, memory loss | Mediators of excitatory neurotransmission | SCLC, breast/ovarian cancer | [133,134] |
9. Miscellaneous Antibodies
9.1. New-Onset Refractory Status Epilepticus
9.2. Predictors of Autoimmune Epilepsy and Responses to Immunotherapy
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM23 | a disintegrin and metalloprotease domain 23 |
| ADLTE | autosomal dominant lateral temporal lobe epilepsy |
| AE | autoimmune encephalitis |
| AEDs | antiepileptic drugs |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ANNA-1 | antineuronal nuclear antibody type 1 |
| APE | antibody prevalence in epilepsy |
| BBB | blood-brain barrier |
| Caspr2 | contactin-associated protein-like 2 |
| CNS | central nervous system |
| CRMP-5 | collapsin response mediator protein-5 |
| DPPX | dipeptidyl-peptidase-like protein 6 |
| FBDSs | faciobrachial dystonic seizures |
| FLAIR | fluid-attenuated inversion recovery |
| GABA | ϒ-aminobutyric acid |
| GAD65 | glutamic acid decarboxylase |
| GEFS+ | generalized epilepsy with febrile seizures plus |
| HMGB1 | high-mobility group box protein 1 |
| HS | hippocampal sclerosis |
| HSV | herpes simplex virus |
| IL-1 | interleukin-1 |
| ILAE | International League Against Epilepsy |
| IFNγ | interferon gamma |
| IVIg | intravenous immunoglobulin |
| LE | limbic encephalitis |
| LGI1 | leucine-rich glioma-inactivated protein 1 |
| LPS | lipopolysaccharide |
| MOG | myelin oligodendrocyte glycoprotein |
| mTOR | mammalian target of rapamycin |
| NMDA | N-methyl-D-aspartate |
| NMDAR | NMDA receptor |
| NORSE | New-onset refractory status epilepticus |
| NR1 | GluN1 |
| PERM | progressive encephalomyelitis with rigidity and myoclonus |
| PET | positron emission tomography |
| PNS | peripheral nervous system |
| PNSs | paraneoplastic neurological syndromes |
| PRE | pharmacoresistant epilepsy |
| PTEN | phosphatase and tensin homolog |
| RITE | response to immunotherapy in epilepsy |
| TLE | temporal lobe epilepsy |
| TLR4 | toll-like receptor 4 |
| TNFα | tumor necrosis factor alpha |
| VGKC | voltage-gated potassium channel |
References
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug resistant epilepsy: A systemic revies and meta-analysis. Epilepsia 2018, 59, 2179–2193. [Google Scholar] [CrossRef] [PubMed]
- Sultana, B.; Panzini, M.A.; Carpentier, A.V.; Comtois, J.; Rioux, B.; Gore, G.; Bauer, P.R.; Kwon, C.S.; Jetté, N.; Josephson, C.B.; et al. Incidence and prevalence of drug-resistant epilepsy: A systemic review and meta-analysis. Neurology 2021, 96, 805–817. [Google Scholar] [CrossRef] [PubMed]
- French, J.A.; Perucca, E. Time to start calling things by their own names? The case for antiseizure medicines. Epilepsy Curr. 2020, 20, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position papter of the ILAE commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Granata, T.; Cross, H.; Theodore, W.; Avanzini, G. Immune-mediated epilepsies. Epilepsia 2011, 52, 5–11. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F.; Rosenblum, M.K.; Posner, J.B. Anti-Hu associated paraneoplastic encephalomyelitis/sensory neuropathy: A clinical study of 71 patients. Medicine 1992, 71, 59–72. [Google Scholar] [CrossRef]
- Dalmau, J.; Gultekin, S.H.; Voltz, R.; Hoard, R.; DesChamps, T.; Balmaceda, C.; Batchelor, T.; Gerstner, E.; Eichen, J.; Frennier, J.; et al. Ma1, a novel neuron- and testis-specific protein, is recognized by the serum of patients with paraneoplastic neurological disorders. Brain 1999, 122, 27–39. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F.; Villarejo, A.; Posner, J.B.; Blumenthal, D.; Thiessen, B.; Saiz, A.; Meneses, P.; Rosenfeld, M.R. Clinical analysis of anti-Ma2-associated encephalitis. Brain 2004, 127, 1831–1844. [Google Scholar] [CrossRef]
- Overeem, S.; Dalmau, J.; Bataller, L.; Nishino, S.; Mignot, E.; Verschuuren, J.; Lammers, G.J. Hypocretin-1 CSF levels in anti-Ma2 associated encephalitis. Neurology 2004, 62, 138–140. [Google Scholar] [CrossRef]
- Dalmau, J. Limbic encephalitis and variants related to neuronal cell membrane autoantigens. Rinsho Shinkeigaku 2008, 48, 871–874. [Google Scholar] [CrossRef][Green Version]
- Dalmau, J.; Tüzün, E.; Wu, H.Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.B.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic antiN-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Saiz, A. Limbic encephalitis: An expanding concept. Neurology 2008, 70, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.S.; Kohane, I.S.; Cai, T.; Gorman, M.P.; Mandl, K.D. Population-level evidence for an autoimmune etiology of epilepsy. JAMA Neurol. 2014, 71, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A.; Wilfong, A.A.; Mays, M.; Talley, C.M.; Agricola, K.; Tudor, C.; Capal, J.; Holland-Bouley, K.; Franz, D.N. Long-term treatment of epilepsy with everolimus in tuberous sclerosis. Neurology 2016, 87, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A. Before epilepsy unfolds: Finding the epileptogenesis switch. Nat. Med. 2012, 18, 1626–1627. [Google Scholar] [CrossRef]
- Curatolo, P.; Moavero, R.; van Scheppingen, J.; Aronica, E. mTOR dysregulation and tuberous sclerosis-related epilepsy. Expert Rev. Neurother. 2018, 18, 185–201. [Google Scholar] [CrossRef]
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol. Rev. 2018, 70, 142–173. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Zuberi, S.M.; Johnson, M.R. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr. Opin. Neurol. 2017, 30, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Yan, L.M.; Su, T.; He, N.; Lin, Z.J.; Wang, J.; Shi, Y.W.; Yi, Y.H.; Liao, W.P. Ion Channel Genes and Epilepsy: Functional Alteration, Pathogenic Potential, and Mechanism of Epilepsy. Neurosci. Bull. 2017, 33, 455–477. [Google Scholar] [CrossRef]
- Catterall, W.A. Forty Years of Sodium Channels: Structure, Function, Pharmacology, and Epilepsy. Neurochem. Res. 2017, 42, 2495–2504. [Google Scholar] [CrossRef]
- Lai, M.C.; Lin, K.M.; Yeh, P.S.; Wu, S.N.; Huang, C.W. The novel effect of immunomodulatory-glatiramer acetate on epileptogenesis and epileptic seizures. Cell. Physiol. Biochem. 2018, 50, 150–168. [Google Scholar] [CrossRef]
- Catterall, W.A.; Kalume, F.; Oakley, J.C. NaV1.1 channels and epilepsy. J. Physiol. 2010, 588, 1849–1859. [Google Scholar] [CrossRef]
- Wu, X.; Hong, L. Calmodulin Interactions with Voltage-Gated Sodium Channels. Int. J. Mol. Sci. 2021, 22, 9798. [Google Scholar] [CrossRef] [PubMed]
- Toledano, M.; Britton, J.W.; McKeon, A.; Shin, C.; Lennon, V.A.; Quek, A.M.L.; So, E.; Worrell, G.A.; Cascino, G.D.; Klein, C.J.; et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmue epilepsy. Neurology 2014, 82, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Crino, P.B. Inflammation in epilepsy: Clinical observations. Epilepsia 2011, 52, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; Lanerolle, N.C.D.; Michael A Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.M.; Hutchison, M.R.; Northcutt, A.; Maier, S.F.; Watkins, L.R.; Barth, D.S. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain 2009, 132, 2478–2486. [Google Scholar] [CrossRef]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.M. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef]
- Represa, A.; Niquet, J.; Pollard, H.; Ben-Ari, Y. Cell death, gliosis, and synaptic remodeling in the hippocampus of epileptic rats. J. Neurobiol. 1995, 26, 413–425. [Google Scholar] [CrossRef]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-a. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Huang, A.M.; Kuo, Y.M.; Wang, S.T.; Chang, Y.Y.; Huang, C.C. Febrile seizures impair memory and cAMP response-element binding protein activation. Ann. Neurol. 2003, 54, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Pitkanen, A.; Kharatishvili, I.; Nissinen, J.; McIntosh, T. Posttraumatic epilepsy induced by lateral fluidpercussion brain injury in rats. In Models of Seizures and Epilepsy; Pitkanen, A., Schwartzkroin, P.A., Moshe, S.L., Eds.; Elsevier Academic Press: Burlington, VT, USA, 2006; pp. 465–476. [Google Scholar]
- Holmes, G.L.; Ben-Ari, Y. Seizures in the developing brain: Perhaps not so benign after all. Neuron 1998, 21, 1231–1234. [Google Scholar] [CrossRef]
- Clarkson, B.D.; Kahoud, R.J.; McCarthy, C.B.; Howe, C.L. Infammatory cytokine-induced changes in neural network activity measured by waveform analysis of high-content calcium imaging in murine cortical neurons. Sci. Rep. 2017, 7, 9037. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Li, H.; Fuentealba, L.C.; Molofsky, A.V.; Taveira-Marques, R.; Zhuang, H.; Tenney, A.; Murnen, A.T.; Fancy, S.P.J.; Merkle, F.; et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 2012, 337, 358–362. [Google Scholar] [CrossRef]
- Diniz, L.P.; Almeida, J.C.; Tortelli, V.; Vargas Lopes, C.; Setti-Perdigão, P.; Stipursky, J.; Kahn, S.A.; Romão, L.F.; de Miranda, J.; Alves-Leon, S.V.; et al. Astrocyte-induced Synaptogenesis Is Mediated by Transforming Growth factor ϐ signaling through modulation of D-serine levels in cerebral cortex neurons. J. Biol. Chem. 2012, 287, 41432–41445. [Google Scholar] [CrossRef]
- Brambilla, R.; Persaud, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic inhibition of astroglial NF-kappaB improves functional outcome in experimental autoimmune encephalomyelitis by supressing chronic central nervous system inflammation. J. Immunol. 2009, 182, 2628–2640. [Google Scholar] [CrossRef]
- Takano, T.; Kang, J.; Jaiswal, J.K.; Simon, S.M.; Lin, J.H.-C.; Yu, Y.; Li, Y.; Yang, J.; Dienel, G.; Zielke, H.R.; et al. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 16466–16471. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef]
- Oby, E.; Janigro, D. The blood-brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006, 129, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, E.; Dreier, J.P.; Ivens, S.; Bechmann, I.; Tomkins, O.; Heinemann, U.; Friedman, A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J. Neurosci. 2004, 24, 7829–7836. [Google Scholar] [CrossRef]
- Gershen, L.D.; Zanotti-Fregonara, P.; Dustin, I.H.; Liow, J.S.; Hirvonen, J.; Kreisl, W.C.; Jenko, K.J.; Inati, S.K.; Fujita, M.; Morse, C.L.; et al. Neuroinflammation in temporal lobe epilepsy measured using positron emission tomographic imaging of tanslocator protein. JAMA Neurol. 2015, 72, 882–888. [Google Scholar] [CrossRef]
- Choi, J.; Nordli, D.R., Jr.; Alden, T.D.; DiPatri, A., Jr.; Laux, L.; Kelley, K.; Rosenow, J.; Schuele, S.U.; Rajaram, V.; Koh, S. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J. Neuroinflamm. 2009, 6, 38. [Google Scholar] [CrossRef]
- Iyer, A.; Zurolo, E.; Spliet, W.G.M.; van Rijen, P.C.; Baayen, J.C.; Gorter, J.A.; Aronica, E. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 2010, 51, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Varadkar, S.; Bien, C.G.; Kruse, C.A.; Jensen, F.E.; Bauer, J.; Pardo, C.A.; Vincent, A.; Mathern, G.W.; Cross, J.H. Rasmussen’s encephalitis: Clinical features, pathobiology, and treatment advances. Lancet Neurol. 2014, 13, 195–205. [Google Scholar] [CrossRef]
- Pruss, H.; Finke, C.; Holtje, M.; Hofmann, J.; Klingbeil, C.; Probst, C.; Borowski, K.; Ahnert-Hilger, G.; Harms, L.; Schwab, J.M.; et al. N-methyl-D-aspartate receptor antibodies in herpes simplex encephalitis. Ann. Neurol. 2012, 72, 902–911. [Google Scholar] [CrossRef]
- Hacohen, Y.; Deiva, K.; Pettingill, P.; Waters, P.; Siddiqui, A.; Chretien, P.; Menson, E.; Lin, J.P.; Tardieu, M.; Vincent, A.; et al. N-methyl-D-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov. Disord. 2014, 29, 90–96. [Google Scholar] [CrossRef]
- Norsadini, M.; Mohammad, S.S.; Coraza, F.; Ruga, E.M.; Kothur, K.; Perilongo, G.; Frigo, A.C.; Toldo, I.; Dale, R.C.; Sartori, S. Herpes simplex virus-induced anti-N-methyl-D-aspartate receptor encephalitis: A systematic literature review with analysis of 43 cases. Dev. Med. Child Neurol. 2017, 59, 796–805. [Google Scholar] [CrossRef]
- Armangue, T.; Spatola, M.; Vlagea, A.; Mattozzi, S.; Cárceles-Cordon, M.; Martinez-Heras, E.; Llufriu, S.; Muchart, J.; Erro, M.E.; Abraira, L.; et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: A prospective observational study and retrospective analysis. Lancet Neurol. 2018, 17, 760–772. [Google Scholar] [CrossRef]
- Enquist, L.W.; Husak, P.J.; Banfield, B.W.; Smith, G.A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus. Res. 1998, 51, 237–347. [Google Scholar] [PubMed]
- Wickham, S.; Lu, B.; Ash, J.; Daniel, J.J.; Carr, D.J. Chemokine receptor deficiency is associated with increased chemokine expression in the peripheral and central nervous systems and increased resistance to herpetic encephalitis. J. Neuroimmunol. 2005, 162, 51–59. [Google Scholar] [CrossRef]
- Luster, A.D. The role of chemokines in linking innate and adaptive immunity. Curr. Opin. Immunol. 2002, 14, 129–135. [Google Scholar] [CrossRef]
- Chan, W.L.; Javanovic, T.; Lukic, M.L. Infiltration of immune T cells in the brain of mice with herpes simplex virus-induced encephalitis. J. Neuroimmunol. 1989, 23, 195–201. [Google Scholar] [CrossRef]
- Hudson, S.J.; Streilein, J.W. Functional cytotoxic T cells are associated with focal lesions in the brains of SJL mice with experimental herpes simplex encephalitis. J. Immunol. 1994, 152, 5540–5547. [Google Scholar] [PubMed]
- Marques, C.P.; Cheeran, M.C.; Palmquist, J.M.; Hu, S.; Urban, S.L.; Lokensgard, J.R. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J. Immunol. 2008, 181, 6417–6426. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, N.; Imai, T.; Shindo, K.; Sato, A.; Fujii, W.; Ichinohe, T.; Takemura, N.; Kakuta, S.; Uematsu, S.; Kiyono, H.; et al. Herpes simplex virus-1 evasion of CD8+ T cell accumulation contributes to viral encephalitis. J. Clin. Investig. 2017, 127, 3784–3795. [Google Scholar] [CrossRef]
- McKeon, A.; Pittock, S.J. Paraneoplastic encephalomyelopathies: Pathology and mechanisms. Acta Neuropathol. 2011, 122, 381–400. [Google Scholar] [CrossRef]
- Roberts, W.K.; Deluca, I.J.; Thomas, A.; Fak, J.; Williams, T.; Buckley, N.; Dousmanis, A.G.; Posner, J.B.; Darnell, R.B. Patients with lung cancer and paraneoplastic Hu syndrome harbor HuD-specific type 2 CD8+ T cells. J. Clin. Investig. 2009, 119, 2042–2051. [Google Scholar] [CrossRef][Green Version]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blümcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J.; et al. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 2012, 135, 1622–1638. [Google Scholar] [CrossRef] [PubMed]
- Chefdeville, A.; Honnorat, J.; Hampe, C.S.; Desestret, V. Neuronal central nervous system syndromes probably mediated by autoantibodies. Eur. J. Neurosci. 2016, 43, 1535–1552. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Graus, F. Antibody-mediated encephalitis. N. Engl. J. Med. 2018, 378, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Al-Diwani, A.; Waters, P.; Irani, S.R. The autoantibody-mediated encephalitides: From clinical observations to molecular pathogenesis. J. Neurol. 2021, 268, 1689–1707. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol. Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef] [PubMed]
- Armangue, T.; Olivé-Cirera, G.; Martínez-Hernandez, E.; Sepulveda, M.; Ruiz-Garcia, R.; Muñoz-Batista, M.; Ariño, H.; González-Álvarez, V.; Felipe-Rucián, A.; Martínez-González, M.J.; et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: A multicentre observational study. Lancet Neurol. 2020, 19, 234–246. [Google Scholar] [CrossRef]
- Vogrig, A.; Gigli, G.L.; Segatti, S.; Corazza, E.; Marini, A.; Bernardini, A.; Valent, F.; Martina Fabris, M.; Curcio, F.; Brigo, F.; et al. Epidemiology of paraneoplastic neurological syndromes: A population-based study. J. Neurol. 2020, 267, 26–35. [Google Scholar] [CrossRef]
- Marrie, R.A.; Reider, N.; Cohen, J.; Trojano, M.; Sorensen, P.S.; Cutter, G.; Reingold, S.; Stuve, O. A systematic review of the incidence and prevalence of sleep disorders and seizure disorders in multiple sclerosis. Mult. Scler. 2015, 21, 342–349. [Google Scholar] [CrossRef]
- Mody, I. Ion channels in epilepsy. Int. Rev. Neurobiol. 1998, 42, 199–226. [Google Scholar]
- Blumenfeld, H. Cellular and network mechanisms of spike-wave seizures. Epilepsia 2005, 46, 21–23. [Google Scholar] [CrossRef]
- Lerche, H.; Shah, M.; Beck, H.; Noebels, J.; Johnston, D.; Vincent, A. Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 2013, 591, 753–764. [Google Scholar] [CrossRef]
- Waszkielewicz, A.M.; Gunia, A.; Szkaradek, N.; Słoczyńska, K.; Krupińska, S.; Marona, H. Ion channels as drug targets in central nervous system disorders. Curr. Med. Chem. 2013, 20, 1241–1285. [Google Scholar] [CrossRef] [PubMed]
- Hargus, N.J.; Nigam, A.; Bertram, E.H., 3rd; Patel, M.K. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J. Neurophysiol. 2013, 110, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Pappas, C.; Dahle, E.J.; Claes, L.R.; Pruess, T.H.; De Jonghe, P.; Thompson, J.; Dixon, M.; Gurnett, C.; Peiffer, A.; et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009, 5, e1000649. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.N.; Gavrilovici, C.; Shah, S.U.; Shaheen, F.; Choudhary, M.I.; Rahman, A.U.; Fahnestock, M.; Simjee, S.U.; Poulter, M.O. A novel anticonvulsant modulates voltage-gated sodium channel inactivation and prevents kindling-induced seizures. J. Neurochem. 2013, 126, 651–661. [Google Scholar] [CrossRef]
- Mulley, J.C.; Hodgson, B.; McMahon, J.M.; Iona, X.; Bellows, S.; Mullen, S.A.; Farrell, K.; Mackay, M.; Sadleir, L.; Bleasel, A.; et al. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia 2013, 54, e122–e126. [Google Scholar] [CrossRef]
- Stafstrom, C.E. The role of the subiculum in epilepsy and epileptogenesis. Epilepsy Curr. 2005, 4, 121–129. [Google Scholar] [CrossRef]
- Yan, B.; Li, P. An integrative view of mechanisms underlying generalized spike-and-wave epileptic seizures and its implication on optimal therapeutic treatments. PLoS ONE 2011, 6, e22440. [Google Scholar] [CrossRef]
- Simeone, K.A.; Matthews, S.A.; Samson, K.K.; Simeone, T.A. Targeting deficiencies in mitochondrial respiratory complex I and functional uncoupling exerts anti-seizure effects in a genetic model of temporal lobe epilepsy and in a model of acute temporal lobe seizures. Exp. Neurol. 2014, 251, 84–90. [Google Scholar] [CrossRef]
- Huang, C.W.; Lin, K.M.; Hung, T.Y.; Chuang, Y.C.; Wu, S.N. Multiple actions of rotenone, an inhibitor of mitochondrial respiratory chain, on ionic currents and miniature end-plate potential in mouse hippocampal (mHippoE-14) neurons. Cell. Physiol. Biochem. 2018, 47, 330–343. [Google Scholar] [CrossRef]
- Lai, M.C.; Wu, S.N.; Huang, C.W. Zingerone modulates neuronal voltage-gated Na+ and L-type Ca2+ currents. Int. J. Mol. Sci. 2022, 23, 3123. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Wali, A.F.; Rashid, S.M.; Alsaffar, R.M.; Ahmad, A.; Jan, B.L.; Paray, B.A.; Alqahtani, S.M.A.; Arafah, A.; Rehman, M.U. Zingerone Targets Status Epilepticus by Blocking Hippocampal Neurodegeneration via Regulation of Redox Imbalance, Inflammation and Apoptosis. Pharmaceuticals 2021, 14, 146. [Google Scholar] [CrossRef] [PubMed]
- Gataullina, S.; Dulac, O. From genotype to phenotype in Dravet disease. Seizure 2017, 44, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.X.; Hong, S.Q.; Li, T.S.; Wang, J.; Xie, L.L.; Han, W.; Jiang, L. Genetic and phenotypic characteristics of SCN1A-related epilepsy in Chinese children. Neuroreport 2019, 30, 671–680. [Google Scholar] [CrossRef]
- Zoidl, G.R.; Spray, D.C. The Roles of Calmodulin and CaMKII in Cx36 Plasticity. Int. J. Mol. Sci. 2021, 22, 4473. [Google Scholar] [CrossRef]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of Immune Responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef]
- Cho, C.H. Frontier of epilepsy research—mTOR signaling pathway. Exp. Mol. Med. 2011, 43, 231–274. [Google Scholar] [CrossRef]
- Moloney, P.B.; Cavalleri, G.L.; Delanty, N. Epilepsy in the mTORopathies: Opportunities for precision medicine. Brain Commun. 2021, 3, fcab222. [Google Scholar] [CrossRef]
- Weng, O.Y.; Li, Y.; Wang, L.Y. Modeling Epilepsy Using Human Induced Pluripotent Stem Cells-Derived Neuronal Cultures Carrying Mutations in Ion Channels and the Mechanistic Target of Rapamycin Pathway. Front. Mol. Neurosci. 2022, 15, 810081. [Google Scholar] [CrossRef]
- Wang, Y.; Tao, J.; Wang, M.; Yang, L.; Ning, F.; Xin, H.; Xu, X.; Cai, H.; Zhang, W.; Yu, K.; et al. Mechanism of regulation of big-conductance Ca2+-activated K+ channels by mTOR complex 2 in podocytes. Front. Physiol. 2019, 10, 167. [Google Scholar] [CrossRef]
- Jurado, S.; Benoist, M.; Lario, A.; Knafo, S.; Petrok, C.N.; Esteban, J.A. PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J. 2010, 29, 2827–2840. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-J.; Wang, X.-L.; Ismail, A.; Ashman, C.J.; Valori, C.F.; Wang, G.; Gao, S.; Higginbottom, A.; Ince, P.G.; Azzouz, M.; et al. PTEN regulates AMPA receptor-mediated cell viability in iPS-derived motor neurons. Cell Death Dis. 2014, 5, e1096. [Google Scholar] [CrossRef] [PubMed]
- Niere, F.; Raab-Graham, K.F. mTORC1 is a local, postsynaptic voltage sensor regulated by positive and negative feedback pathways. Front. Cell. Neurosci. 2017, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Anderson, A.E. mTOR-dependent alterations of Kv1.1 subunit expression in the neuronal subset-specific Pten knockout mouse model of cortical dysplasia with epilepsy. Sci. Rep. 2018, 8, 3568. [Google Scholar] [CrossRef]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef]
- Cellucci, T.; Van Mater, H.; Graus, F.; Muscal, E.; Gallentine, W.; Klein-Gitelman, M.S.; Benseler, S.M.; Frankovich, J.; Gorman, M.P.; Van Haren, K.; et al. Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e663. [Google Scholar] [CrossRef] [PubMed]
- Cabezudo- García, P.; Mena-Vázquez, N.; Villagrán-García, M.; Serrano-Castro, P.J. Efficacy of antiepileptic drugs in autoimmune epilepsy: A systematic review. Seizure 2018, 59, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; Pittock, S.J.; Kelly, C.R.; McKeon, A.; Lopez-Chiriboga, A.S.; Lennon, V.A.; Gadoth, A.; Smith, C.Y.; Bryant, S.C.; Klein, C.J.; et al. Autoimmune encephalitis epidemiology and a comparison to Infectious encephalitis. Ann. Neurol. 2018, 83, 166–177. [Google Scholar] [CrossRef]
- Byun, J.I.; Lee, S.T.; Jung, K.H.; Sunwoo, J.S.; Moon, J.; Lim, J.A.; Lee, D.Y.; Shin, Y.W.; Kim, T.J.; Lee, K.J.; et al. Effct of immunotherapy on seizure outcome in patients with autoimmune encephalitis: A propective observational registry study. PLoS ONE 2016, 11, e0146455. [Google Scholar] [CrossRef]
- De Bruijn, M.A.A.M.; van Sonderen, A.; van Coevorden-Hameete, M.H.; Bastiaansen, A.E.M.; Schreurs, M.W.J.; Rouhl, R.P.W.; van Donselaar, C.A.; Majoie, M.H.J.M.; Neuteboom, R.F.; Sillevis Smitt, P.A.E.; et al. Evaluation of seizure treatment in anti-LGI1, anti-NMDAR and anti GABABR encephalitis. Neurology 2019, 92, e2185–e2196. [Google Scholar] [CrossRef]
- Laube, B.; Hirai, H.; Sturgess, M.; Kuhse, J. Molecular determinants of agonist discrimination by NMDA receptor subunits: Analysis of the glutamate binding site on the NR2B subunit. Neuron 1997, 18, 493–503. [Google Scholar] [CrossRef]
- Li, F.; Tsien, J.Z. Memory and the NMDA receptors. N. Engl. J. Med. 2009, 361, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangué, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol. 2013, 12, 157–165. [Google Scholar] [CrossRef]
- Quek, A.M.L.; O’Toole, O. Autoimmune Epilepsy: The Evolving Science of Neural Autoimmunity and Its Impact on Epilepsy Management. Semin. Neurol. 2018, 38, 290–302. [Google Scholar] [PubMed]
- Prüss, H.; Dalmau, J.; Harms, L.; Höltje, M.; Ahnert-Hilger, G.; Borowski, K.; Stoecker, W.; Wandinger, K.P. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknown origin. Neurology 2010, 75, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Sagane, K.; Ishihama, Y.; Sugimotor, H. LGI1 and LGI4 bind to ADAM22, ADAM 23 and ADAM11. Int. J. Biol. Sci. 2008, 4, 387–396. [Google Scholar] [CrossRef]
- Fukata, Y.; Lovero, K.L.; Iwanaga, T.; Watanabe, A.; Yokoi, N.; Tabuchi, K.; Shigemoto, R.; Nicoll, R.A.; Fukata, M. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc. Natl. Acad. Sci. USA 2010, 107, 3799–3804. [Google Scholar] [CrossRef]
- Van Sonderen, A.; Petit-Pedrol, M.; Dalmau, J.; Titulaer, M.J. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat. Rev. Neurol. 2017, 13, 290–301. [Google Scholar] [CrossRef]
- Lai, M.; Huijbers, M.G.; Lancaster, E.; Graus, F.; Bataller, L.; Balice-Gordon, R.; Cowell, J.K.; Dalmau, J. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: A case series. Lancet Neurol. 2010, 9, 776–785. [Google Scholar] [CrossRef]
- Morante-Redolat, J.M.; Gorostidi-Pagola, A.; Piquer-Sirerol, S.; Sáenz, A.; Poza, J.J.; Galán, J.; Gesk, S.; Sarafidou, T.; Mautner, V.F.; Binelli, S.; et al. Mutations in the LGI1/Epitemptin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum. Mol. Genet. 2002, 11, 1119–1128. [Google Scholar] [CrossRef]
- Irani, S.R.; Michell, A.W.; Lang, B.; Pettingill, P.; Waters, P.; Johnson, M.R.; Schott, J.M.; Armstrong, R.J.E.; Zagami, A.S.; Bleasel, A.; et al. Faciobrachial dystonic seizures precede LGI1 antibody limbic encephalitis. Ann. Neurol. 2011, 69, 892–900. [Google Scholar] [CrossRef]
- Van Sonderen, A.; Thijs, R.D.; Coenders, E.C.; Jiskoot, L.C.; Sanchez, E.; de Bruijn, M.A.A.M.; van Coevorden-Hameete, M.H.; Wirtz, P.W.; Schreurs, M.W.J.; Sillevis Smitt, P.A.E.; et al. Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up. Neurology 2016, 87, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.R.; Stagg, C.J.; Schott, J.M.; Rosenthal, C.R.; Schneider, S.A.; Pettingill, P.; Pettingill, R.; Waters, P.; Thomas, A.; Voets, N.L.; et al. Faciobrachial dystonic seizures: The influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013, 136, 3151–3162. [Google Scholar] [CrossRef]
- Boronat, A.; Sabater, L.; Saiz, A.; Dalmau, J.; Graus, F. GABAB receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology 2011, 76, 795–800. [Google Scholar] [CrossRef]
- Hoftberger, R.; Titulaer, M.J.; Sabater, L.; Dome, B.; Rózsás, A.; Hegedus, B.; Hoda, M.A.; Laszlo, V.; Ankersmit, H.J.; Harms, L.; et al. Encephalitis and GABAB receptor antibodies: Novel findings in a new case series of 20 patients. Neurology 2013, 81, 1500–1506. [Google Scholar] [CrossRef]
- Jeffery, O.J.; Lennon, V.A.; Pittock, S.J.; Gregory, J.K.; Britton, J.W.; McKeon, A. GABAB receptor autoantibody frequency in service serologic evaluation. Neurology 2013, 81, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, E.; Lai, M.; Peng, X.; Hughes, E.; Constantinescu, R.; Raizer, J.; Friedman, D.; Skeen, M.B.; Grisold, W.; Kimura, A.; et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: Case series and characterisation of the antigen. Lancet Neurol. 2010, 9, 67–76. [Google Scholar] [CrossRef]
- Benarroch, E.E. GABAA receptor heterogeneity, function, and implications for epilepsy. Neurology 2007, 68, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Petit-Pedrol, M.; Armangue, T.; Peng, X.; Bataller, L.; Cellucci, T.; Davis, R.; McCracken, L.; Martinez-Hernandez, E.; Mason, W.P.; Kruer, M.C.; et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: A case series, characterization of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014, 13, 276–286. [Google Scholar] [CrossRef]
- Ohkawa, T.; Satake, S.; Yokoi, N.; Miyazaki, Y.; Ohshita, T.; Sobue, G.; Takashima, H.; Watanabe, O.; Fukata, Y.; Fukata, M. Identification and characterization of GABAA receptor autoantibodies in autoimmune encephalitis. J. Neurosci. 2014, 34, 8151–8163. [Google Scholar] [CrossRef]
- Spatola, M.; Petit-Pedrol, M.; Simabukuro, M.M.; Armangue, T.; Castro, F.J.; Barcelo Artigues, M.I.; Julià Benique, M.R.; Benson, L.; Gorman, M.; Felipe, A.; et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology 2017, 88, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Poliak, S.; Gollan, L.; Martinez, R.; Custer, A.; Einheber, S.; Salzer, J.L.; Trimmer, J.S.; Shrager, P.; Peles, E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 1999, 24, 1037–1047. [Google Scholar] [CrossRef]
- Joubert, B.; Saint-Martin, M.; Noraz, N.; Picard, G.; Rogemond, V.; Ducray, F.; Desestret, V.; Psimaras, D.; Delattre, J.Y.; Antoine, J.C.; et al. Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol. 2016, 73, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, J.S.; Lee, S.T.; Byun, J.I.; Moon, J.; Shin, J.W.; Jeong, D.E.; Lee, G.H.; Jeong, S.H.; Shin, Y.W.; Jung, K.H.; et al. Clinical manifestations of patients with CASPR2 antibodies. J. Neuroimmunol. 2015, 281, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Van Sonderen, A.; Ariño, H.; Petit-Pedrol, M.; Leypoldt, F.; Körtvélyessy, P.; Wandinger, K.P.; Lancaster, E.; Wirtz, P.W.; Schreurs, M.W.J.; Sillevis Smitt, P.A.E.; et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016, 87, 521–528. [Google Scholar] [CrossRef]
- Lancaster, E.; Huijbers, M.G.; Bar, V.; Boronat, A.; Wong, A.; Martinez-Hernandez, E.; Wilson, C.; Jacobs, D.; Lai, M.; Walker, R.W.; et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann. Neurol. 2011, 69, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Boronat, A.; Gelfand, J.M.; Gresa-Arribas, N.; Jeong, H.Y.; Walsh, M.; Roberts, K.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R.; Graus, F.; et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann. Neurol. 2013, 73, 120–128. [Google Scholar] [CrossRef]
- Hara, M.; Ariño, H.; Petit-Pedrol, M.; Sabater, L.; Titulaer, M.J.; Martinez-Hernandez, E.; Schreurs, M.W.J.; Rosenfeld, M.R.; Graus, F.; Dalmau, J. DPPX antibody-associated encephalitis: Main syndrome and antibody effects. Neurology 2017, 88, 1340–1348. [Google Scholar] [CrossRef]
- Balint, B.; Jarius, S.; Nagel, S.; Haberkorn, U.; Probst, C.; Blöcker, I.M.; Bahtz, R.; Komorowski, L.; Stöcker, W.; Kastrup, A.; et al. Progressive encephalomyelitis with rigidity and myoclonus: A new variant with DPPX antibodies. Neurology 2014, 82, 1521–1528. [Google Scholar] [CrossRef]
- Piepgras, J.; Höltje, M.; Michel, K.; Li, Q.; Otto, C.; Drenckhahn, C.; Probst, C.; Schemann, M.; Jarius, S.; Stöcker, W.; et al. Anti-DPPX encephalitis: Pathogenic effects of antibodies on gut and brain neurons. Neurology 2015, 85, 890–897. [Google Scholar] [CrossRef]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Hughes, E.G.; Peng, X.; Zhou, L.; Gleichman, A.J.; Shu, H.; Matà, S.; Kremens, D.; Vitaliani, R.; Geschwind, M.D.; et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann. Neurol. 2009, 65, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Höftberger, R.; van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Titulaer, M.J.; et al. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology 2015, 84, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.; Kerschen, P.; Zekeridou, A.; Desestret, V.; Rogemond, V.; Chaffois, M.O.; Ducray, F.; Larrue, V.; Daubail, B.; Idbaih, A.; et al. Clinical spectrum of encephalitis associated with antibodies against the α-Amino-3-Hydroxy-5-Methyl-4-isoxazolepropionic acid receptor: Case series and review of the literature. JAMA Neurol. 2015, 72, 1163–1169. [Google Scholar] [CrossRef]
- Walikonis, J.E.; Lennon, V.A. Radioimmunoassay for glutamic acid decarboxylase (GAD65) autoantibodies as a diagnostic aid for stiff-man syndrome and a correlate of susceptibility to type 1 diabetes mellitus. Mayo. Clin. Proc. 1998, 73, 1161–1166. [Google Scholar] [CrossRef]
- Muñoz-Lopetegi, A.; de Bruijn, M.A.A.M.; Boukhrissi, S.; Bastiaansen, A.E.M.; Nagtzaam, M.M.P.; Hulsenboom, E.S.P.; Boon, A.J.W.; Neuteboom, R.F.; de Vries, J.M.; Sillevis Smitt, P.A.E.; et al. Neurologic syndromes related to anti-GAD65: Clinical and serologic response to treatment. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e696. [Google Scholar] [CrossRef]
- Pittock, S.J.; Yoshikawa, H.; Ahlskog, J.E.; Tisch, S.H.; Benarroch, E.E.; Kryzer, T.J.; Lennon, V.A. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin. Proc. 2006, 81, 1207–1214. [Google Scholar] [CrossRef]
- Budhram, A.; Sechi, E.; Flanagan, E.P.; Dubey, D.; Zekeridou, A.; Shah, S.S.; Gadoth, A.; Naddaf, E.; McKeon, A.; Pittock, S.J.; et al. Clinical Specturm of high-titer GAD 65 antibodies. J. Neurol. Neurosurg. Psychiatry 2021, 92, 645–654. [Google Scholar] [CrossRef]
- Lilleker, J.B.; Biswas, V.; Mohanraj, R. Glutamic acid decarboxylase (GAD) antibodies in epilepsy: Diagnostic yield and therapeutic implications. Seizure 2014, 23, 598–602. [Google Scholar] [CrossRef]
- Dubey, D.; Toledano, M.; McKeon, A. Clinical presentation of autoimmune and viral encephalitides. Curr. Opin. Crit. Care 2018, 24, 80–90. [Google Scholar] [CrossRef]
- Kashyap, P.; Farrugia, G. Enteric autoantibodies and gut motility disorder. Gastroenterol. Clin. N. Am. 2008, 37, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, L.A.; Pittock, S.J.; McKeon, A.; Lennon, V.A.; Britton, J.W. Extratemporal EEG and MRI findings in ANNA-1 (anti-Hu) encephalitis. Epilepsy Res. 2011, 95, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Shavit, Y.B.; Graus, F.; Probst, A.; Rene, R.; Steck, A.J. Epilepsia partialis continua: A new manifestation of anti-Hu-associated paraneoplastic encephalomyelitis. Ann. Neurol. 1999, 45, 255–258. [Google Scholar] [CrossRef]
- Keime-Guibert, F.; Graus, F.; Fleury, A.; René, R.; Honnorat, J.; Broet, P.; Delattre, J.Y. Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide and methylprednisolone. J. Neurol. Neurosurg. Psychiatry 2000, 68, 479–482. [Google Scholar] [CrossRef]
- Hoffmann, L.A.; Jarius, S.; Pellkofer, H.L.; Schueller, M.; Krumbholz, M.; Koenig, F.; Johannis, W.; la Fougere, C.; Newman, T.; Vincent, A.; et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J. Neurol. Neurosurg. Psychiatry 2008, 79, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Sahashi, K.; Sakai, K.; Mano, K.; Hirose, G. Anti-Ma2 antibody related paraneoplastic limbic/brain stem encephalitis associated with breast cancer expressing Ma1, Ma2, and Ma3 mRNAs. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1332–1335. [Google Scholar] [CrossRef]
- Ortega Suero, G.; Sola-Valls, N.; Escudero, D.; Saiz, A.; Graus, F. Anti-Ma and anti-Ma2-associated paraneoplastic neurological syndromes. Neurologia 2018, 33, 18–27. [Google Scholar] [CrossRef]
- Yu, Z.; Kryzer, T.J.; Griesmann, G.E.; Kim, K.; Benarroch, E.E.; Lennon, V.A. CRMP-5 neuronal autoantibody: Marker of lung cancer and thymoma-related autoimmunity. Ann. Neurol. 2001, 49, 146–154. [Google Scholar] [CrossRef]
- Vernino, S.; Tuite, P.; Adler, C.H.; Meschia, J.F.; Boeve, B.F.; Boasberg, P.; Parisi, J.E.; Lennon, V.A. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann. Neurol. 2002, 51, 625–630. [Google Scholar] [CrossRef]
- Dubey, D.; Lennon, V.A.; Gadoth, A.; Pittock, S.J.; Flanagan, E.P.; Schmeling, J.E.; McKeon, A.; Klein, C.J. Autoimmune CRMP5 neuropathy phenotype and outcome defined from 105 cases. Neurology 2018, 90, e103–e110. [Google Scholar] [CrossRef]
- Quek, A.M.L.; Britton, J.W.; Mckeon, A.; So, E.; Lennon, V.A.; Shin, C.; Klein, C.; Watson, R.E., Jr.; Kotsenas, A.L.; Lagerlund, T.D.; et al. Autoimmune epilepsy: Clinical characteristics and response to immunotherapy. Arch. Neurol. 2012, 69, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Sculie, C.; Gaspard, N. New onset status epilepticus (NORSE). Seizure 2019, 68, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Foreman, B.P.; Alvarez, V.; Cabrera Kang, C.; Probasco, J.C.; Jongeling, A.C.; Meyers, E.; Espinera, A.; Haas, K.F.; Schmitt, S.E.; et al. New-onset refractory status epilepticus: Etiology, clinical features, and outcome. Neurology 2015, 85, 1604–1613. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Hirsch, L.J.; Sculie, C.; Loddenkemper, T.; van Baalen, A.; Lancrenon, J.; Emmery, M.; Specchio, N.; Farias-Moeller, R.; Wong, N.; et al. New-onset refractory status epilepticus (NORSE) and febrile infection-related epilepsy syndrome (FIRES): State of the art and perspectives. Epilepsia 2018, 59, 745–752. [Google Scholar] [CrossRef]
- Dubey, D.; Singh, J.; Britton, J.W.; Pittock, S.J.; Flanagan, E.P.; Lennon, V.A.; Tillema, J.M.; Wirrell, E.; Shin, C.; So, E.; et al. Predictive models in the diagnosis and treatment of autoimmune epilepsy. Epilepsia 2017, 58, 1181–1189. [Google Scholar] [CrossRef]
- Husari, K.S.; Dubey, D. Autoimmune Epilepsy. Neurotherapeutics 2019, 16, 685–702. [Google Scholar] [CrossRef]
- Abboud, H.; Probasco, J.C.; Irani, S.; Ances, B.; Benavides, D.R.; Bradshaw, M.; Christo, P.P.; Dale, R.C.; Fernandez-Fournier, M.; Flanagan, E.P.; et al. Autoimmune encephalitis: Proposed best practice recommendations for diagnosis and acute management. J. Neurol. Neurosurg. Psychiatry 2021, 92, 757–768. [Google Scholar] [CrossRef]
- Mc Ginty, R.; Handel, A.; Moloney, T.; Ramesh, A.; Fower, A.; Emma Torzillo, E.; Kramer, H.; Howell, S.; Waters, P.; Adcock, J.; et al. Clinical features which predictneuronal surface autoantibodies in new-onset focal epilepsy: Implications for immunotherapies. J. Neurol. Neurosurg. Psychiatry 2021, 92, 291–294. [Google Scholar] [CrossRef]
- Seidling, V.; Hoffmann, J.H.; Enk, A.H.; Hadaschik, E.N. Analysis of high-dose intravenous immunoglobulin therapy in 16 patients with refractory autoimmune blistering skin disease: High efficacy and no serious adverse events. Acta Derm. Venereol. 2013, 93, 346–349. [Google Scholar] [CrossRef]
- Bichuetti-Silva, D.C.; Furlan, F.P.; Nobre, F.A.; Pereira, C.T.; Gonçalves, T.R.; Gouveia-Pereira, M.; Rota, R.; Tavares, L.; Mazzucchelli, J.T.; Costa-Carvalho, B.T. Immediate infusion-related adverse reactions to intravenous immunoglobulin in a prospective cohort of 1765 infusions. Int. Immunopharmacol. 2014, 23, 442–446. [Google Scholar] [CrossRef]
- Daniel, G.W.; Menis, M.; Sridhar, G.; Scott, D.; Wallace, A.E.; Ovanesov, M.V.; Golding, B.; Anderson, S.A.; Epstein, J.; Martin, D.; et al. Immune globulins and thrombotic adverse events as recorded in a large administrative database in 2008 through 2010. Transfusion 2012, 52, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, E.; Romero-Garrido, J.A.; López-Granados, E.; Borobia, A.M.; Pérez, T.; Medrano, N.; Rueda, C.; Tong, H.Y.; Herrero, A.; Frías, J. Symptomatic thromboembolic events in patients treated with intravenous-immunoglobulins: Results from a retrospective cohort study. Thromb. Res. 2014, 133, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Kasi, P.M.; Tawbi, H.A.; Oddis, C.V.; Kulkarni, H.S. Clinical review: Serious adverse events associated with the use of rituximab—A critical care perspective. Crit. Care 2012, 16, 231. [Google Scholar] [CrossRef] [PubMed]
- McAtee, C.L.; Lubega, J.; Underbrink, K.; Curry, K.; Msaouel, P.; Barrow, M.; Muscal, E.; Lotze, T.; Srivaths, P.; Forbes, L.R.; et al. Association of Rituximab Use with Adverse Events in Children, Adolescents, and Young Adults. JAMA Netw. Open 2021, 4, e2036321. [Google Scholar] [CrossRef]
- Barnes, H.; Holland, A.E.; Westall, G.P.; Goh, N.S.; Glaspole, I.N. Cyclophosphamide for connective tissue disease-associated interstitial lung disease. Cochrane Database Syst. Rev. 2018, 1, CD010908. [Google Scholar] [CrossRef]
- Von Rhein, B.; Wagner, J.; Widman, G.; Malter, M.P.; Elger, C.E.; Helmstaedter, C. Suspected antibody negative autoimmune limbic encephalitis: Outcome of immunotherapy. Acta Neurol. Scand. 2017, 135, 134–141. [Google Scholar] [CrossRef]
- Rüegg, S. Antineuronal antibodies and epilepsy: Treat the patient, not the lab. J. Neurol. Neurosurg. Psychiatry 2021, 92, 230. [Google Scholar] [CrossRef]

| APE Score and Components | |
|---|---|
| Clinical Characteristics | Score |
| New-onset, rapidly progressive mental status changes that developed over 1–6 weeks or new-onset seizure activity (within 1 year of evaluation) | 1 |
| Neuropsychiatry changes; agitation, aggressiveness, emotional lability | 1 |
| Autonomic dysfunction (sustained atrial tachycardia or bradycardia, orthostatic hypotension (≥20 mm Hg fall in systolic pressure or ≥10 mm Hg fall in diastolic pressure within 3 min of standing), hyperhidrosis, persistently labile blood pressure, ventricular tachycardia, or cardiac asystole) | 1 |
| Viral prodrome (rhinorrhea, sore throat, low-grade fever) in the absence of underlying malignancy | 2 |
| Facial dyskinesias or faciobrachial dystonic movements | 2 |
| Refractory seizure | 2 |
| CSF: signs of inflammation (elevated CSF protein >50 mg/dL and/or lymphocytic pleocytosis >5 cells/dL if the total number of CSF RBC is <1000 cells/dL a | 2 |
| Brain MRI findings indicate limbic encephalitis (medial temporal T2/FLAIR signal changes) a | 2 |
| Underlying malignancy (excluding cutaneous squamous cell carcinoma and basal cell carcinoma) | 2 |
| a: No brain MRI or CSF analyses were scored zero. | Total max: 15 |
| APE2 Score and Components | |
| Clinical Characteristics | Score |
| New-onset, rapidly progressive mental status changes that developed over 1–6 weeks or new-onset seizure activity (within 1 year of evaluation) | 1 |
| Neuropsychiatry changes: agitation, aggressiveness, emotional lability | 1 |
| Autonomic dysfunction (sustained atrial tachycardia or bradycardia, orthostatic hypotension, hyperhidrosis, persistently labile blood pressure, ventricular tachycardia, cardiac asystole, or gastrointestinal dysmotility) * | 1 |
| Viral prodrome (rhinorrhea, sore throat, low-grade fever) in the absence of underlying systemic malignancy within 5 years of neurological symptom onset | 2 |
| Faciobrachial dystonic seizures | 3 |
| Facial dyskinesias in the absence of faciobrachial dystonic seizures | 2 |
| Seizure refractory to at least two antiseizure medications | 2 |
| CSF signs of inflammation # (elevated CSF protein >50 mg/dL and/or lymphocytic pleocytosis >5 cells/dL if the total number of CSF RBC is <1000 cells/dL | 2 |
| Brain MRI suggesting encephalitis (T2/FLAIR hyperintensity restricted to one or both medial temporal lobes or multifocal in gray matter, white matter, or both compatible with demyelination or inflammation) | 2 |
| Systemic cancer diagnosed within 5 years of neurological symptom onset (excluding cutaneous squamous cell carcinoma, basal cell carcinoma, brain tumor, cancer with brain metastasis) | 2 |
| * Scored only if no history of autonomic dysfunction prior to onset of suspected autoimmune syndrome and the autonomic dysfunction not attributable to medications, hypovolemia, plasmapheresis, or infection # Patients scored zero if brain MRI or CSF analysis not performed | Total max: 18 |
| Clinical Characteristics | Score |
|---|---|
| Rapid progressive mental change within 6 weeks or new-onset seizure within one year | 1 |
| Neuropsychiatry symptoms: agitation, aggressiveness, emotional lability | 1 |
| Autonomic dysfunction (sustained atrial tachycardia or bradycardia, orthostatic hypotension (≥20 mm Hg fall in systolic pressure or ≥10 mm Hg fall in diastolic pressure within 3 min of standing), hyperhidrosis, persistently labile blood pressure, ventricular tachycardia, or cardiac asystole) | 1 |
| Viral prodrome (rhinorrhea, sore throat, low-grade fever) in the absence of underlying malignancy | 2 |
| Facial dyskinesias or faciobrachial dystonic movements | 2 |
| Refractory seizure | 2 |
| CSF: signs of inflammation (elevated CSF protein >50 mg/dL and/or lymphocytic pleocytosis >5 cells/dL if the total number of CSF RBC is <1000 cells/dL a | 2 |
| Brain MRI findings indicate limbic encephalitis (medial temporal T2/FLAIR signal changes) a | 2 |
| Underlying malignancy (excluding cutaneous squamous cell carcinoma, basal cell carcinoma) | 2 |
| Initiation of immunotherapy within 6 month of symptom onset | 2 |
| Presence of neural plasma membrane autoantibody (e.g., NMDA-R Ab, GABAA-R Ab, GABAB-R AMPA-R Ab, DPPX, GAD-65, LGI-1 Ab, or CASPR-2 Ab) | 2 |
| a: No brain MRI or CSF analyses were scored zero. | Max: 19 |
| New-onset, rapidly progressive mental status changes that developed over 1–6 weeks or new-onset seizure activity (within 1 year of evaluation) | 1 |
| Neuropsychiatry symptoms: agitation, aggressiveness, emotional lability | 1 |
| Autonomic dysfunction (sustained atrial tachycardia or bradycardia, orthostatic hypotension, hyperhidrosis, persistently labile blood pressure, ventricular tachycardia, cardiac asystole, or gastrointestinal dysmotility) * | 1 |
| Viral prodrome (rhinorrhea, sore throat, low-grade fever) only to be scored in the absence of underlying malignancy within 5 years of neurological symptom onset | 2 |
| Faciobrachial dystonic seizures | 3 |
| Facial dyskinesias in the absence of faciobrachial dystonic seizures | 2 |
| Seizure refractory to at least two antiseizure medications | 2 |
| CSF signs of inflammation # (elevated CSF protein >50 mg/dL and/or lymphocytic pleocytosis >5 cells/dL if the total number of CSF RBC is <1000 cells/dL | 2 |
| Brain MRI suggesting encephalitis (T2/FLAIR hyperintensity restricted to one or both medial temporal lobes or multifocal in gray matter, white matter, or both compatible with demyelination or inflammation) | 2 |
| Systemic cancer diagnosed within 5 years of neurological symptom onset (excluding cutaneous squamous cell carcinoma, basal cell carcinoma, brain tumor, cancer with brain metastasis) | 2 |
| Initiation of immunotherapy within 6 month of symptom onset | 2 |
| Neural plasma membrane autoantibody detected (NMDA-R, GABAAR, GABABR, AMPA-R, DPPX, mGluR1, mGluR5, LGI1, CASPR-2, neurexin-3α, MOG) | 2 |
| * Scored only if no history of autonomic dysfunction prior to onset of suspected autoimmune syndrome and the autonomic dysfunction is not attributable to medications, hypovolemia, plasmapheresis, or infection # Patients scored zero if brain MRI or CSF analyses not performed | Max: 22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-S.; Lai, M.-C.; Huang, H.-Y.I.; Wu, S.-N.; Huang, C.-W. Immunity, Ion Channels and Epilepsy. Int. J. Mol. Sci. 2022, 23, 6446. https://doi.org/10.3390/ijms23126446
Chen T-S, Lai M-C, Huang H-YI, Wu S-N, Huang C-W. Immunity, Ion Channels and Epilepsy. International Journal of Molecular Sciences. 2022; 23(12):6446. https://doi.org/10.3390/ijms23126446
Chicago/Turabian StyleChen, Tsang-Shan, Ming-Chi Lai, Huai-Ying Ingrid Huang, Sheng-Nan Wu, and Chin-Wei Huang. 2022. "Immunity, Ion Channels and Epilepsy" International Journal of Molecular Sciences 23, no. 12: 6446. https://doi.org/10.3390/ijms23126446
APA StyleChen, T.-S., Lai, M.-C., Huang, H.-Y. I., Wu, S.-N., & Huang, C.-W. (2022). Immunity, Ion Channels and Epilepsy. International Journal of Molecular Sciences, 23(12), 6446. https://doi.org/10.3390/ijms23126446