
Antamanide Analogs as Potential Inhibitors of Tyrosinase

 , ,
, ,  , , ,
, , ,
Abstract
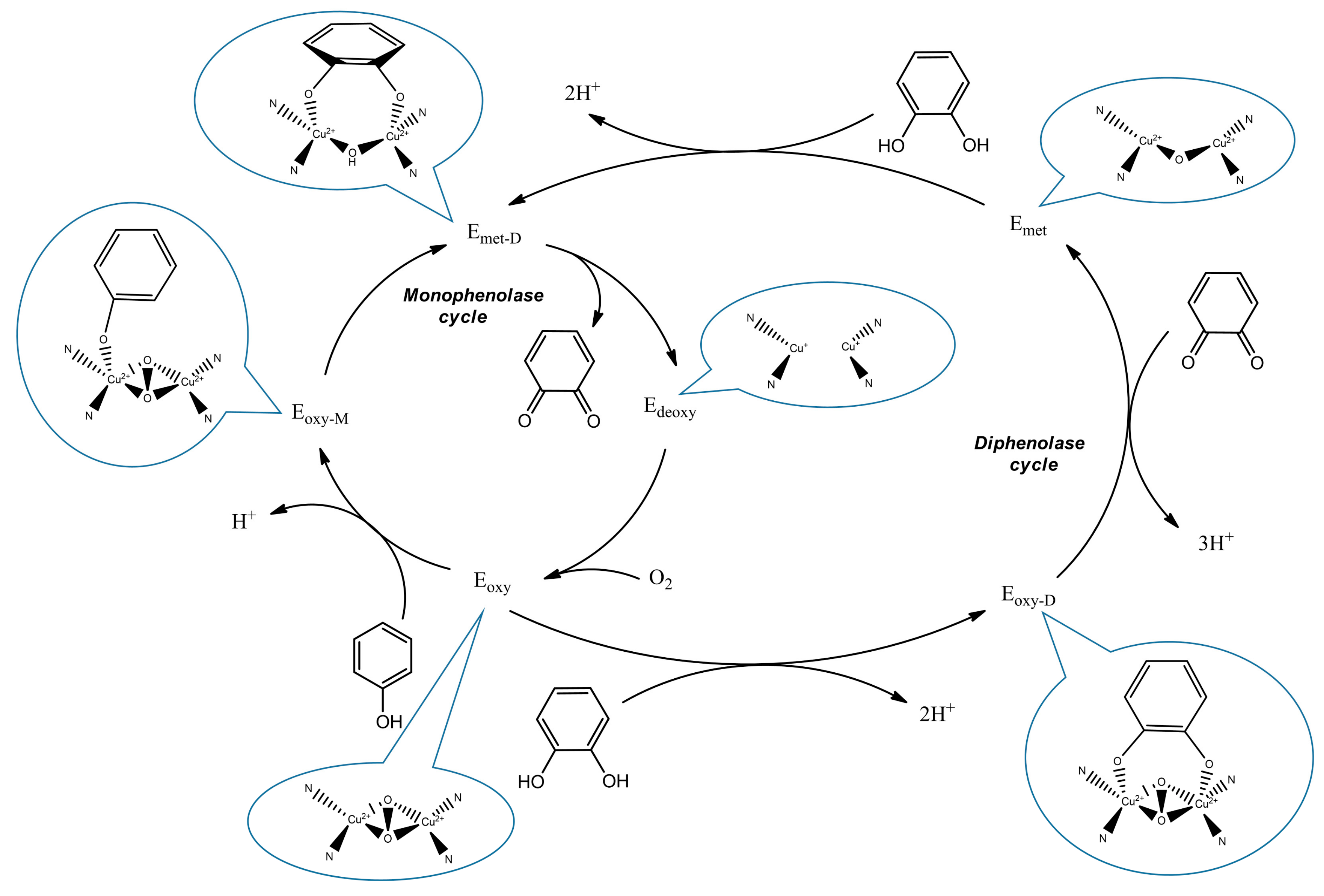
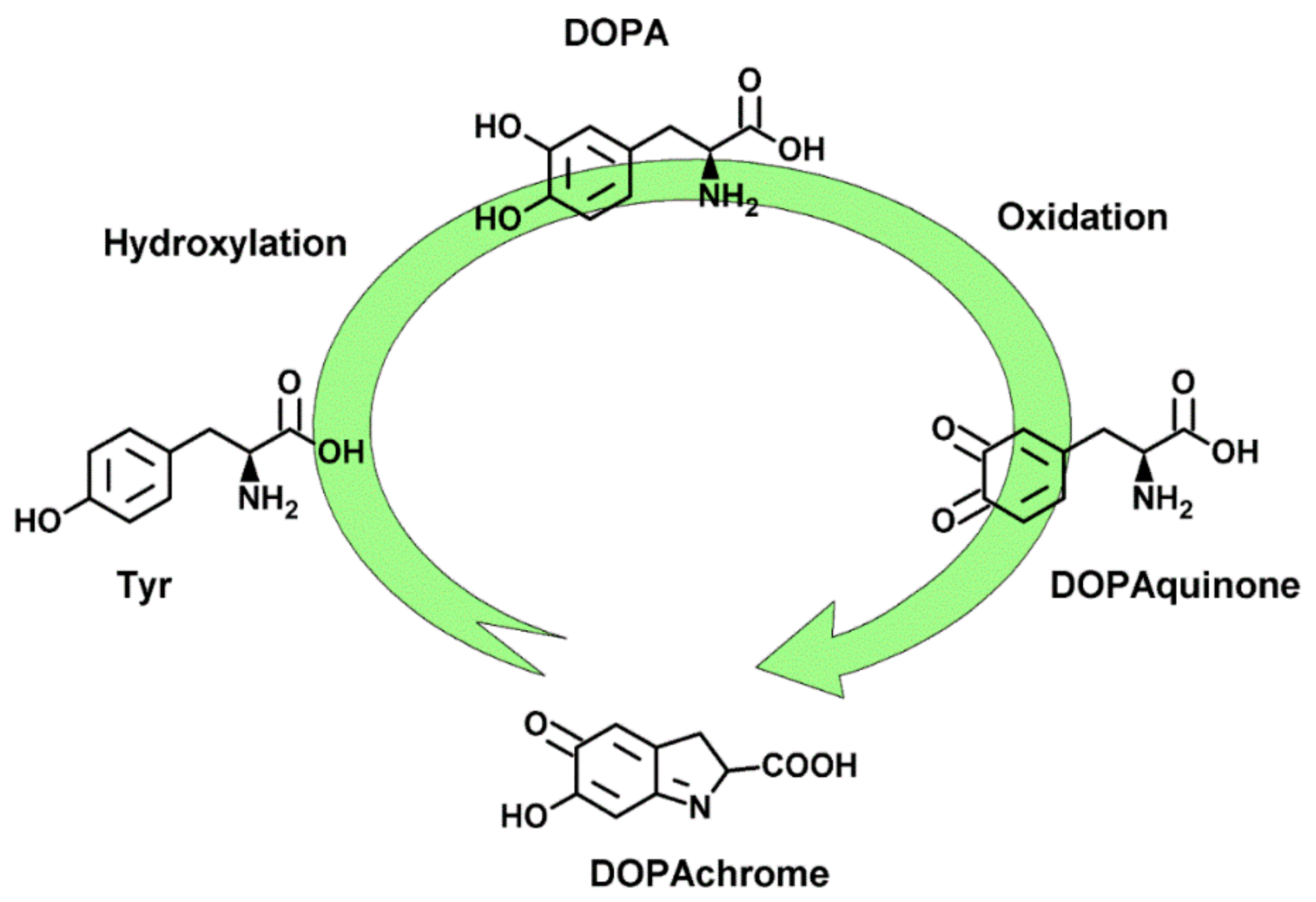
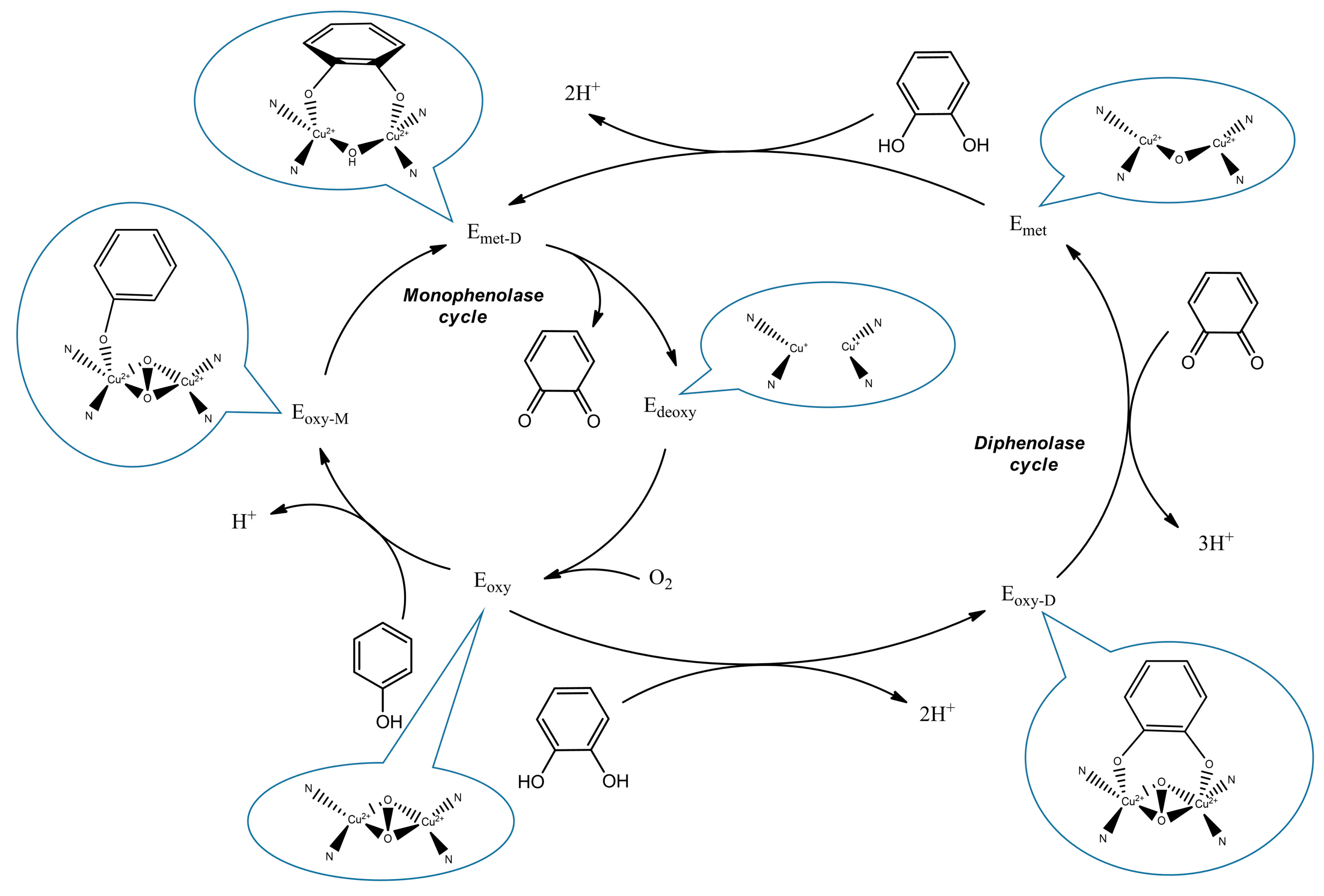
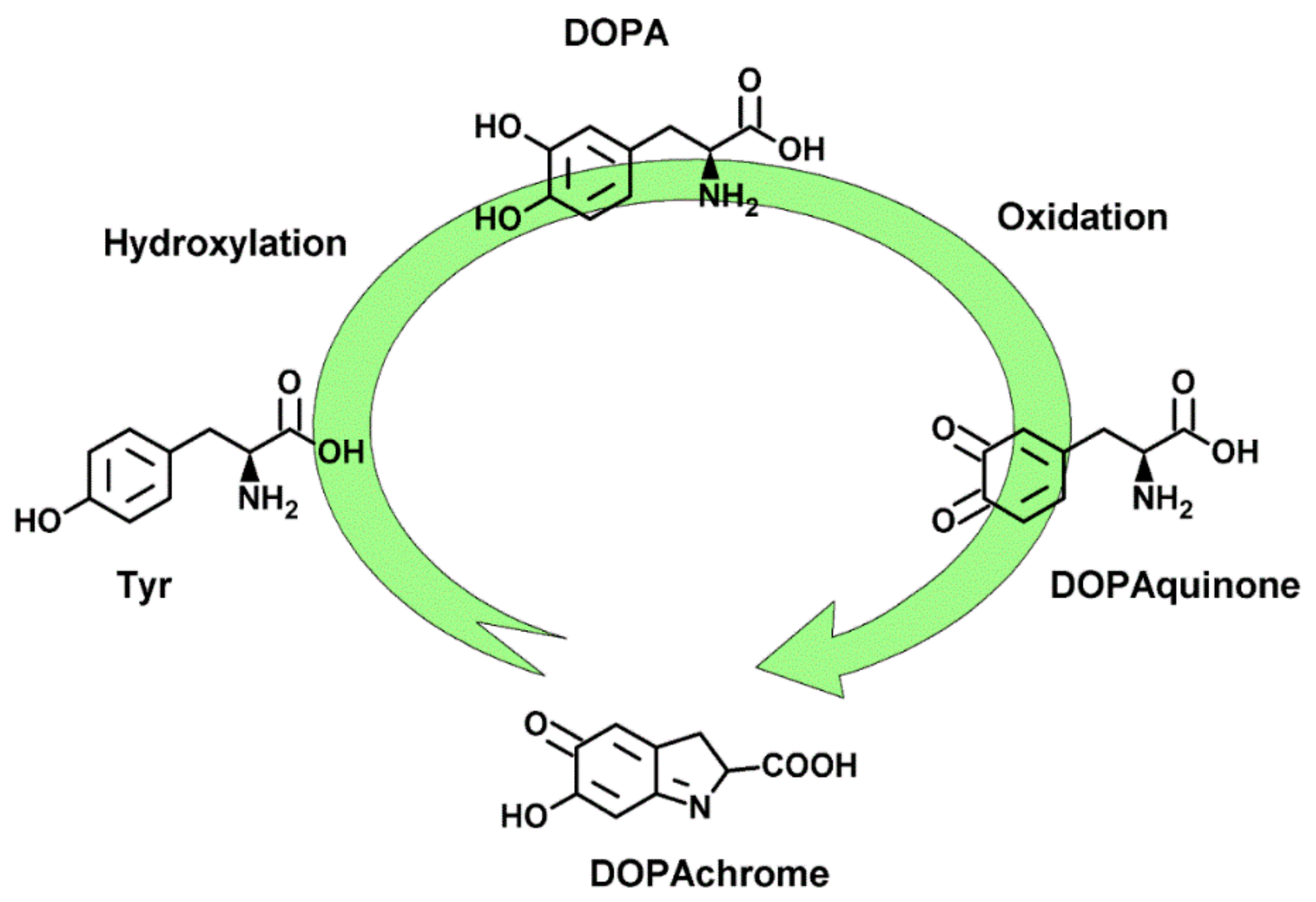
:1. Introduction
2. Results
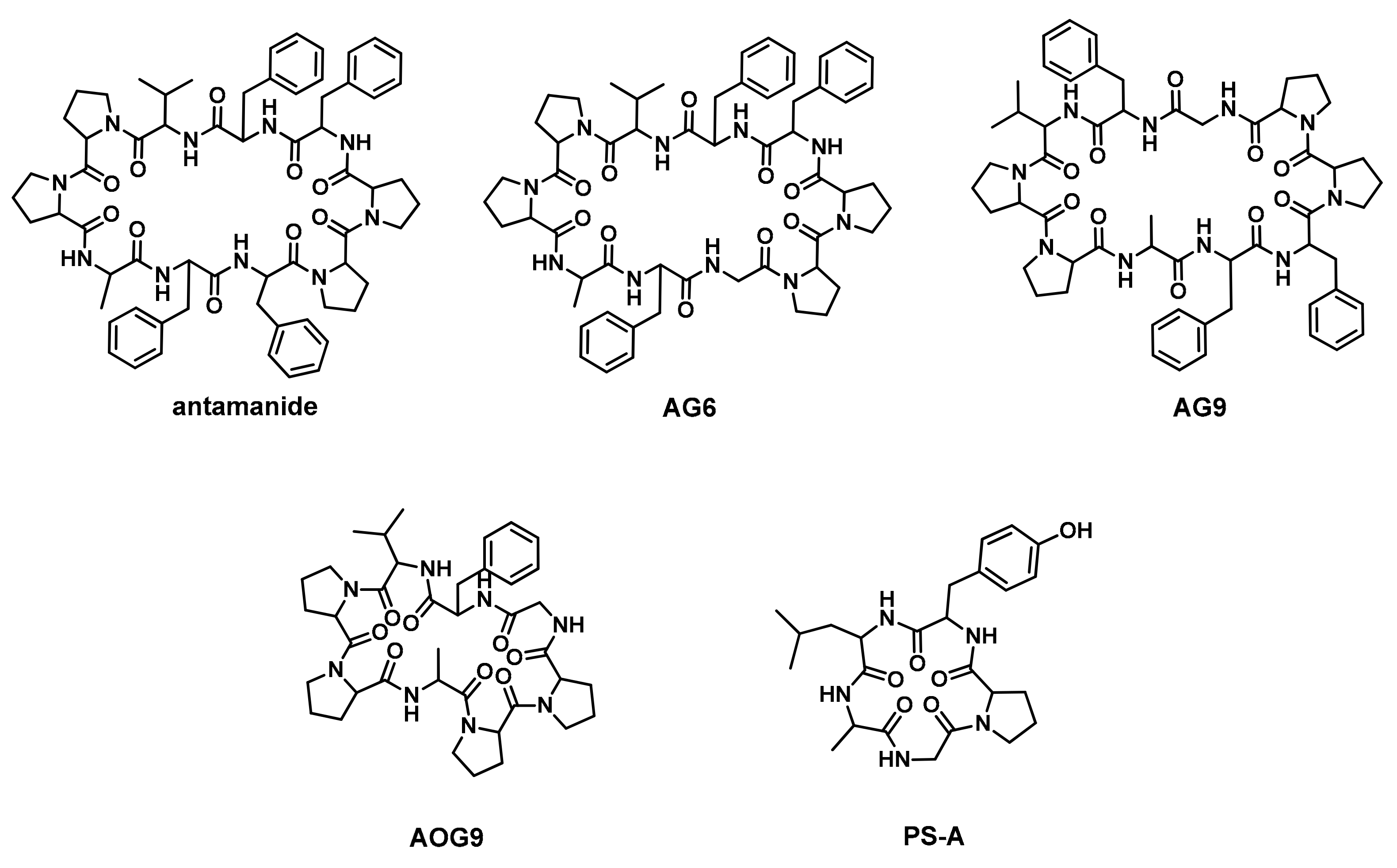
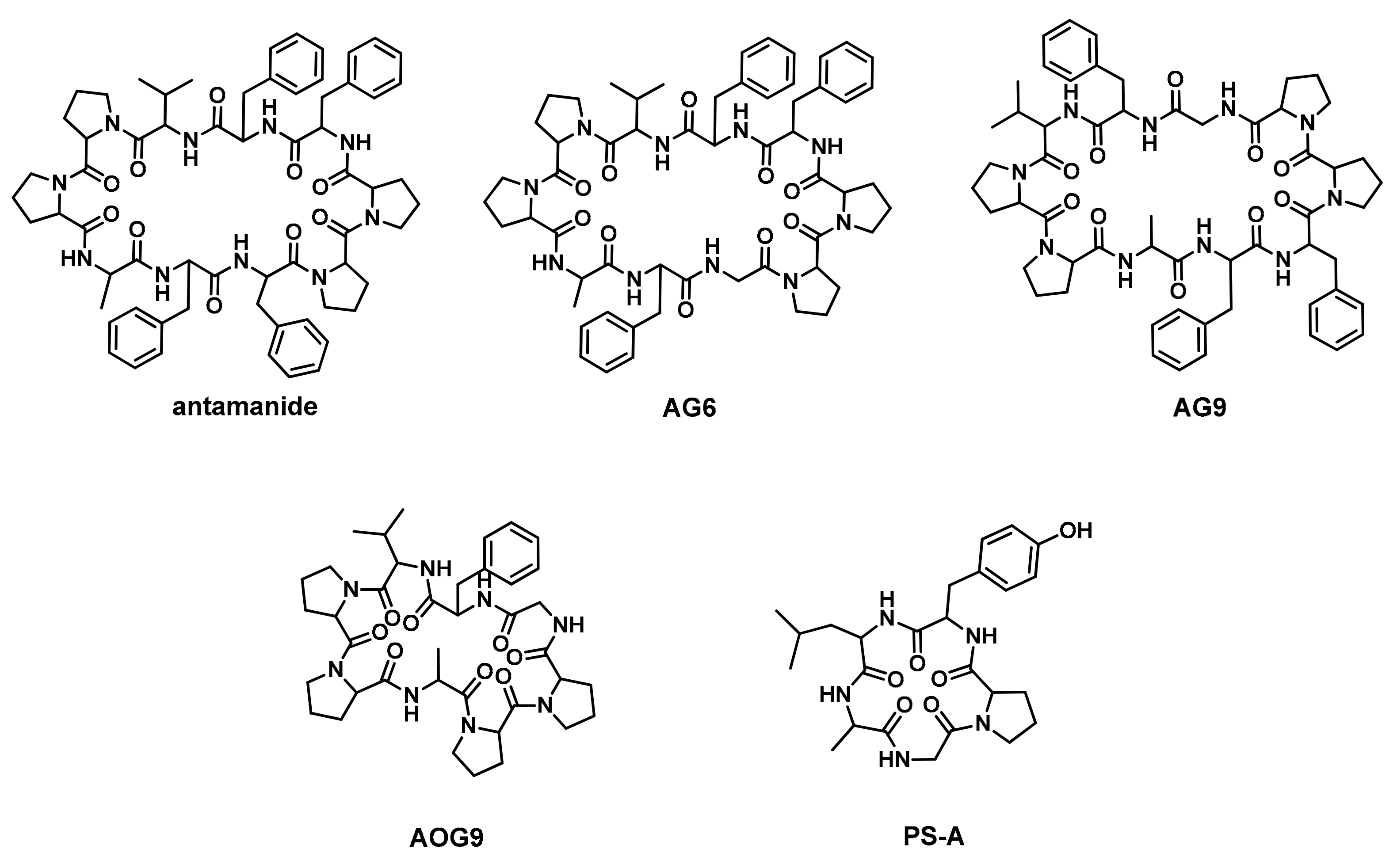
2.1. Peptide Design and Synthesis
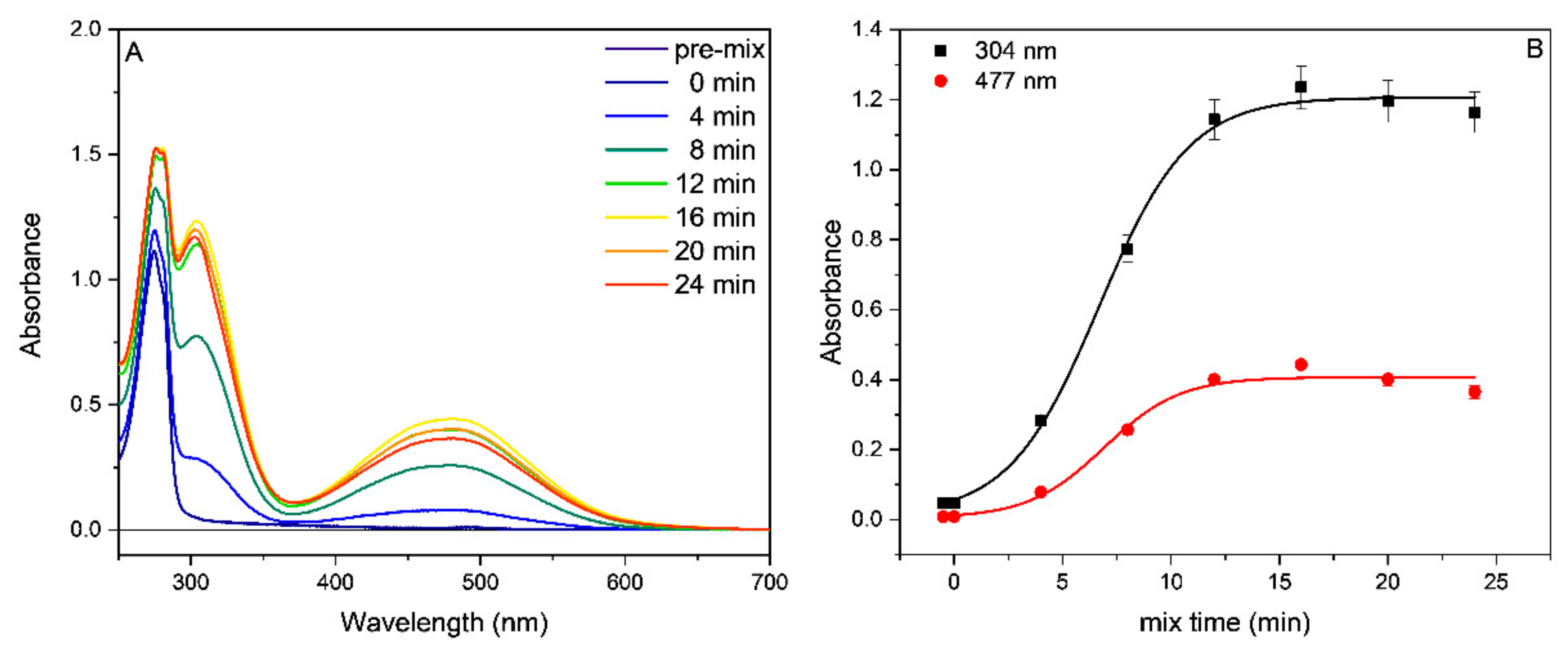
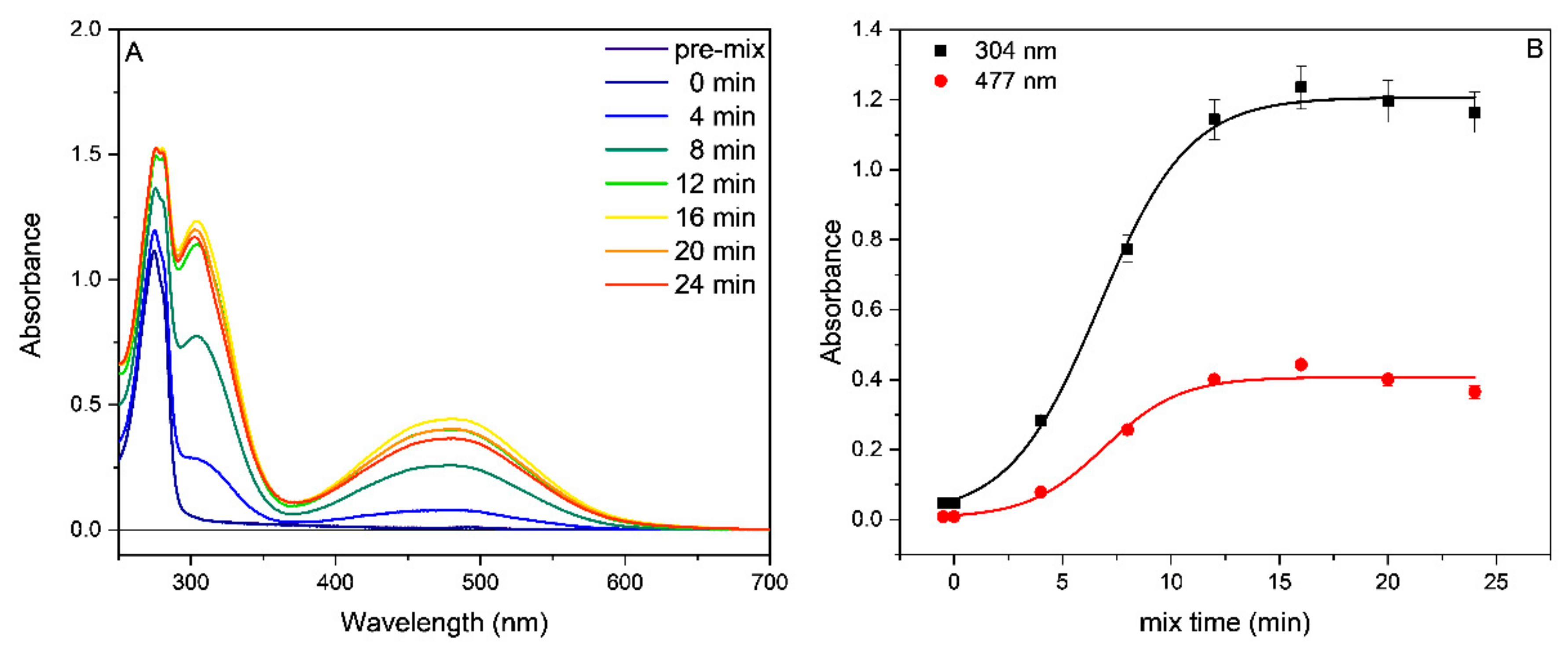
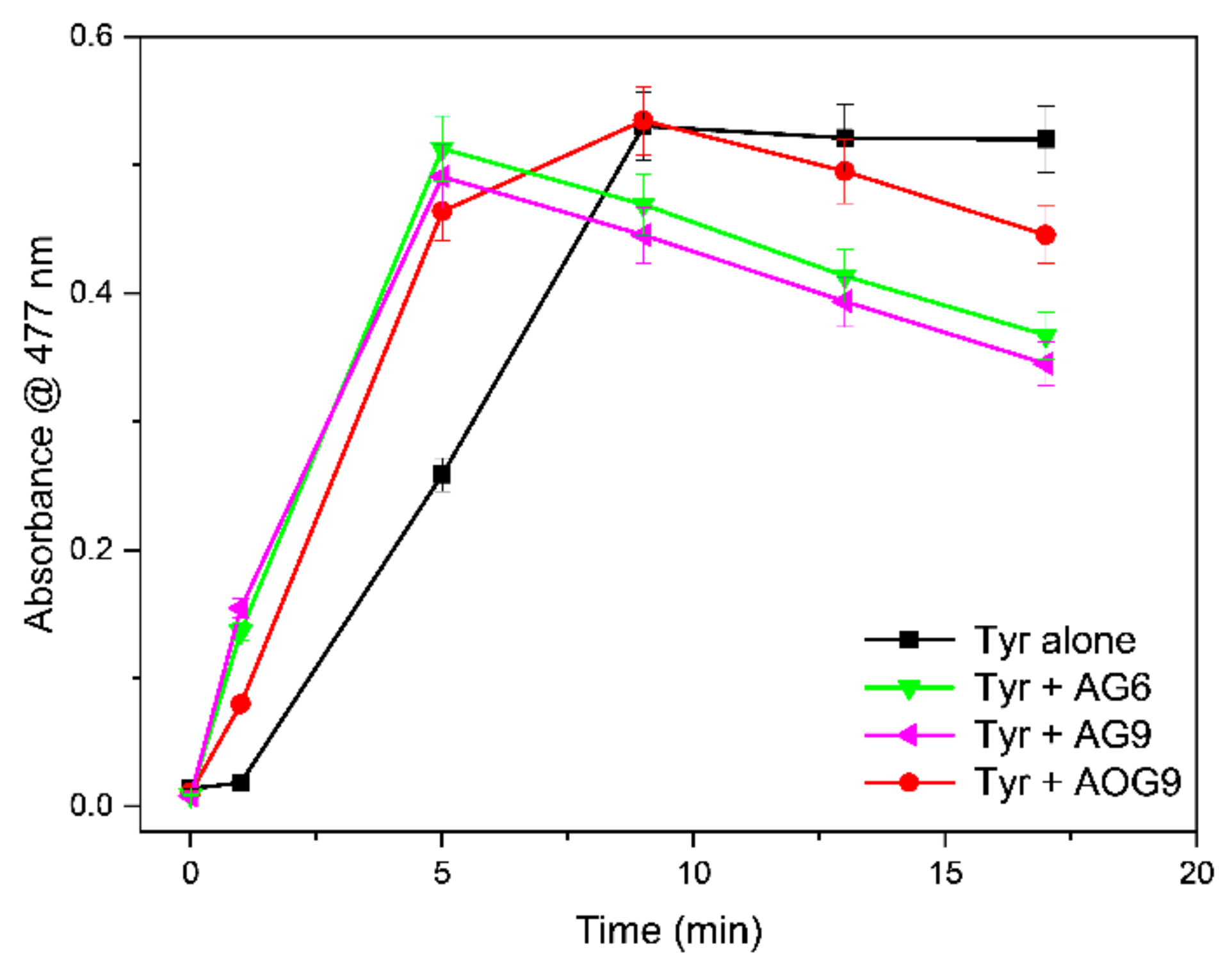
2.2. Tyrosinase Inhibition
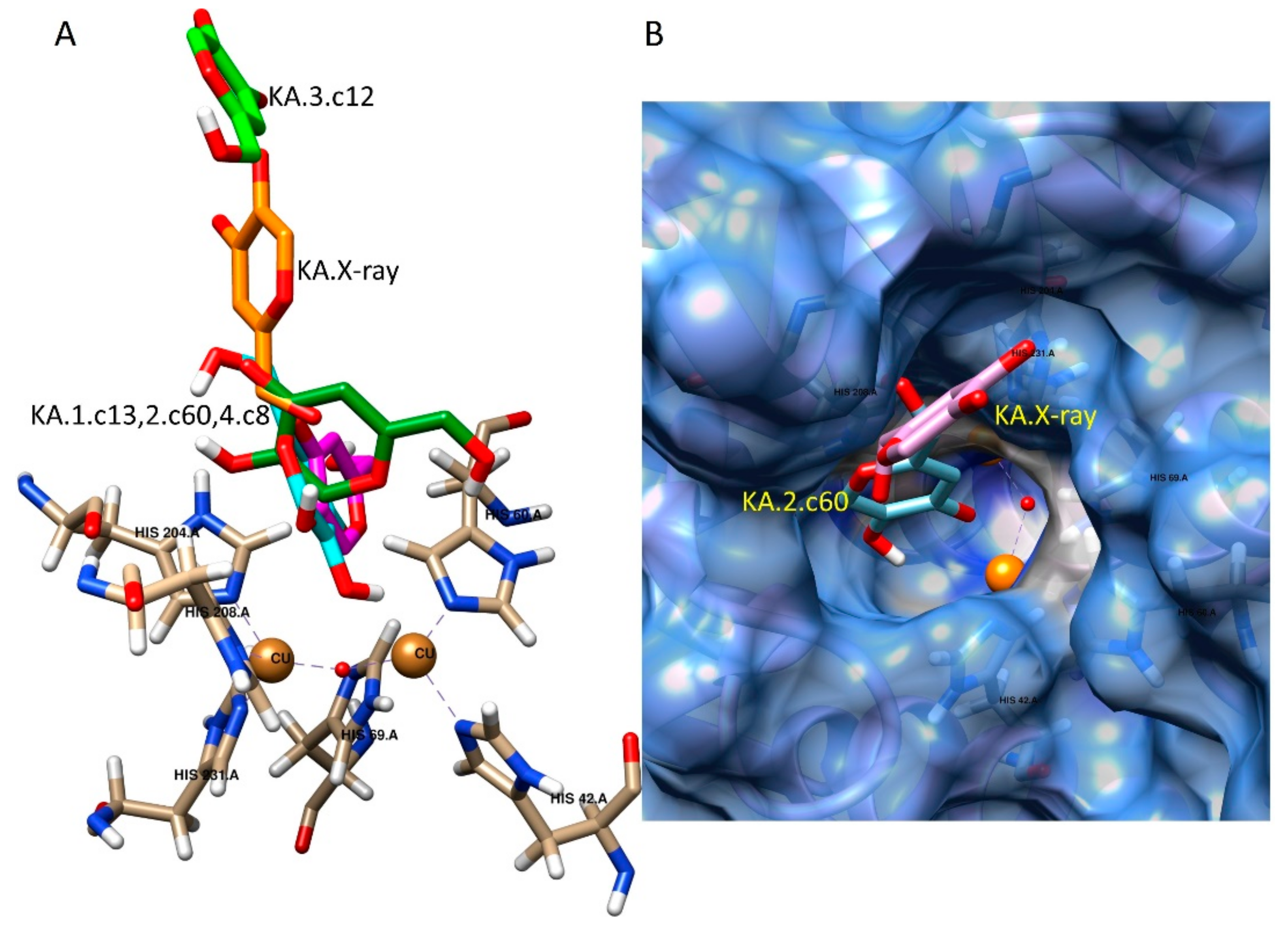
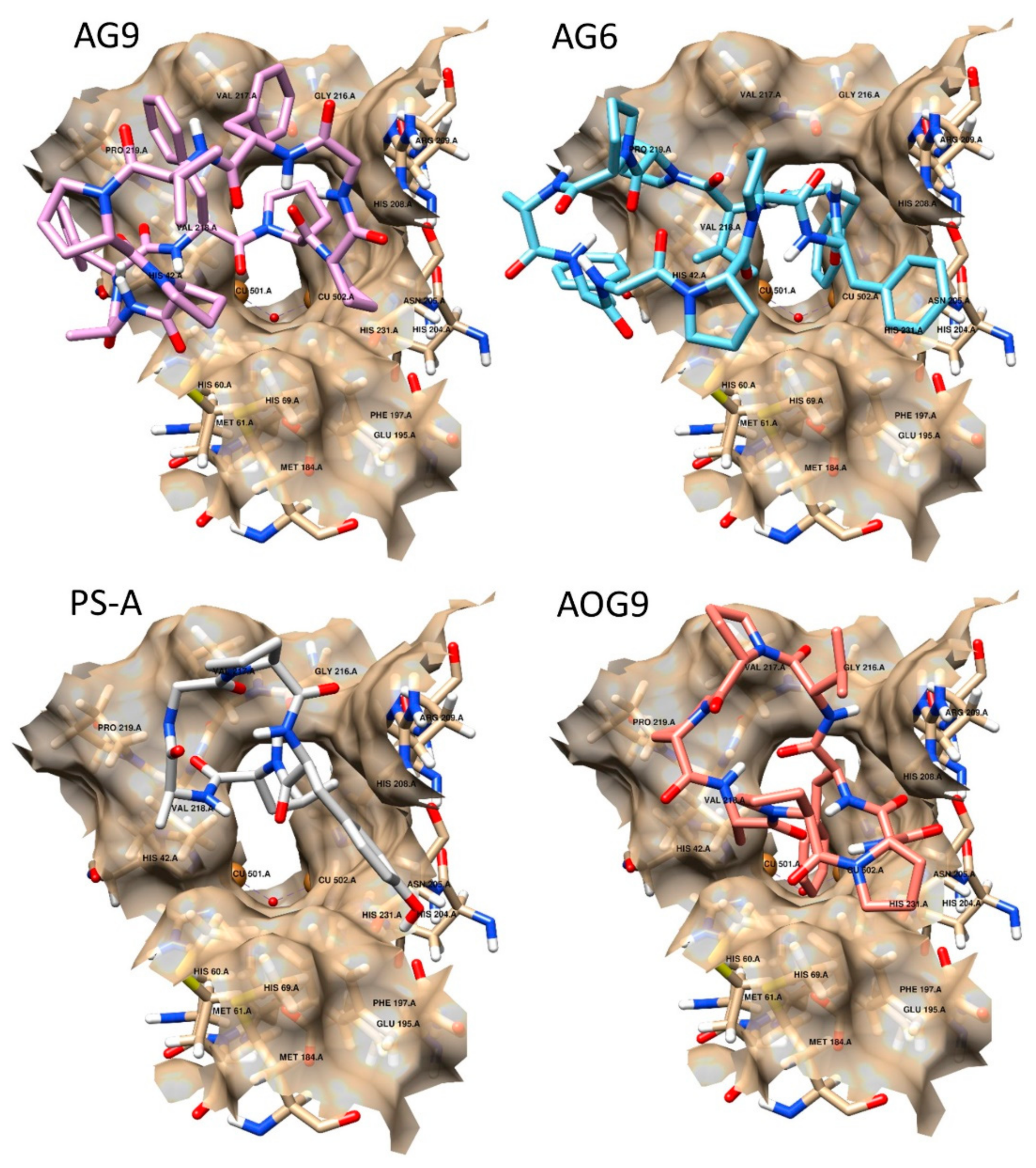
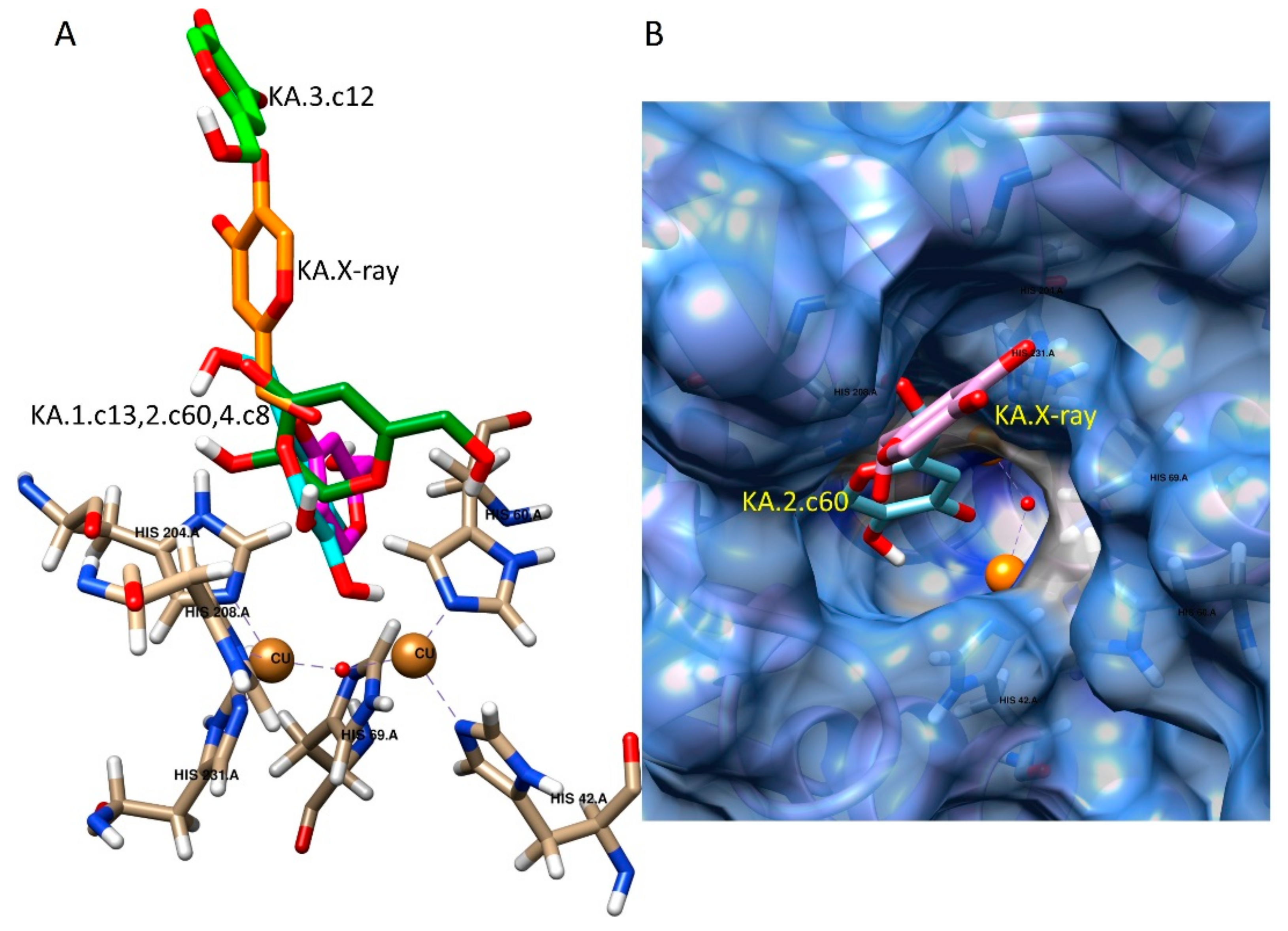
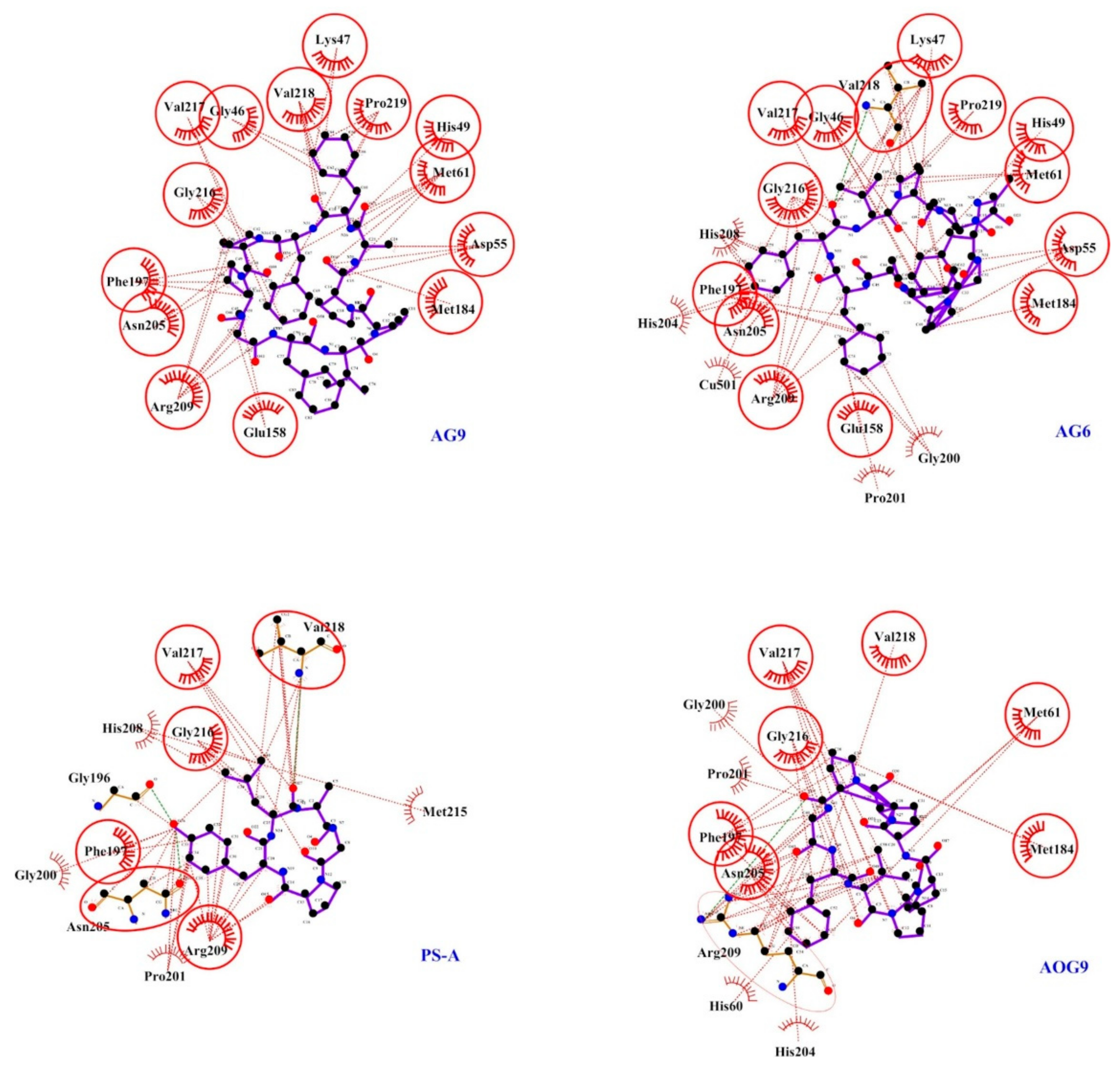
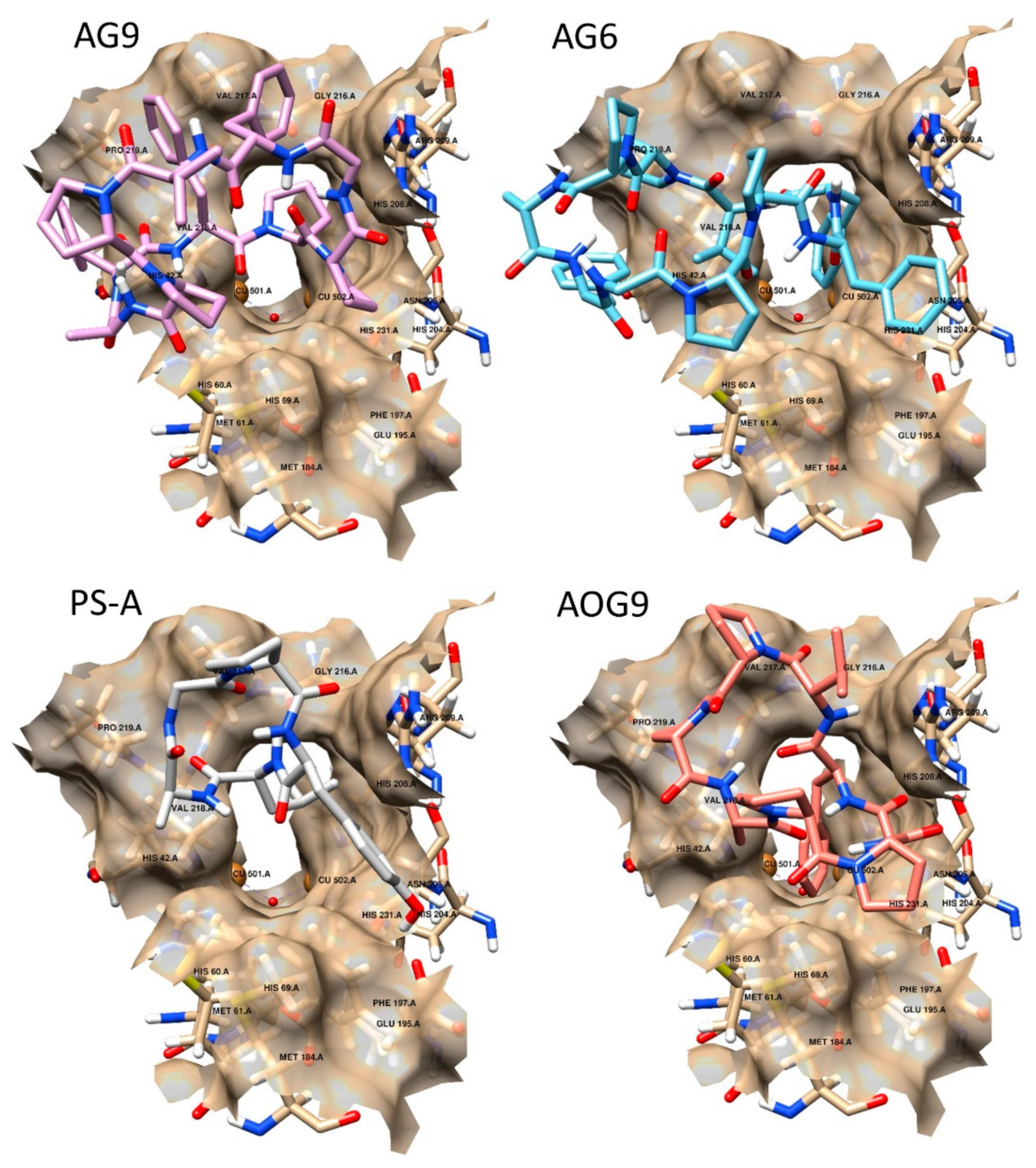
2.3. Docking Studies
3. Materials and Methods
3.1. Peptide Synthesis
3.2. Tyrosinase Inhibition
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, R.; Liu, Z.K.; Zhang, Y.N.; Wong, J.H.; Ng, T.B.; Liu, F. Research Progress of Bioactive Proteins from the Edible and Medicinal Mushrooms. Curr. Protein Pept. Sci. 2019, 20, 196–219. [Google Scholar] [CrossRef] [PubMed]
- Landi, N.; Clemente, A.; Pedone, P.V.; Ragucci, S.; Di Maro, A. An Updated Review of Bioactive Peptides from Mushrooms in a Well-Defined Molecular Weight Range. Toxins 2022, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Wieland, T. The Toxic Peptides of Amanita Phalloides. In Fortschritte der Chemie Organischer Naturstoffe; Zechmeister, L., Ed.; Springer: Vienna, Austria, 1967; pp. 214–250. [Google Scholar]
- Makrlík, E.; Böhm, S.; Vaňura, P.; Ruzza, P. Complexation of the calcium cation with antamanide: An experimental and theoretical study. Mol. Phys. 2015, 113, 1472–1477. [Google Scholar] [CrossRef]
- Makrlík, E.; Böhm, S.; Vaňura, P.; Ruzza, P. Complexation of Li+ with antamanide: An experimental and theoretical study. Monatsh. Chem. 2014, 145, 1051–1054. [Google Scholar] [CrossRef]
- Makrlík, E.; Böhm, S.; Vaňura, P.; Ruzza, P. Experimental and theoretical study on interaction of the barium cation with antamanide. J. Mol. Struct. 2014, 1065–1066, 61–64. [Google Scholar] [CrossRef]
- Makrlík, E.; Vaňura, P.; Böhm, S.; Ruzza, P. Extraction and theoretical study on complexation of the strontium cation with antamanide. J. Radioanal. Nucl. Chem. 2014, 300, 1291–1294. [Google Scholar] [CrossRef]
- Makrlík, E.; Böhm, S.; Vaňura, P.; Ruzza, P. Interaction of the univalent thallium cation with antamanide: Experimental and theoretical study. J. Mol. Struct. 2014, 1064, 107–110. [Google Scholar] [CrossRef]
- Ruzza, P.; Calderan, A.; Biondi, B.; Carrara, M.; Tancredi, T.; Borin, G. Ion-binding and pharmacological properties of Tyr6 and Tyr9 antamanide analogs. J. Pept. Res. 1999, 53, 442–452. [Google Scholar] [CrossRef]
- Siemion, I.Z.; Pedyczak, A.; Trojnar, J.; Zimecki, M.; Wieczorek, Z. Immunosuppressive activity of antamanide and some of its analogues. Peptides 1992, 13, 1233–1237. [Google Scholar] [CrossRef]
- Azzolin, L.; Antolini, N.; Calderan, A.; Ruzza, P.; Sciacovelli, M.; Marin, O.; Mammi, S.; Bernardi, P.; Rasola, A. Antamanide, a Derivative of Amanita phalloides, Is a Novel Inhibitor of the Mitochondrial Permeability Transition Pore. PLoS ONE 2011, 6, e16280. [Google Scholar] [CrossRef] [Green Version]
- Welbourn, R.; Goldman, G.; Kobzik, L.; Valeri, C.R.; Hechtman, H.B.; Shepro, D. Attenuation of IL-2-induced multisystem organ edema by phalloidin and antamanide. J. Appl. Physiol. 1991, 70, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O. Antamanide Antagonizes Phalloidin-induced Human Lymphocyte Aggregation and Prevents Leukaemic Mice from Death: A Pilot Study. Acta Pharmacol. Toxicol. 1986, 59, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Ruzza, P.; Calderan, A.; Carrara, M.; Tancredi, T.; Borin, G. Antamanide and its Gly or Tyr-analogues: Synthesis and pharmacological properties. Curr. Top. Pept. Protein Res. 1997, 2, 21–32. [Google Scholar]
- Revythis, A.; Shah, S.; Kutka, M.; Moschetta, M.; Ozturk, M.A.; Pappas-Gogos, G.; Ioannidou, E.; Sheriff, M.; Rassy, E.; Boussios, S. Unraveling the Wide Spectrum of Melanoma Biomarkers. Diagnostics 2021, 11, 1341. [Google Scholar] [CrossRef] [PubMed]
- Hamann, J.N.; Rolff, M.; Tuczek, F. Monooxygenation of an appended phenol in a model system of tyrosinase: Implications on the enzymatic reaction mechanism. Dalton Trans. 2015, 44, 3251–3258. [Google Scholar] [CrossRef]
- García-Molina, P.; García-Molina, F.; Teruel-Puche, J.A.; Rodríguez-López, J.N.; García-Cánovas, F.; Muñoz-Muñoz, J.L. Considerations about the kinetic mechanism of tyrosinase in its action on monophenols: A review. Mol. Catal. 2022, 518, 112072. [Google Scholar] [CrossRef]
- Bourquelot, E.; Bertrand, G. Le bleuissement et le noircissement des champignons. Comp. Rend. Soc. Biol. 1895, 47, 582–584. [Google Scholar]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A comprehensive review on tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.-J.; Shakerian, F.; Zhao, J.; Li, S.-P. Chemistry, pharmacology and analysis of Pseudostellaria heterophylla: A mini-review. Chin. Med. 2019, 14, 21. [Google Scholar] [CrossRef] [Green Version]
- Pangavhane, S.D.; Makrlík, E.; Ruzza, P.; Kašička, V. Affinity capillary electrophoresis employed for determination of stability constants of antamanide complexes with univalent and divalent cations in methanol. Electrophoresis 2019, 40, 2321–2328. [Google Scholar] [CrossRef]
- Morita, H.; Kobata, H.; Takeya, K.; Itokawa, H. Pseudostellarin G, a new tyrosinase inhibitory cyclic octapeptide from Pseudostellaria heterophylla. Tetrahedron Lett. 1994, 21, 3563–3564. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, J.R. Classification of Spectra of Cata-Condensed Hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Pignataro, M.F.; Herrera, M.G.; Dodero, V.I. Evaluation of Peptide/Protein Self-Assembly and Aggregation by Spectroscopic Methods. Molecules 2020, 25, 4854. [Google Scholar] [CrossRef] [PubMed]
- Sendovski, M.; Kanteev, M.; Ben-Yosef, V.S.; Adir, N.; Fishman, A. First structures of an active bacterial tyrosinase reveal copper plasticity. J. Mol. Biol. 2011, 405, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Matoba, Y.; Oda, K.; Muraki, Y.; Masuda, T. The basicity of an active-site water molecule discriminates between tyrosinase and catechol oxidase activity. Int. J. Biol. Macromol. 2021, 183, 1861–1870. [Google Scholar] [CrossRef]
- Deri, B.; Kanteev, M.; Goldfeder, M.; Lecina, D.; Guallar, V.; Adir, N.; Fishman, A. The unravelling of the complex pattern of tyrosinase inhibition. Sci. Rep. 2016, 6, 34993. [Google Scholar] [CrossRef] [Green Version]
- Goldfeder, M.; Kanteev, M.; Isaschar-Ovdat, S.; Adir, N.; Fishman, A. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 2014, 5, 4505. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Shioiri, T.; Ninomiya, K.; Yamada, S. Diphenylphosphoryl azide. New convenient reagent for a modified Curtius reaction and for peptide synthesis. J. Am. Chem. Soc. 1972, 94, 6203–6205. [Google Scholar] [CrossRef] [PubMed]
- Dettori, M.A.; Fabbri, D.; Dessì, A.; Dallocchio, R.; Carta, P.; Honisch, C.; Ruzza, P.; Farina, D.; Migheli, R.; Serra, P.A.; et al. Synthesis and Studies of the Inhibitory Effect of Hydroxylated Phenylpropanoids and Biphenols Derivatives on Tyrosinase and Laccase Enzymes. Molecules 2020, 25, 2709. [Google Scholar] [CrossRef] [PubMed]
- Honisch, C.; Osto, A.; Dupas de Matos, A.; Vincenzi, S.; Ruzza, P. Isolation of a tyrosinase inhibitor from unripe grapes juice: A spectrophotometric study. Food Chem. 2020, 305, 125506. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Scheraga, H.A. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 6611–6615. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09 (Revision A02); Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Nielsen, A.B.; Holder, A.J. Gauss View 5.0, User’s Reference; Gaussian Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Antamanide | V | P | P | A | F | F | P | P | F | F |
| AG6 | V | P | P | A | F | G | P | P | F | F |
| AG9 | V | P | P | A | F | F | P | P | G | F |
| AOG9 | V | P | P | A | - | - | P | P | G | F |
| PS-A | G | P | Y | L | A | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honisch, C.; Gazziero, M.; Dallocchio, R.; Dessì, A.; Fabbri, D.; Dettori, M.A.; Delogu, G.; Ruzza, P. Antamanide Analogs as Potential Inhibitors of Tyrosinase. Int. J. Mol. Sci. 2022, 23, 6240. https://doi.org/10.3390/ijms23116240
Honisch C, Gazziero M, Dallocchio R, Dessì A, Fabbri D, Dettori MA, Delogu G, Ruzza P. Antamanide Analogs as Potential Inhibitors of Tyrosinase. International Journal of Molecular Sciences. 2022; 23(11):6240. https://doi.org/10.3390/ijms23116240
Chicago/Turabian StyleHonisch, Claudia, Matteo Gazziero, Roberto Dallocchio, Alessandro Dessì, Davide Fabbri, Maria Antonietta Dettori, Giovanna Delogu, and Paolo Ruzza. 2022. "Antamanide Analogs as Potential Inhibitors of Tyrosinase" International Journal of Molecular Sciences 23, no. 11: 6240. https://doi.org/10.3390/ijms23116240
APA StyleHonisch, C., Gazziero, M., Dallocchio, R., Dessì, A., Fabbri, D., Dettori, M. A., Delogu, G., & Ruzza, P. (2022). Antamanide Analogs as Potential Inhibitors of Tyrosinase. International Journal of Molecular Sciences, 23(11), 6240. https://doi.org/10.3390/ijms23116240