
Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects



Abstract
1. Introduction
2. Vascular Cognitive Impairment and Dementia
2.1. Etiological Study of VCID
2.2. Pathogenesis of VCID
2.2.1. Neuroinflammation
2.2.2. Oxidative Stress
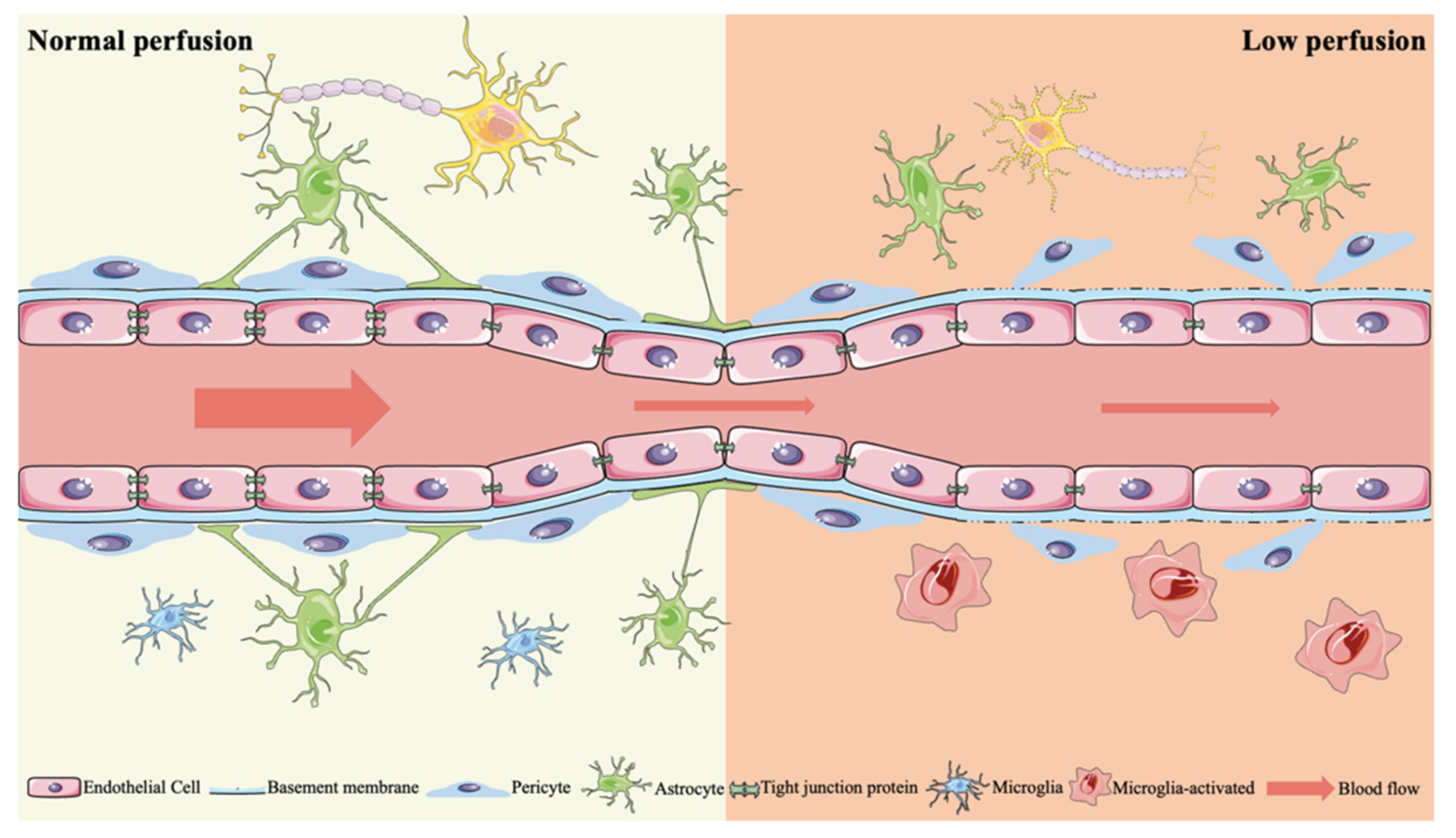
2.2.3. BBB Dysfunction
2.2.4. Neurovascular Nutrient Uncoupling
2.2.5. Others
3. Relationship between the Etiology of VCID and Neuroinflammation
3.1. Neuroinflammation
3.2. Neuroinflammation Induced by Ischemia
3.3. Neuroinflammation Induced by Hypoxia
4. Research Progress of Neuroinflammation in VCID
4.1. Immune Cells in VCID
4.2. Inflammatory Mediators in VCID
4.3. Inflammatory Pathways in VCID
5. Relationship between AD and VCID
6. Relationship between Other Tauopathic Syndromes and VCID
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Primers 2018, 4, 18003. [Google Scholar] [CrossRef] [PubMed]
- Krzosek, P.; Madetko, N.; Migda, A.; Migda, B.; Jaguś, D.; Alster, P. Differential Diagnosis of Rare Subtypes of Progressive Supranuclear Palsy and PSP-Like Syndromes-Infrequent Manifestations of the Most Common Form of Atypical Parkinsonism. Front. Aging Neurosci. 2022, 14, 804385. [Google Scholar] [CrossRef] [PubMed]
- Dunalska, A.; Pikul, J.; Schok, K.; Wiejak, K.A.; Alster, P. The Significance of Vascular Pathogenesis in the Examination of Corticobasal Syndrome. Front. Aging Neurosci. 2021, 13, 668614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; Decarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- Stebbins, G.T.; Nyenhuis, D.L.; Wang, C.; Cox, J.L.; Freels, S.; Bangen, K.; de Toledo-Morrell, L.; Sripathirathan, K.; Moseley, M.; Turner, D.A.; et al. Gray matter atrophy in patients with ischemic stroke with cognitive impairment. Stroke 2008, 39, 785–793. [Google Scholar] [CrossRef]
- Cao, Q.; Tan, C.C.; Xu, W.; Hu, H.; Cao, X.P.; Dong, Q.; Tan, L.; Yu, J.T. The Prevalence of Dementia: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2020, 73, 1157–1166. [Google Scholar] [CrossRef]
- Romay, M.C.; Toro, C.; Iruela-Arispe, M.L. Emerging molecular mechanisms of vascular dementia. Curr. Opin. Hematol. 2019, 26, 199–206. [Google Scholar] [CrossRef]
- Sha, S.; Tan, J.; Miao, Y.; Zhang, Q. The Role of Autophagy in Hypoxia-Induced Neuroinflammation. DNA Cell Biol 2021, 40, 733–739. [Google Scholar] [CrossRef]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Mamun, A.A.; Barreto, G.E.; Rashid, M.; Perveen, A.; Ashraf, G.M. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int. Immunopharmacol. 2020, 84, 106479. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S369–S379. [Google Scholar] [CrossRef] [PubMed]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef] [PubMed]
- Stonesifer, C.; Corey, S.; Ghanekar, S.; Diamandis, Z.; Acosta, S.A.; Borlongan, C.V. Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog. Neurobiol. 2017, 158, 94–131. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Iadecola, C.; Duering, M.; Hachinski, V.; Joutel, A.; Pendlebury, S.T.; Schneider, J.A.; Dichgans, M. Vascular Cognitive Impairment and Dementia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2019, 73, 3326–3344. [Google Scholar] [CrossRef]
- Skrobot, O.A.; Black, S.E.; Chen, C.; DeCarli, C.; Erkinjuntti, T.; Ford, G.A.; Kalaria, R.N.; O’Brien, J.; Pantoni, L.; Pasquier, F.; et al. Progress toward standardized diagnosis of vascular cognitive impairment: Guidelines from the Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement. 2018, 14, 280–292. [Google Scholar] [CrossRef]
- Mast, H.; Tatemichi, T.K.; Mohr, J.P. Chronic brain ischemia: The contributions of Otto Binswanger and Alois Alzheimer to the mechanisms of vascular dementia. J. Neurol. Sci. 1995, 132, 4–10. [Google Scholar] [CrossRef]
- Zhao, H.; Alam, A.; Soo, A.P.; George, A.J.T.; Ma, D. Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine 2018, 28, 31–42. [Google Scholar] [CrossRef]
- Chen, Y.M.; He, X.Z.; Wang, S.M.; Xia, Y. δ-Opioid Receptors, microRNAs, and Neuroinflammation in Cerebral Ischemia/Hypoxia. Front. Immunol. 2020, 11, 421. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.C.; Lee, M.H.H.; Wu, C.Y.C.; Couto, E.S.A.; Possoit, H.E.; Hsieh, T.H.; Minagar, A.; Lin, H.W. Cerebral ischemia and neuroregeneration. Neural. Regen. Res. 2018, 13, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Acker, T.; Acker, H. Cellular oxygen sensing need in CNS function: Physiological and pathological implications. J. Exp. Biol. 2004, 207, 3171–3188. [Google Scholar] [CrossRef]
- Thompson, J.K.; Peterson, M.R.; Freeman, R.D. Single-neuron activity and tissue oxygenation in the cerebral cortex. Science 2003, 299, 1070–1072. [Google Scholar] [CrossRef]
- Corcoran, A.; O’Connor, J.J. Hypoxia-inducible factor signalling mechanisms in the central nervous system. Acta Physiol. 2013, 208, 298–310. [Google Scholar] [CrossRef]
- Fajersztajn, L.; Veras, M.M. Hypoxia: From Placental Development to Fetal Programming. Birth Defects Res. 2017, 109, 1377–1385. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Meijer, J.; Kiliaan, A.; Ruitenberg, A.; van Swieten, J.C.; Stijnen, T.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Inflammatory proteins in plasma and the risk of dementia: The rotterdam study. Arch. Neurol. 2004, 61, 668–672. [Google Scholar] [CrossRef]
- Gervois, P.; Lambrichts, I. The Emerging Role of Triggering Receptor Expressed on Myeloid Cells 2 as a Target for Immunomodulation in Ischemic Stroke. Front. Immunol 2019, 10, 1668. [Google Scholar] [CrossRef]
- Farkas, E.; Luiten, P.G.; Bari, F. Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 2007, 54, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, B.; Lu, K.; Deng, J.; Zhao, F.; Zhao, B.Q.; Zhao, Y. Prevention of Hippocampal Neuronal Damage and Cognitive Function Deficits in Vascular Dementia by Dextromethorphan. Mol. Neurobiol. 2016, 53, 3494–3502. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Bang, J.H.; Lee, J.; Han, J.S.; Kang, H.W.; Jeon, W.K. Fructus mume Ethanol Extract Prevents Inflammation and Normalizes the Septohippocampal Cholinergic System in a Rat Model of Chronic Cerebral Hypoperfusion. J. Med. Food 2016, 19, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Miyamoto, N.; Hayakawa, K.; Pham, L.D.; Maki, T.; Ayata, C.; Kim, K.W.; Lo, E.H.; Arai, K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Investig. 2013, 123, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Du, S.Q.; Wang, X.R.; Xiao, L.Y.; Tu, J.F.; Zhu, W.; He, T.; Liu, C.Z. Molecular Mechanisms of Vascular Dementia: What Can Be Learned from Animal Models of Chronic Cerebral Hypoperfusion? Mol. Neurobiol. 2017, 54, 3670–3682. [Google Scholar] [CrossRef]
- Back, S.A.; Kroenke, C.D.; Sherman, L.S.; Lawrence, G.; Gong, X.; Taber, E.N.; Sonnen, J.A.; Larson, E.B.; Montine, T.J. White matter lesions defined by diffusion tensor imaging in older adults. Ann. Neurol. 2011, 70, 465–476. [Google Scholar] [CrossRef]
- Lee, J.C.; Won, M.H. Neuroprotection of antioxidant enzymes against transient global cerebral ischemia in gerbils. Anat. Cell Biol. 2014, 47, 149–156. [Google Scholar] [CrossRef]
- Lee, W.L.A.; Michael-Titus, A.T.; Shah, D.K. Hypoxic-Ischaemic Encephalopathy and the Blood-Brain Barrier in Neonates. Dev. Neurosci. 2017, 39, 49–58. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, K.J.; Qi, Z. Occludin regulation of blood-brain barrier and potential therapeutic target in ischemic stroke. Brain Circ. 2020, 6, 152–162. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Thompson, J.; Taheri, S.; Grossetete, M.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Wills, J.; Rosenberg, G.A. Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke 2011, 42, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Fernando, M.S.; Clark, L.; Ince, P.G.; Matthews, F.; Forster, G.; O’Brien, J.T.; Barber, R.; Kalaria, R.N.; Brayne, C.; et al. White matter lesions in an unselected cohort of the elderly: Astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol. Appl. Neurobiol. 2007, 33, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Tomimoto, H.; Akiguchi, I.; Wakita, H.; Sakamoto, H. Blood-brain barrier disruption in white matter lesions in a rat model of chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 2002, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef]
- Guo, Z.; Wu, X.; Fan, W. Clarifying the effects of diabetes on the cerebral circulation: Implications for stroke recovery and beyond. Brain Res. Bull. 2021, 171, 67–74. [Google Scholar] [CrossRef]
- Benkhalifa, M.; Ferreira, Y.J.; Chahine, H.; Louanjli, N.; Miron, P.; Merviel, P.; Copin, H. Mitochondria: Participation to infertility as source of energy and cause of senescence. Int. J. Biochem. Cell Biol. 2014, 55, 60–64. [Google Scholar] [CrossRef]
- Duan, W.; Gui, L.; Zhou, Z.; Liu, Y.; Tian, H.; Chen, J.F.; Zheng, J. Adenosine A2A receptor deficiency exacerbates white matter lesions and cognitive deficits induced by chronic cerebral hypoperfusion in mice. J. Neurol. Sci. 2009, 285, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Ransom, B.R. Roles of white matter in central nervous system pathophysiologies. ASN Neuro 2012, 4, e00079. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Tanaka, K.I.; Kato-Negishi, M. Copper as a Collaborative Partner of Zinc-Induced Neurotoxicity in the Pathogenesis of Vascular Dementia. Int. J. Mol. Sci. 2021, 22, 7242. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Alterations in immune cells and mediators in the brain: It’s not always neuroinflammation! Brain Pathol. 2014, 24, 623–630. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Bonte, M.A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.C. Glycosphingolipids and neuroinflammation in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 59. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Int. J. Mol. Sci. 2021, 23, 14. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, K.; Li, H.; Dong, X.; Tan, G.; Chai, Y.; Wang, W.; Bi, X. Potential of serum metabolites for diagnosing post-stroke cognitive impairment. Mol. Biosyst. 2015, 11, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Alkhalil, M. A promising tool to tackle the risk of cerebral vascular disease, the emergence of novel carotid wall imaging. Brain Circ. 2020, 6, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Fidahic, M.; Jelicic Kadic, A.; Radic, M.; Puljak, L. Celecoxib for rheumatoid arthritis. Cochrane Database Syst. Rev. 2017, 6, Cd012095. [Google Scholar] [CrossRef] [PubMed]
- Tahsili-Fahadan, P.; Farrokh, S.; Geocadin, R.G. Hypothermia and brain inflammation after cardiac arrest. Brain Circ. 2018, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418789854. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. 2016, 5, 213. [Google Scholar] [CrossRef]
- Guo, K.; Mou, X.; Huang, J.; Xiong, N.; Li, H. Trans-caryophyllene suppresses hypoxia-induced neuroinflammatory responses by inhibiting NF-κB activation in microglia. J. Mol. Neurosci. 2014, 54, 41–48. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Simon, M.C. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 2010, 345, 105–120. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Jalal, F.Y.; Yang, Y.; Thompson, J.F.; Roitbak, T.; Rosenberg, G.A. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. J. Cereb. Blood Flow Metab. 2015, 35, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.L.; Desai, R.A.; Bloomfield, P.S.; McIntosh, P.R.; Chapple, K.J.; Linington, C.; Fairless, R.; Diem, R.; Kasti, M.; Murphy, M.P.; et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann. Neurol. 2013, 74, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M. The Interplay of MicroRNAs in the Inflammatory Mechanisms Following Ischemic Stroke. J. Neuropathol. Exp. Neurol. 2017, 76, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Ramasamy, K.; Bouamar, H.; Lin, A.P.; Jiang, D.; Aguiar, R.C. MicroRNAs miR-125a and miR-125b constitutively activate the NF-κB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc. Natl. Acad. Sci. USA 2012, 109, 7865–7870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.; Wang, Q.Q.; Yang, Q.Q.; Gu, H.Y.; Yin, Y.Q.; Li, Y.D.; Hou, J.C.; Chen, R.; Sun, Q.Q.; Sun, Y.F.; et al. NG2 glia regulate brain innate immunity via TGF-β2/TGFBR2 axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hamanaka, G.; Lo, E.H.; Arai, K. Heterogeneity of microglia and their differential roles in white matter pathology. CNS Neurosci. Ther. 2019, 25, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B.; et al. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 2017, 157, 247–272. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Pan, J.; Mamtilahun, M.; Zhu, Y.; Wang, L.; Venkatesh, A.; Shi, R.; Tu, X.; Jin, K.; Wang, Y.; et al. Microglia exacerbate white matter injury via complement C3/C3aR pathway after hypoperfusion. Theranostics 2020, 10, 74–90. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Deneen, B. Astrocyte development: A Guide for the Perplexed. Glia 2015, 63, 1320–1329. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Tsai, H.H.; Li, H.; Fuentealba, L.C.; Molofsky, A.V.; Taveira-Marques, R.; Zhuang, H.; Tenney, A.; Murnen, A.T.; Fancy, S.P.; Merkle, F.; et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 2012, 337, 358–362. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Kelley, K.W.; Tsai, H.H.; Redmond, S.A.; Chang, S.M.; Madireddy, L.; Chan, J.R.; Baranzini, S.E.; Ullian, E.M.; Rowitch, D.H. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 2014, 509, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Walz, W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 2000, 36, 291–300. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Powell, D.K.; Smith, C.D.; Greenstein, A.; Wilcock, D.M. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Price, B.R.; Norris, C.M.; Sompol, P.; Wilcock, D.M. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 2018, 144, 644–650. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Warf, B.C.; Fok-Seang, J.; Miller, R.H. Evidence for the ventral origin of oligodendrocyte precursors in the rat spinal cord. J. Neurosci. 1991, 11, 2477–2488. [Google Scholar] [CrossRef]
- Dean, J.M.; van de Looij, Y.; Sizonenko, S.V.; Lodygensky, G.A.; Lazeyras, F.; Bolouri, H.; Kjellmer, I.; Huppi, P.S.; Hagberg, H.; Mallard, C. Delayed cortical impairment following lipopolysaccharide exposure in preterm fetal sheep. Ann. Neurol. 2011, 70, 846–856. [Google Scholar] [CrossRef]
- Nave, K.A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Utz, S.G.; See, P.; Mildenberger, W.; Thion, M.S.; Silvin, A.; Lutz, M.; Ingelfinger, F.; Rayan, N.A.; Lelios, I.; Buttgereit, A.; et al. Early Fate Defines Microglia and Non-parenchymal Brain Macrophage Development. Cell 2020, 181, 557–573.e518. [Google Scholar] [CrossRef]
- Fabriek, B.O.; Van Haastert, E.S.; Galea, I.; Polfliet, M.M.; Döpp, E.D.; Van Den Heuvel, M.M.; Van Den Berg, T.K.; De Groot, C.J.; Van Der Valk, P.; Dijkstra, C.D. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia 2005, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Mato, M.; Ookawara, S.; Sakamoto, A.; Aikawa, E.; Ogawa, T.; Mitsuhashi, U.; Masuzawa, T.; Suzuki, H.; Honda, M.; Yazaki, Y.; et al. Involvement of specific macrophage-lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc. Natl. Acad. Sci. USA 1996, 93, 3269–3274. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef]
- Thanopoulou, K.; Fragkouli, A.; Stylianopoulou, F.; Georgopoulos, S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 20816–20821. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Uekawa, K.; Garcia-Bonilla, L.; Koizumi, K.; Murphy, M.; Pistik, R.; Younkin, L.; Younkin, S.; Zhou, P.; Carlson, G.; et al. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ. Res. 2017, 121, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Pedragosa, J.; Salas-Perdomo, A.; Gallizioli, M.; Cugota, R.; Miró-Mur, F.; Briansó, F.; Justicia, C.; Pérez-Asensio, F.; Marquez-Kisinousky, L.; Urra, X.; et al. CNS-border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol. Commun. 2018, 6, 76. [Google Scholar] [CrossRef]
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Miró-Mur, F.; Pedragosa, J.; Zawadzka, M.; Pilanc, P.; Planas, A.M.; Kaminska, B. Defining molecular identity and fates of CNS-border associated macrophages after ischemic stroke in rodents and humans. Neurobiol. Dis. 2020, 137, 104722. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, J.; Guo, L.; Xu, Y.; Sun, L.; Wang, S.; Feng, Y.; Gou, L.; Zhang, L.; Liu, Y. Protective role of luteolin against cognitive dysfunction induced by chronic cerebral hypoperfusion in rats. Pharmacol. Biochem. Behav. 2014, 126, 122–130. [Google Scholar] [CrossRef]
- Yoshizaki, K.; Adachi, K.; Kataoka, S.; Watanabe, A.; Tabira, T.; Takahashi, K.; Wakita, H. Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp. Neurol. 2008, 210, 585–591. [Google Scholar] [CrossRef]
- Hou, X.; Liang, X.; Chen, J.F.; Zheng, J. Ecto-5′-nucleotidase (CD73) is involved in chronic cerebral hypoperfusion-induced white matter lesions and cognitive impairment by regulating glial cell activation and pro-inflammatory cytokines. Neuroscience 2015, 297, 118–126. [Google Scholar] [CrossRef]
- Kitamura, A.; Fujita, Y.; Oishi, N.; Kalaria, R.N.; Washida, K.; Maki, T.; Okamoto, Y.; Hase, Y.; Yamada, M.; Takahashi, J.; et al. Selective white matter abnormalities in a novel rat model of vascular dementia. Neurobiol Aging 2012, 33, 1012.e25–1012.e35. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Bang, J.; Kim, B.Y.; Lee, I.S.; Han, J.S.; Hwang, B.Y.; Jeon, W.K. Fructus mume alleviates chronic cerebral hypoperfusion-induced white matter and hippocampal damage via inhibition of inflammation and downregulation of TLR4 and p38 MAPK signaling. BMC Complement Altern. Med. 2015, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.B.; Hoda, M.N.; Vaibhav, K.; Giri, S.; Wang, P.; Waller, J.L.; Ergul, A.; Dhandapani, K.M.; Fagan, S.C.; Hess, D.C. Remote ischemic postconditioning: Harnessing endogenous protection in a murine model of vascular cognitive impairment. Transl. Stroke Res. 2015, 6, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Won, J.S.; Kim, J.; Annamalai, B.; Shunmugavel, A.; Singh, I.; Singh, A.K. Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J. Alzheimers Dis. 2013, 34, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, M.; Zetterberg, H.; Edman, Å.; Blennow, K.; Wallin, A.; Andreasson, U. Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Narantuya, D.; Nagai, A.; Sheikh, A.M.; Wakabayashi, K.; Shiota, Y.; Watanabe, T.; Masuda, J.; Kobayashi, S.; Kim, S.U.; Yamaguchi, S. Microglia transplantation attenuates white matter injury in rat chronic ischemia model via matrix metalloproteinase-2 inhibition. Brain Res. 2010, 1316, 145–152. [Google Scholar] [CrossRef]
- Reimer, M.M.; McQueen, J.; Searcy, L.; Scullion, G.; Zonta, B.; Desmazieres, A.; Holland, P.R.; Smith, J.; Gliddon, C.; Wood, E.R.; et al. Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J. Neurosci. 2011, 31, 18185–18194. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Rahimifard, M.; Maqbool, F.; Moeini-Nodeh, S.; Niaz, K.; Abdollahi, M.; Braidy, N.; Nabavi, S.M.; Nabavi, S.F. Targeting the TLR4 signaling pathway by polyphenols: A novel therapeutic strategy for neuroinflammation. Ageing Res. Rev. 2017, 36, 11–19. [Google Scholar] [CrossRef]
- Wang, L.; Yang, J.W.; Lin, L.T.; Huang, J.; Wang, X.R.; Su, X.T.; Cao, Y.; Fisher, M.; Liu, C.Z. Acupuncture Attenuates Inflammation in Microglia of Vascular Dementia Rats by Inhibiting miR-93-Mediated TLR4/MyD88/NF-κB Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 8253904. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.Y.; Yan, Y.; Chen, R. Minocycline reduces astrocytic reactivation and neuroinflammation in the hippocampus of a vascular cognitive impairment rat model. Neurosci. Bull. 2010, 26, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Seo, Y.H.; Jang, J.H.; Jeong, C.H.; Lee, S.; Park, B. Asiatic acid attenuates methamphetamine-induced neuroinflammation and neurotoxicity through blocking of NF-kB/STAT3/ERK and mitochondria-mediated apoptosis pathway. J. Neuroinflamm. 2017, 14, 240. [Google Scholar] [CrossRef] [PubMed]
- Saggu, R.; Schumacher, T.; Gerich, F.; Rakers, C.; Tai, K.; Delekate, A.; Petzold, G.C. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol. Commun. 2016, 4, 76. [Google Scholar] [CrossRef]
- Kapasi, A.; DeCarli, C.; Schneider, J.A. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017, 134, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: A cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [Google Scholar] [CrossRef]
- Roher, A.E.; Esh, C.; Kokjohn, T.A.; Kalback, W.; Luehrs, D.C.; Seward, J.D.; Sue, L.I.; Beach, T.G. Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2055–2062. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Klomparens, K.; Ding, Y. Updates on the association of brain injury and Alzheimer’s disease. Brain Circ. 2020, 6, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s Disease and Vascular Aging: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 942–951. [Google Scholar] [CrossRef]
- Roher, A.E.; Esh, C.; Rahman, A.; Kokjohn, T.A.; Beach, T.G. Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke 2004, 35, 2623–2627. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G. Resolving atherosclerosis and Alzheimer disease. Nat. Rev. Cardiol. 2019, 16, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Iadecola, C. Impaired Aβ clearance: A potential link between atherosclerosis and Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 115. [Google Scholar] [CrossRef]
- Pellegrini, C.; D’Antongiovanni, V.; Fornai, M.; Duranti, E.; Baldacci, F.; Bernardini, N.; Taddei, S.; Virdis, A.; Blandizzi, C.; Masi, S.; et al. Donepezil improves vascular function in a mouse model of Alzheimer’s disease. Pharmacol. Res. Perspect. 2021, 9, e00871. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef]
- Stamelou, M.; Respondek, G.; Giagkou, N.; Whitwell, J.L.; Kovacs, G.G.; Höglinger, G.U. Evolving concepts in progressive supranuclear palsy and other 4-repeat tauopathies. Nat. Rev. Neurol. 2021, 17, 601–620. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Mulroy, E.; Jaunmuktane, Z.; Balint, B.; Erro, R.; Latorre, A.; Bhatia, K.P. Some New and Unexpected Tauopathies in Movement Disorders. Mov. Disord. Clin. Pract. 2020, 7, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Ruiz, C.; Wang, J.; Ksiezak-Reding, H.; Ho, L.; Qian, X.; Humala, N.; Thomas, S.; Martínez-Martín, P.; Pasinetti, G.M. Role of Hypertension in Aggravating Abeta Neuropathology of AD Type and Tau-Mediated Motor Impairment. Cardiovasc. Psychiatry Neurol. 2009, 2009, 107286. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, S.; Klafki, H.W.; Plesnila, N.; Hübinger, G.; Obermeier, A.; Sahagún, H.; Monse, B.; Seneci, P.; Lewis, J.; Eriksen, J.; et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9673–9678. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef]
- Kwasny, M.J.; Oleske, D.M.; Zamudio, J.; Diegidio, R.; Höglinger, G.U. Clinical Features Observed in General Practice Associated With the Subsequent Diagnosis of Progressive Supranuclear Palsy. Front. Neurol. 2021, 12, 637176. [Google Scholar] [CrossRef]
- Alster, P.; Dunalska, A.; Migda, B.; Madetko, N.; Królicki, L. The Rate of Decrease in Brain Perfusion in Progressive Supranuclear Palsy and Corticobasal Syndrome May Be Impacted by Glycemic Variability-A Pilot Study. Front. Neurol. 2021, 12, 767480. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim Biophys Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer’s disease. PLoS ONE 2012, 7, e32792. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gong, C.X. From chronic cerebral hypoperfusion to Alzheimer-like brain pathology and neurodegeneration. Cell Mol. Neurobiol. 2015, 35, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.A.; Green, K.N.; Blurton-Jones, M.; Laferla, F.M. Oligemic hypoperfusion differentially affects tau and amyloid-β. Am. J. Pathol. 2010, 177, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Alster, P.; Madetko, N.; Koziorowski, D.; Friedman, A. Microglial Activation and Inflammation as a Factor in the Pathogenesis of Progressive Supranuclear Palsy (PSP). Front. Neurosci. 2020, 14, 893. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Subject | Immune Cells | Immune Mediators | Immune Signaling Pathways | References | |
|---|---|---|---|---|---|
| Patient | Vascular cognitive impairment dementia | / | ACT, IL-6 | / | [29] |
| Animal models | Bilateral Common Carotid Artery Stenosis (BCAS) | Activation of microglia and astrocytes | IL-1β, IL-6, TNF-α | / | [110] |
| Activation of microglia and astrocytes | ICAM-1, VCAM-1 | / | [113] | ||
| Oligodendrocyte precursor cell activation | MMP-9 | / | [34] | ||
| Axonal-glial integrity destruction | / | JAK/STAT | [117] | ||
| Activation of microglia and astrocytes; Demyelination | IL-1β, IL-6 | NF-κB/STAT3 | [124] | ||
| 2-Vessel Gradual Occlusion (2-VGO) | Activation of microglia and astrocytes | TNF-α, MCP-1 | / | [111] | |
| Bilateral Common Carotid Artery Occlusion (BCAO) | Activation of microglia; Demyelination | IL-1β, IL-6, TNF-α | C3-C3aR/ITGAM | [86] | |
| Activation of microglia and astrocytes | IL-1β, IL-6, COX-2 | TLR4/MyD88/p38/MAPK | [112] | ||
| / | ICAM-1, VCAM-1 | / | [114] | ||
| Activation of microglia and astrocytes | IL-6, TNF-α, MMP-2 | / | [116] | ||
| Activation of microglia | IL-6, TNF-α | TLR4/MyD88/NF-κB | [121] | ||
| Asymmetric Common Carotid Artery surgery (ACAS) | / | IL-6 IL-4, IL-10 | / | [109] | |
| Hyperhomocysteinemia (Hhcy) | Activation of microglia | MMP-2, MMP-9 | [94] | ||
| Activation of microglia; Activation of astrocytes, pseudopodia rupture and AQP4 channel decrease | IL-1β, IL-6, IL-12, TNF-α, MMP-2, MMP-9 | / | [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Z.; Ji, X.; Liu, J. Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects. Int. J. Mol. Sci. 2022, 23, 6224. https://doi.org/10.3390/ijms23116224
Tian Z, Ji X, Liu J. Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects. International Journal of Molecular Sciences. 2022; 23(11):6224. https://doi.org/10.3390/ijms23116224
Chicago/Turabian StyleTian, Zhengming, Xunming Ji, and Jia Liu. 2022. "Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects" International Journal of Molecular Sciences 23, no. 11: 6224. https://doi.org/10.3390/ijms23116224
APA StyleTian, Z., Ji, X., & Liu, J. (2022). Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects. International Journal of Molecular Sciences, 23(11), 6224. https://doi.org/10.3390/ijms23116224