
Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein–Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases



Abstract
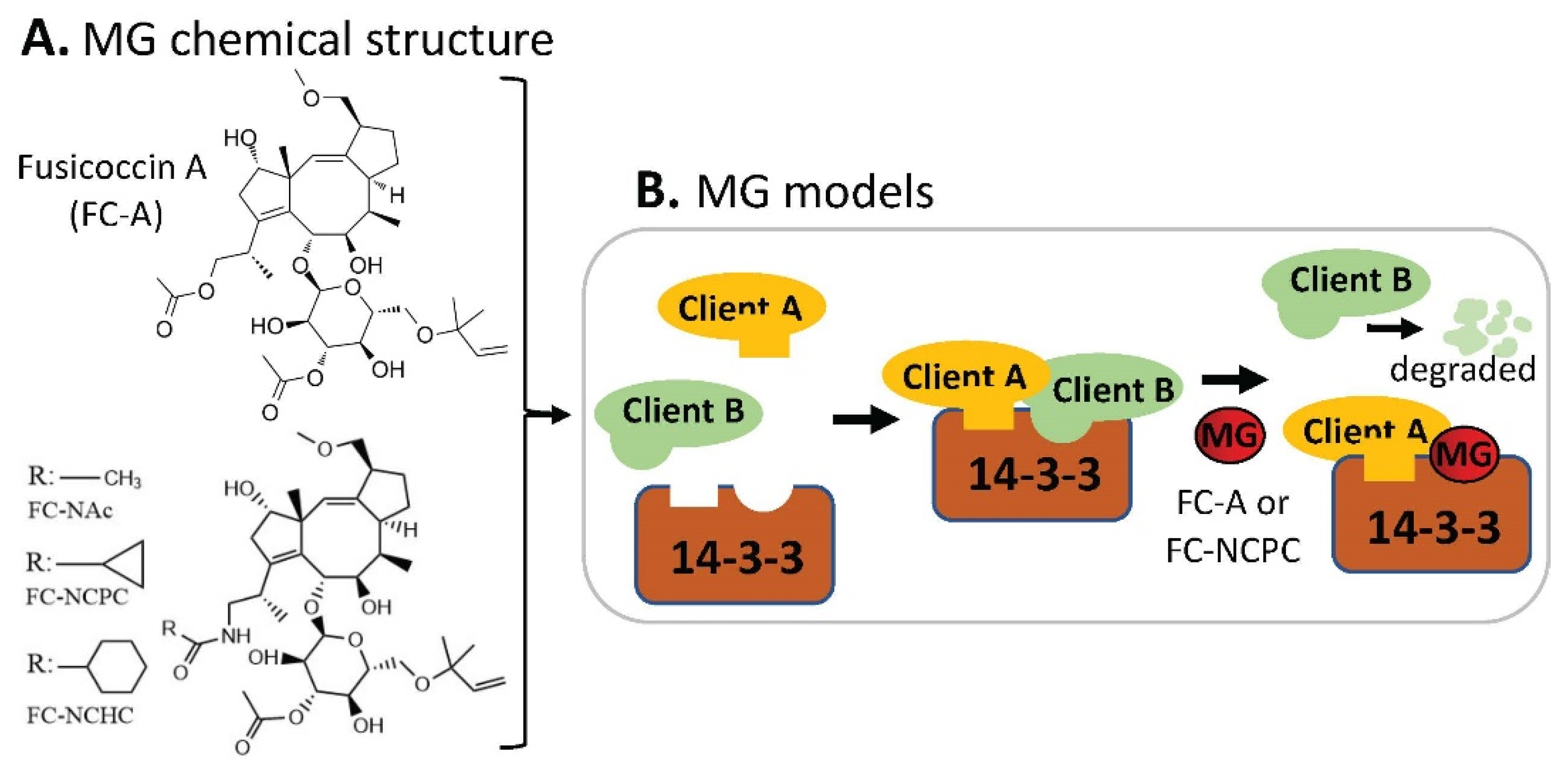
:1. Introduction
2. Molecular Glues for Cancer Treatment
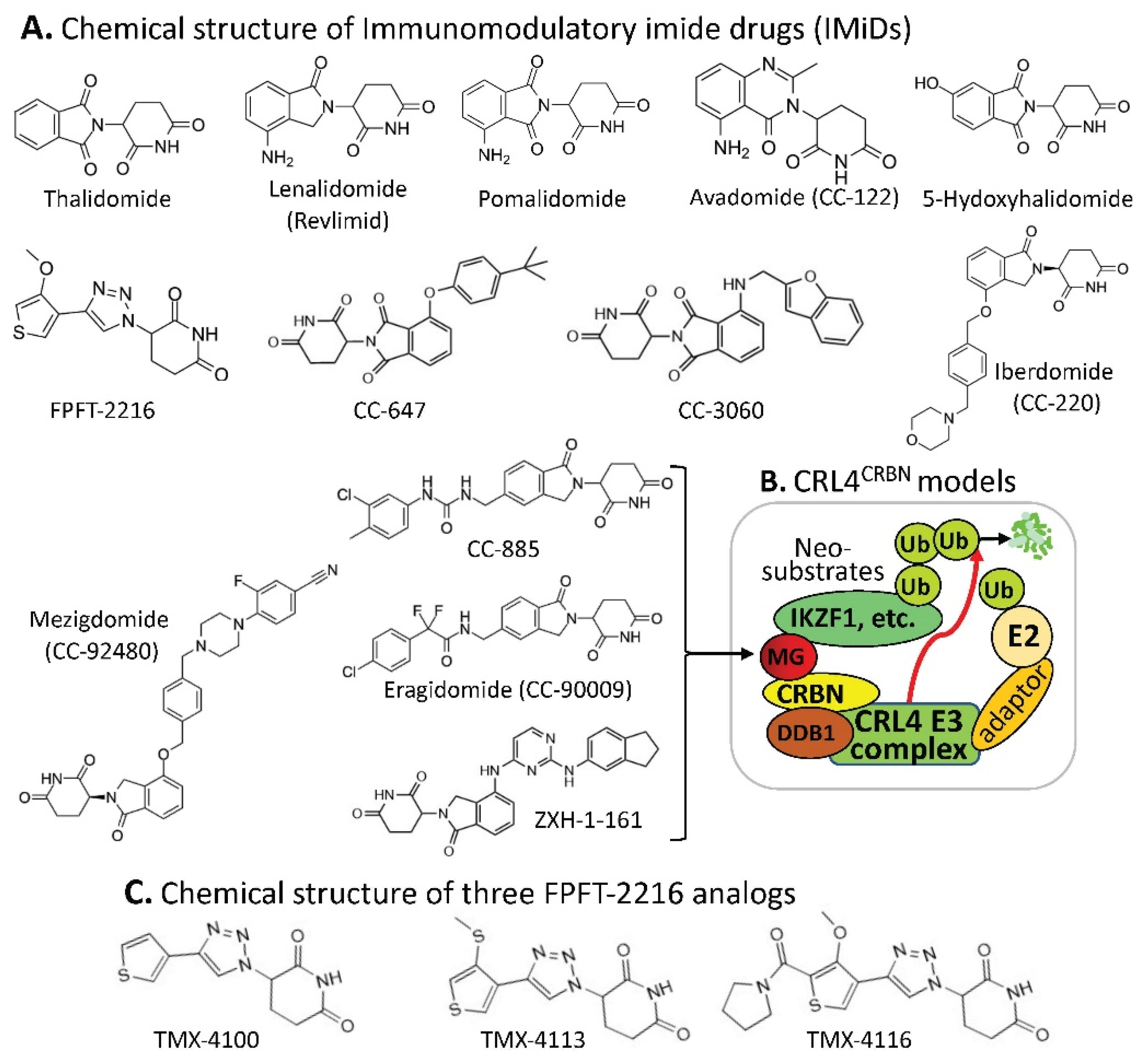
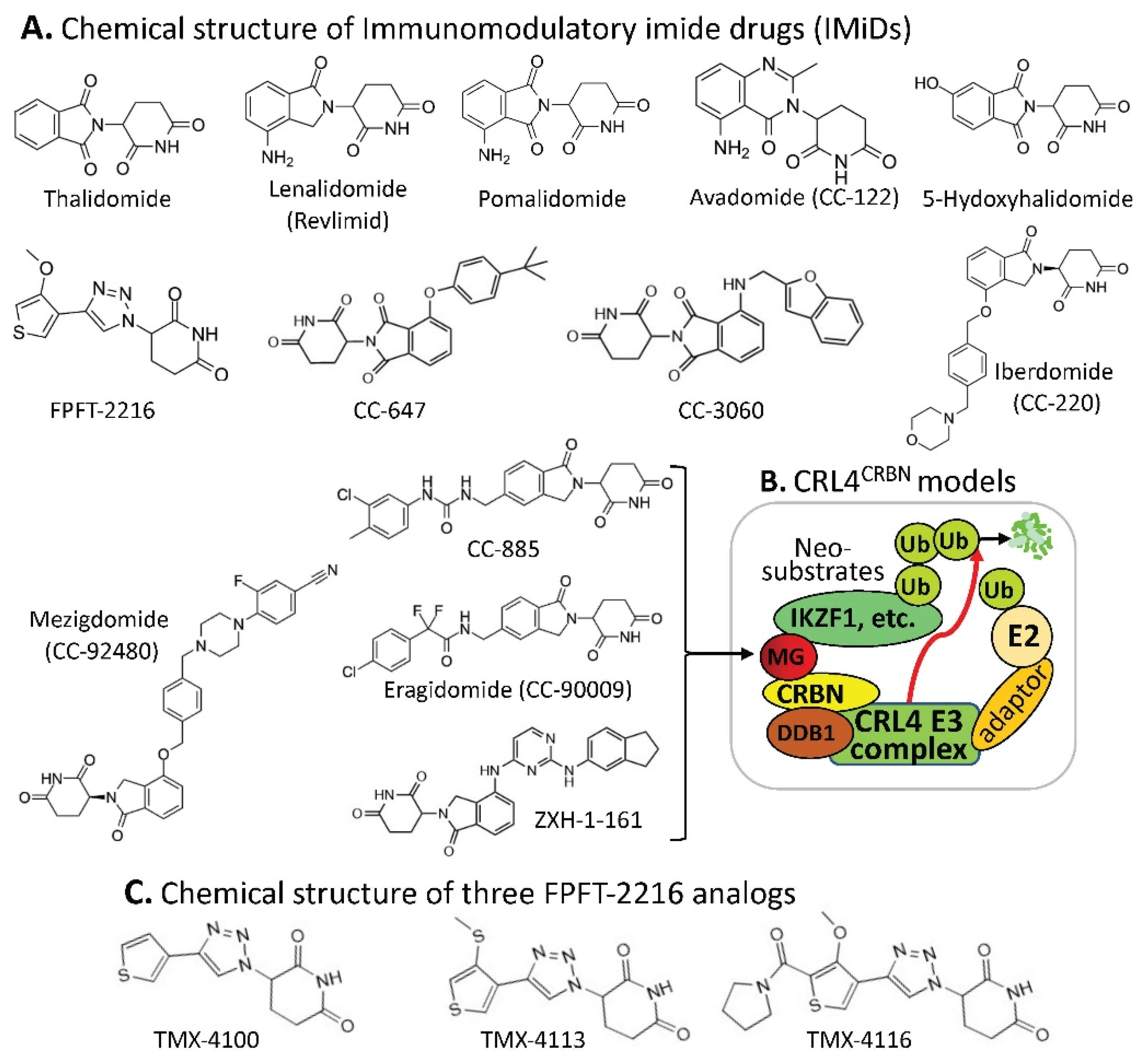
2.1. CRBN-Involved CRL4 Complex-Mediated Degradation of MG-Targeted Proteins
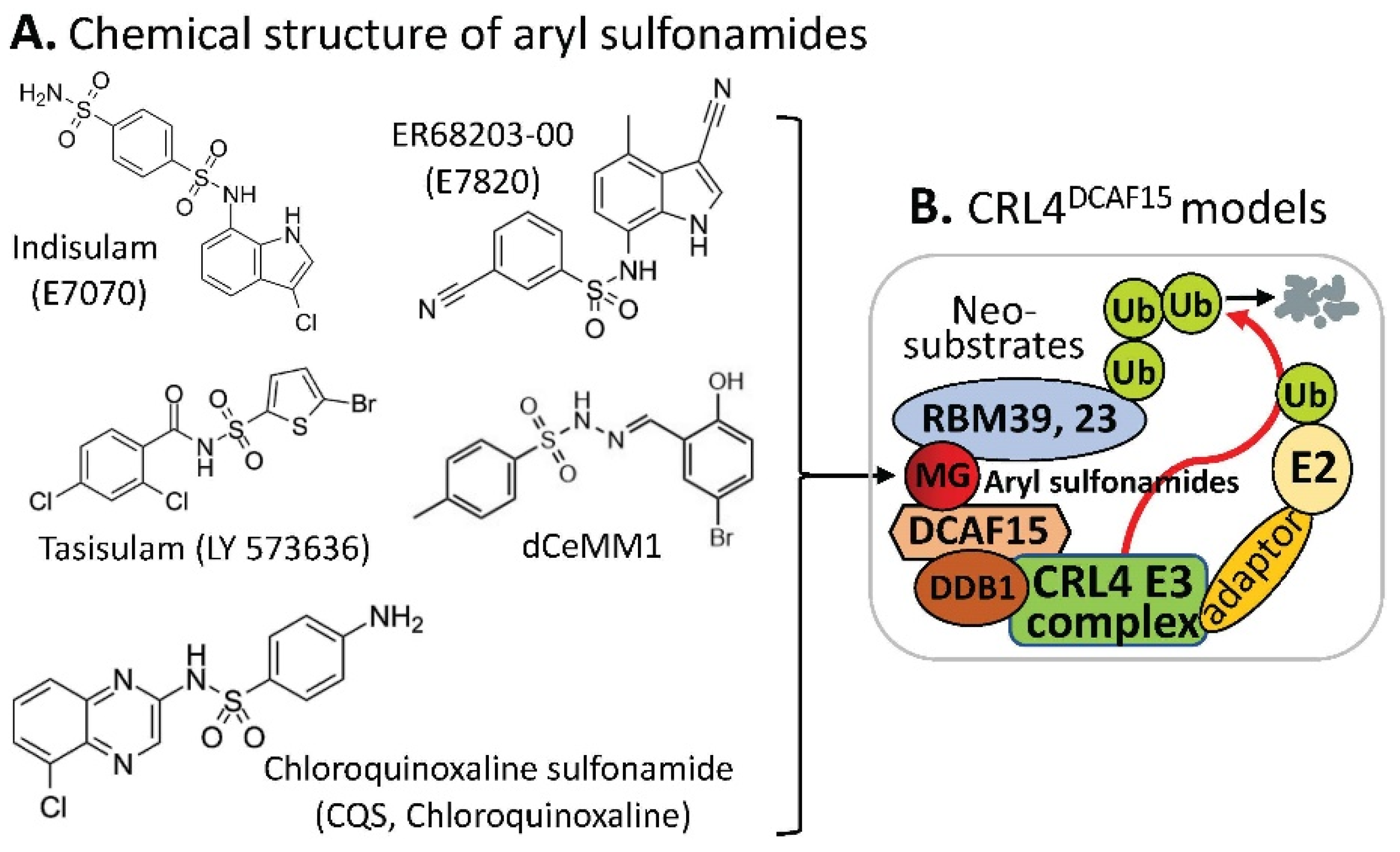
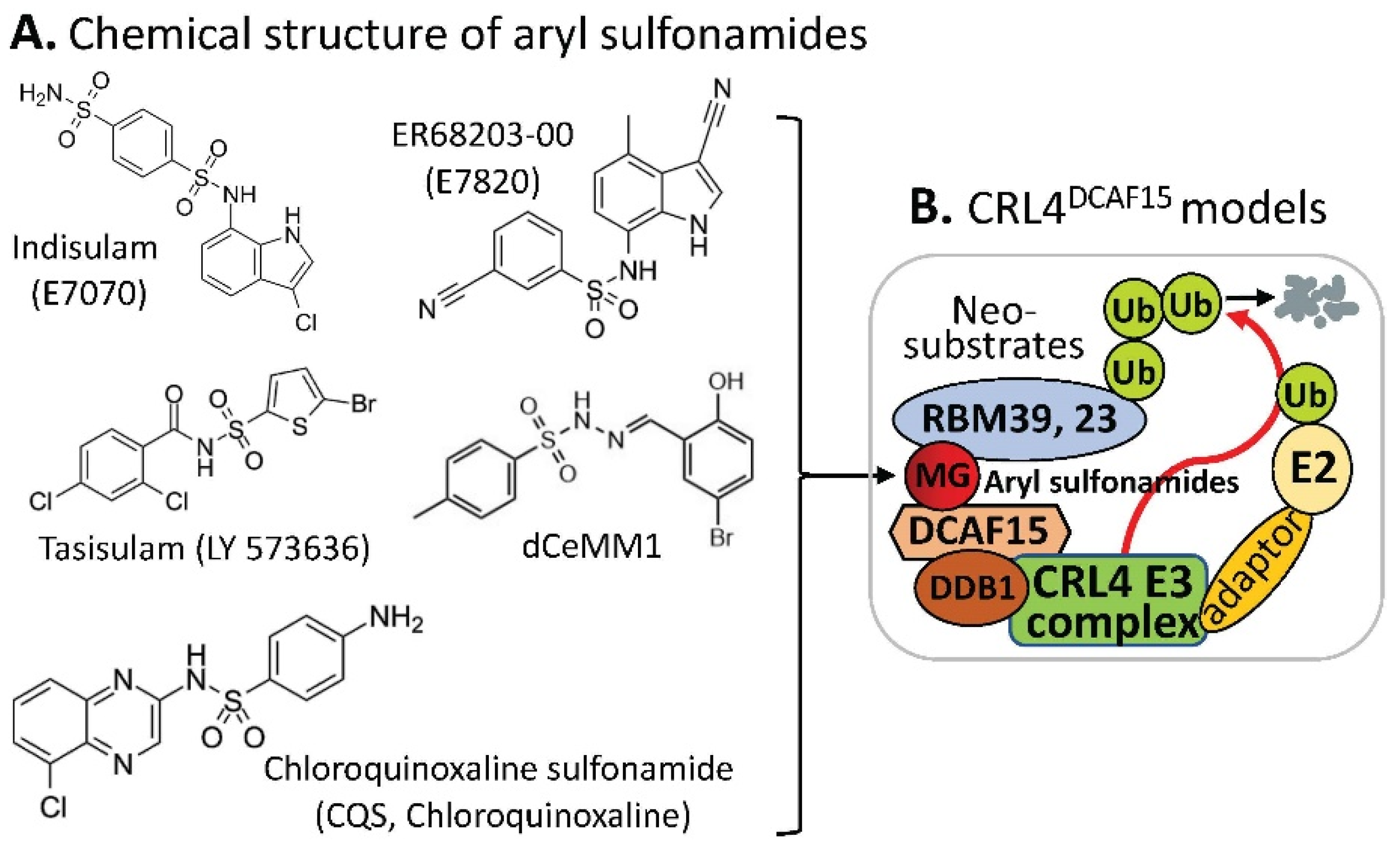
2.2. DCAF15-Involved CRL4 Complex-Mediated Degradation of MG-Targeted Proteins
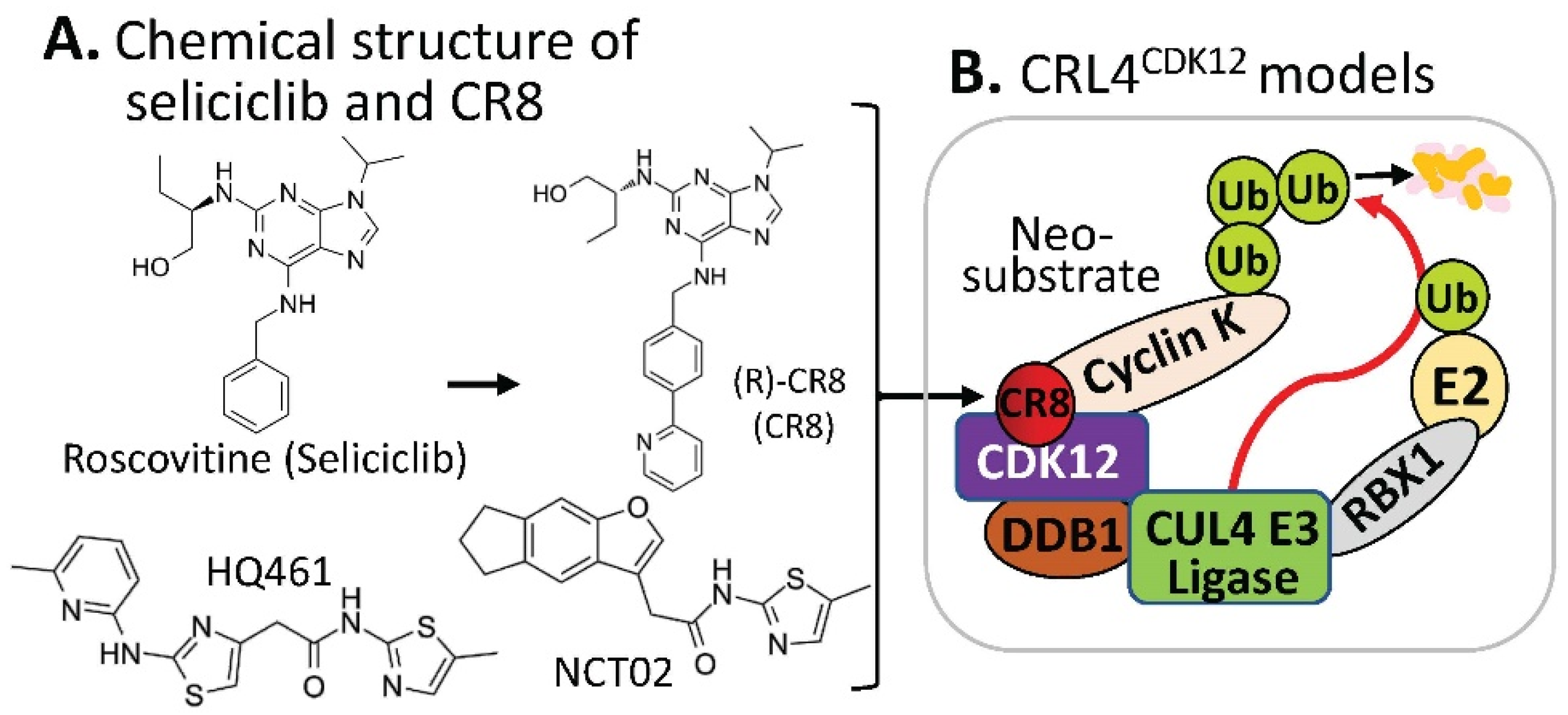
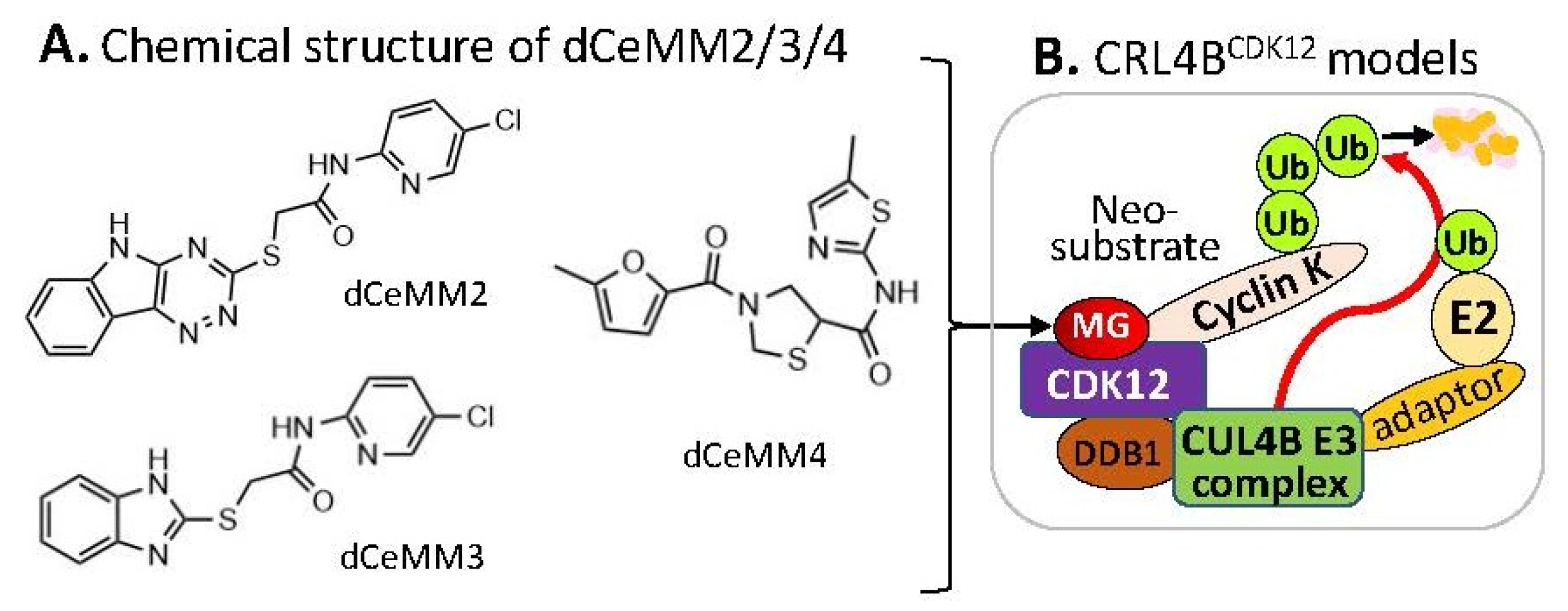
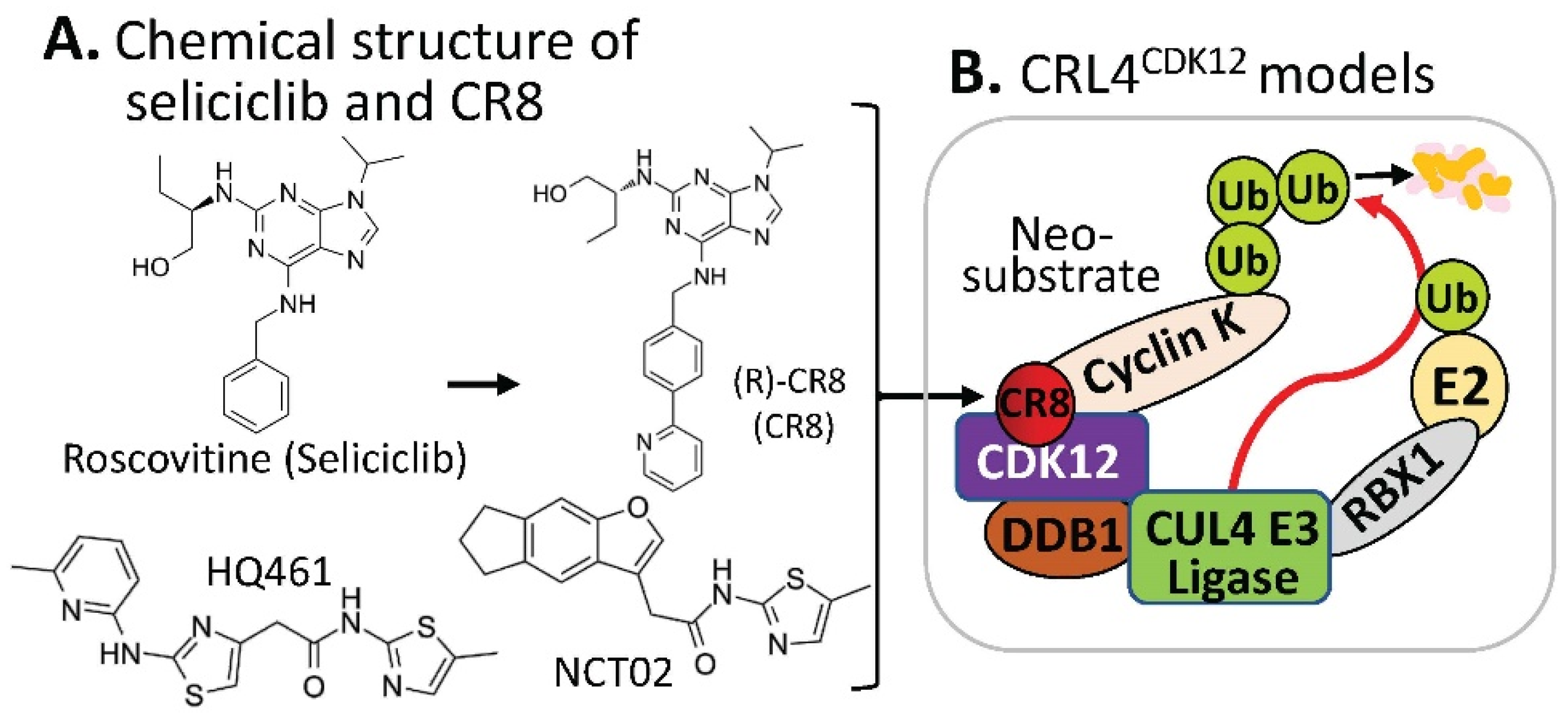
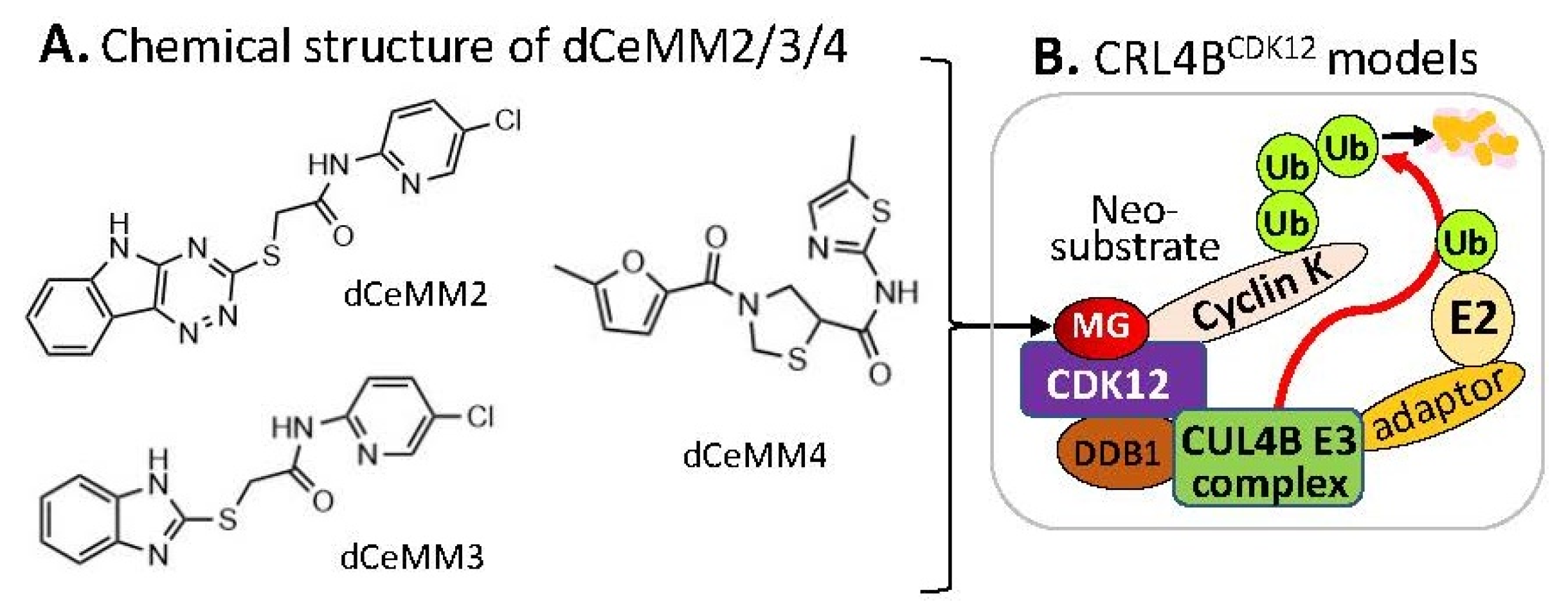
2.3. Substrate Receptor-Independent E3 Ligase-Mediated Degradation of Cyclin K by MG
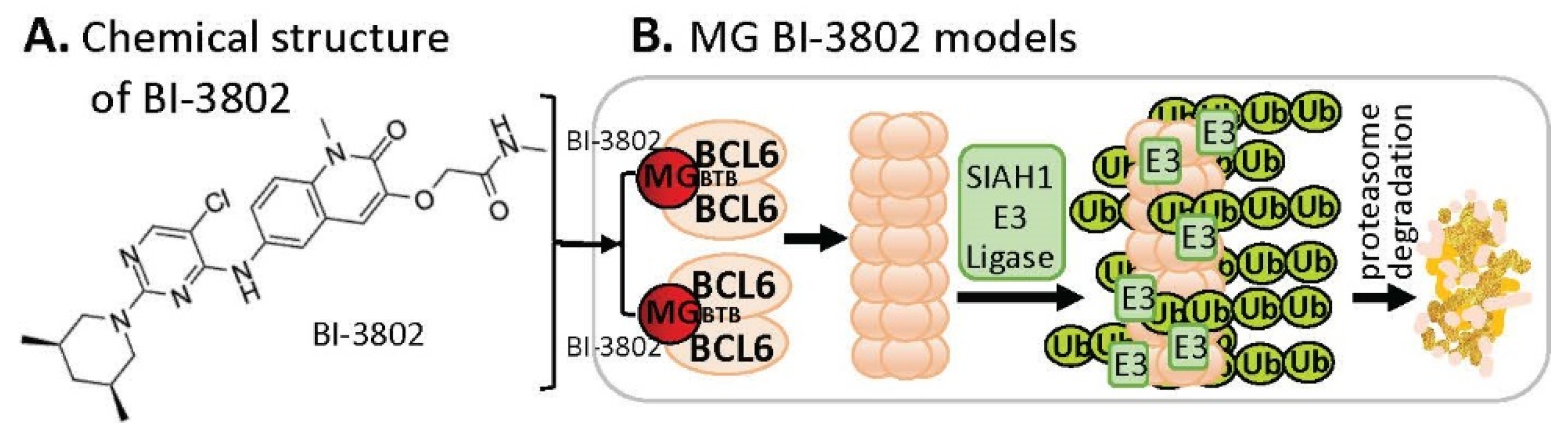
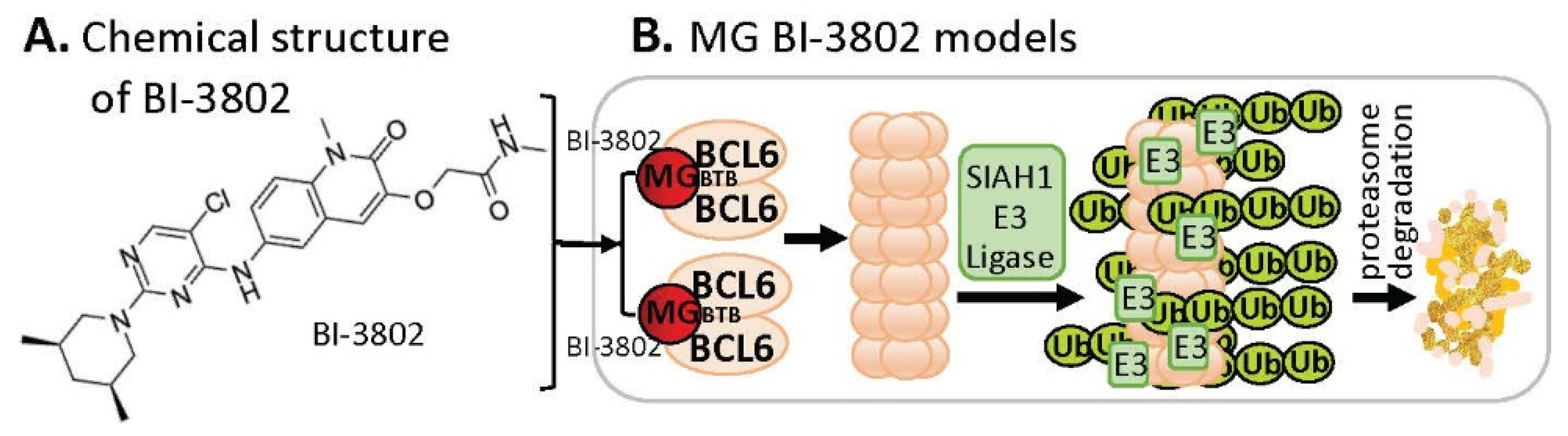
2.4. MG-Mediated Protein Homodimerization-Induced Protein Degradation
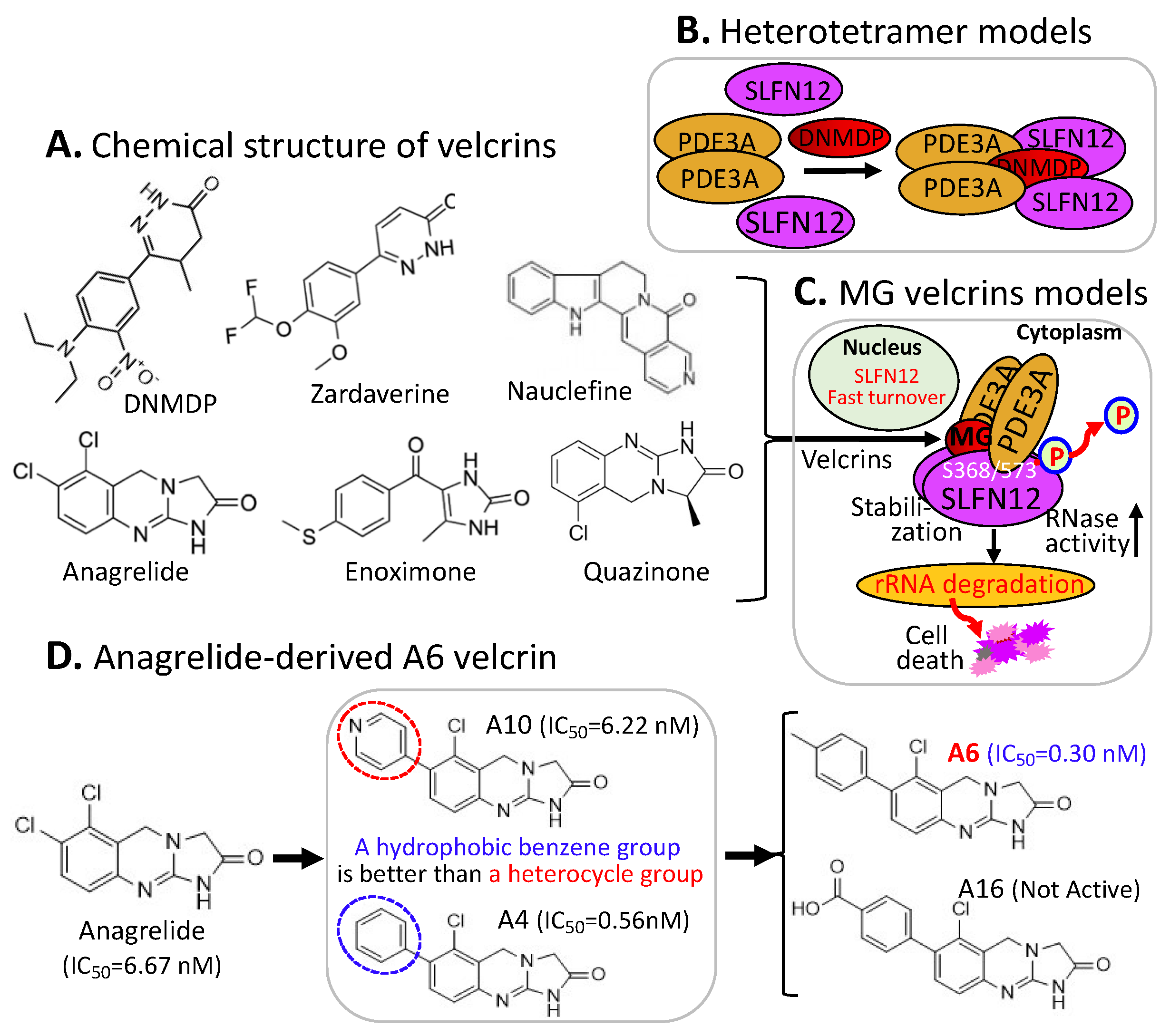
2.5. MG-Mediated Protein Heterodimerization-Induced Protein Stabilization for Cell Death

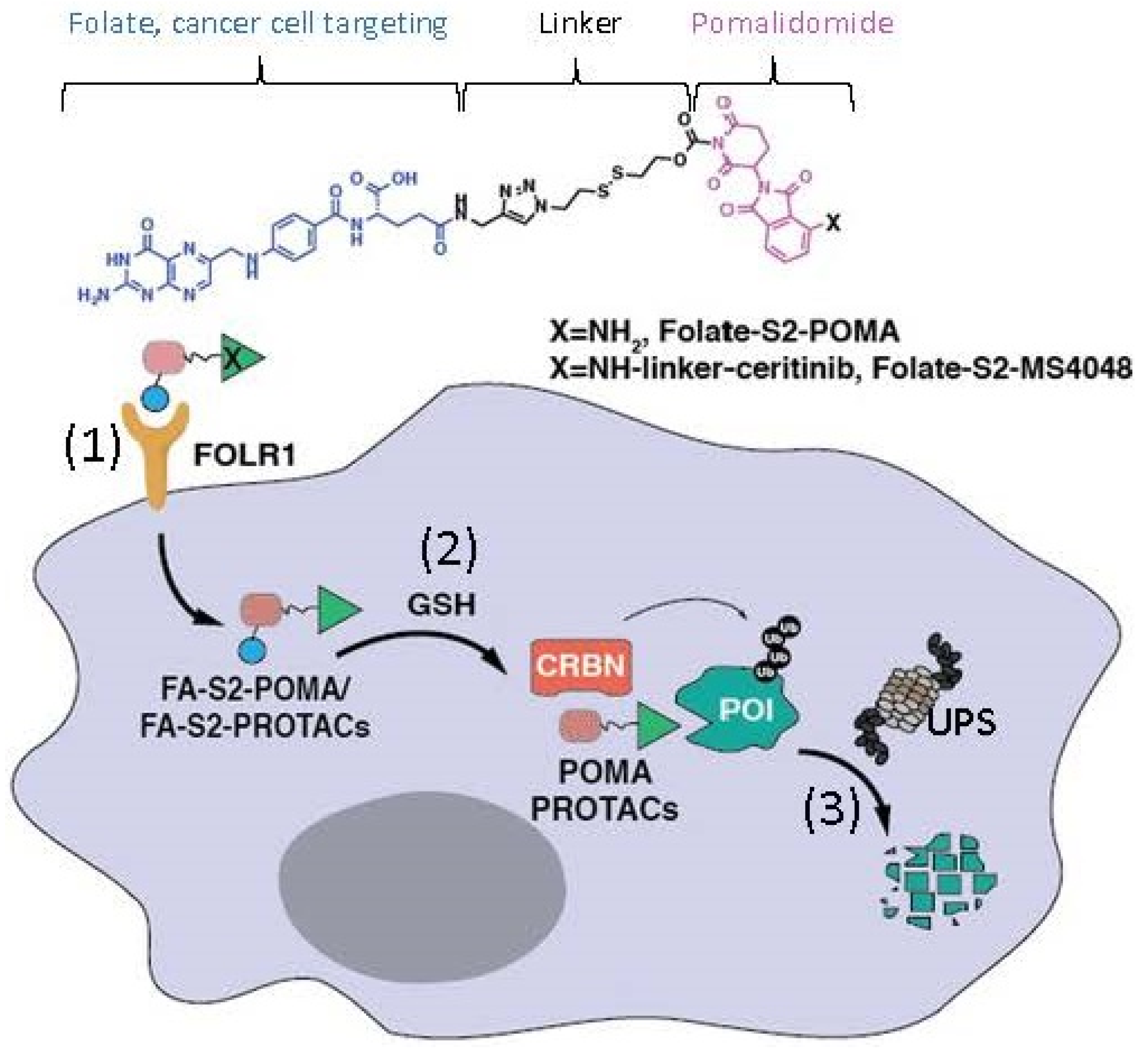
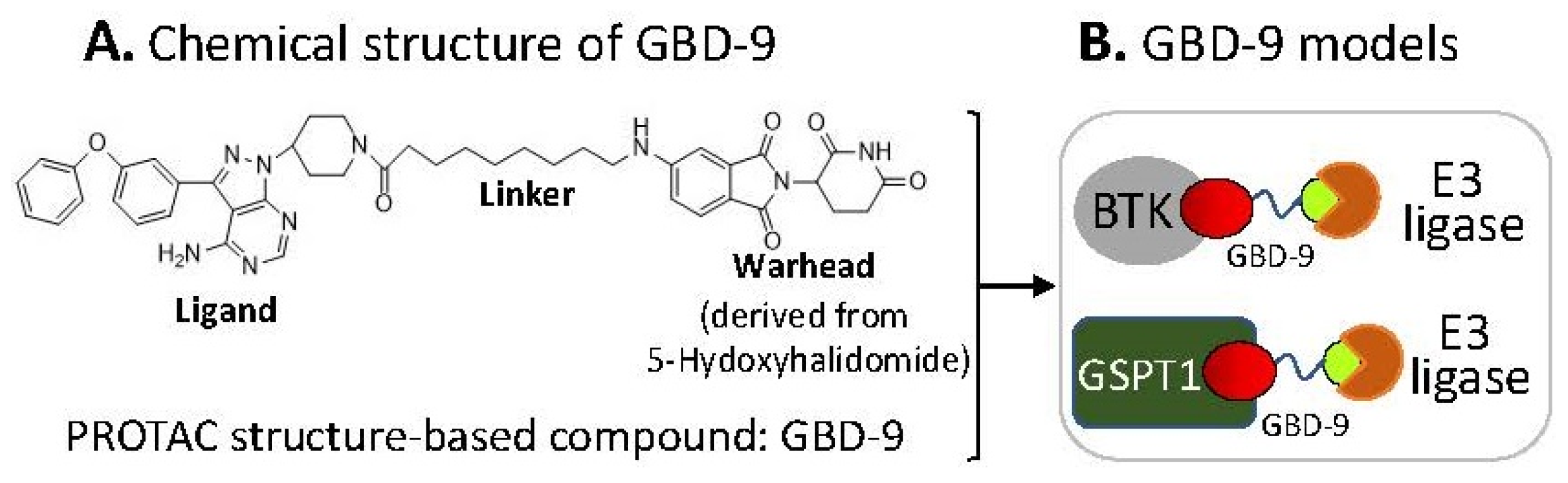
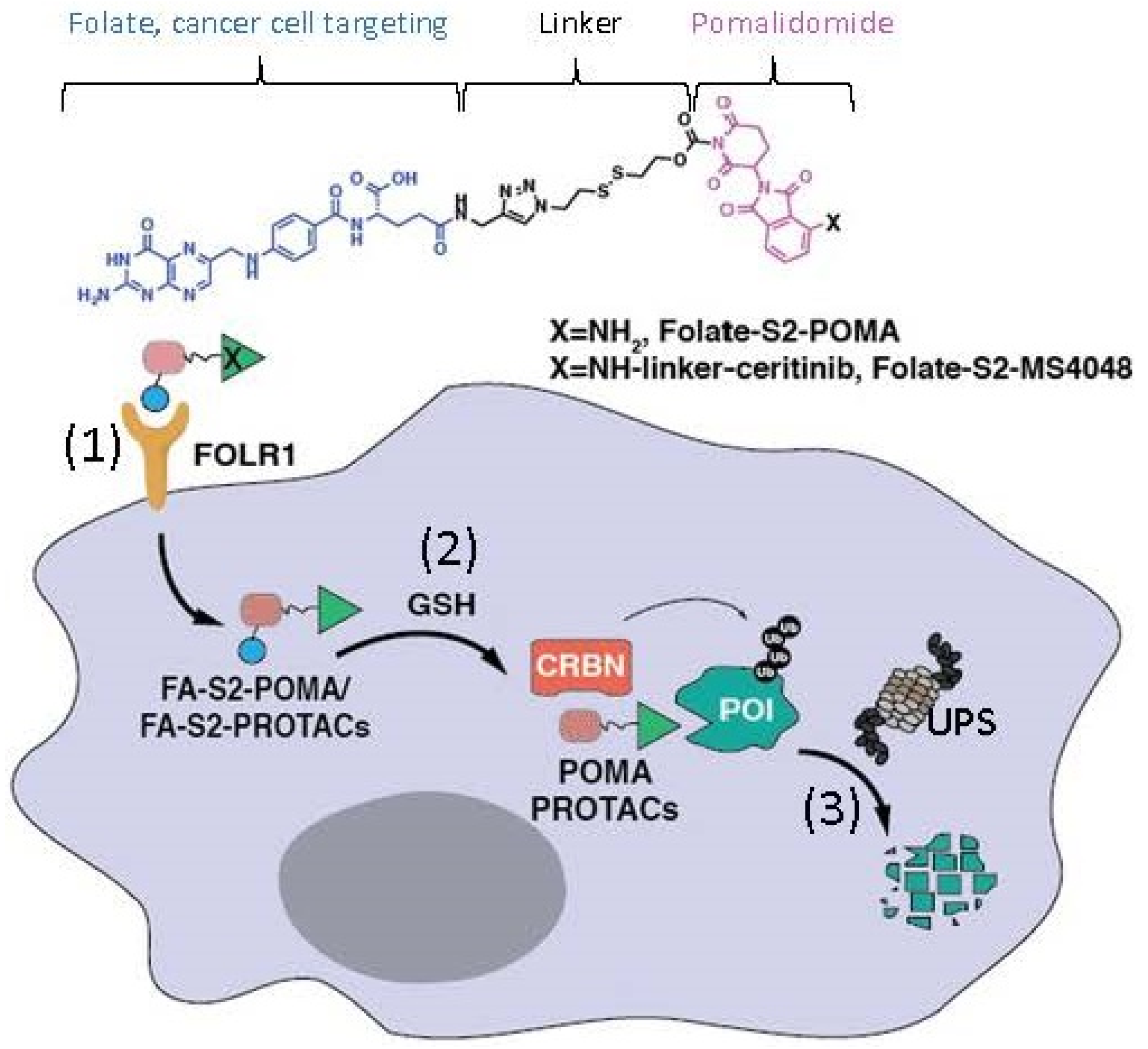
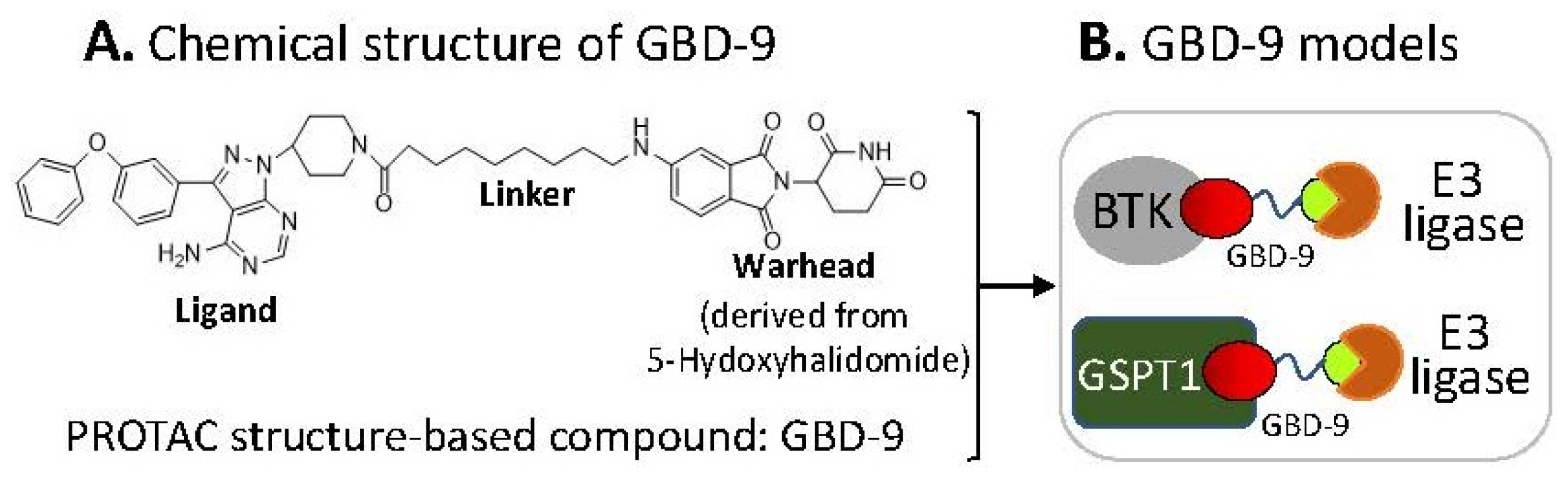
2.6. The Ligand of a PROTAC-Type MG Binding to More than One Protein for Degradation
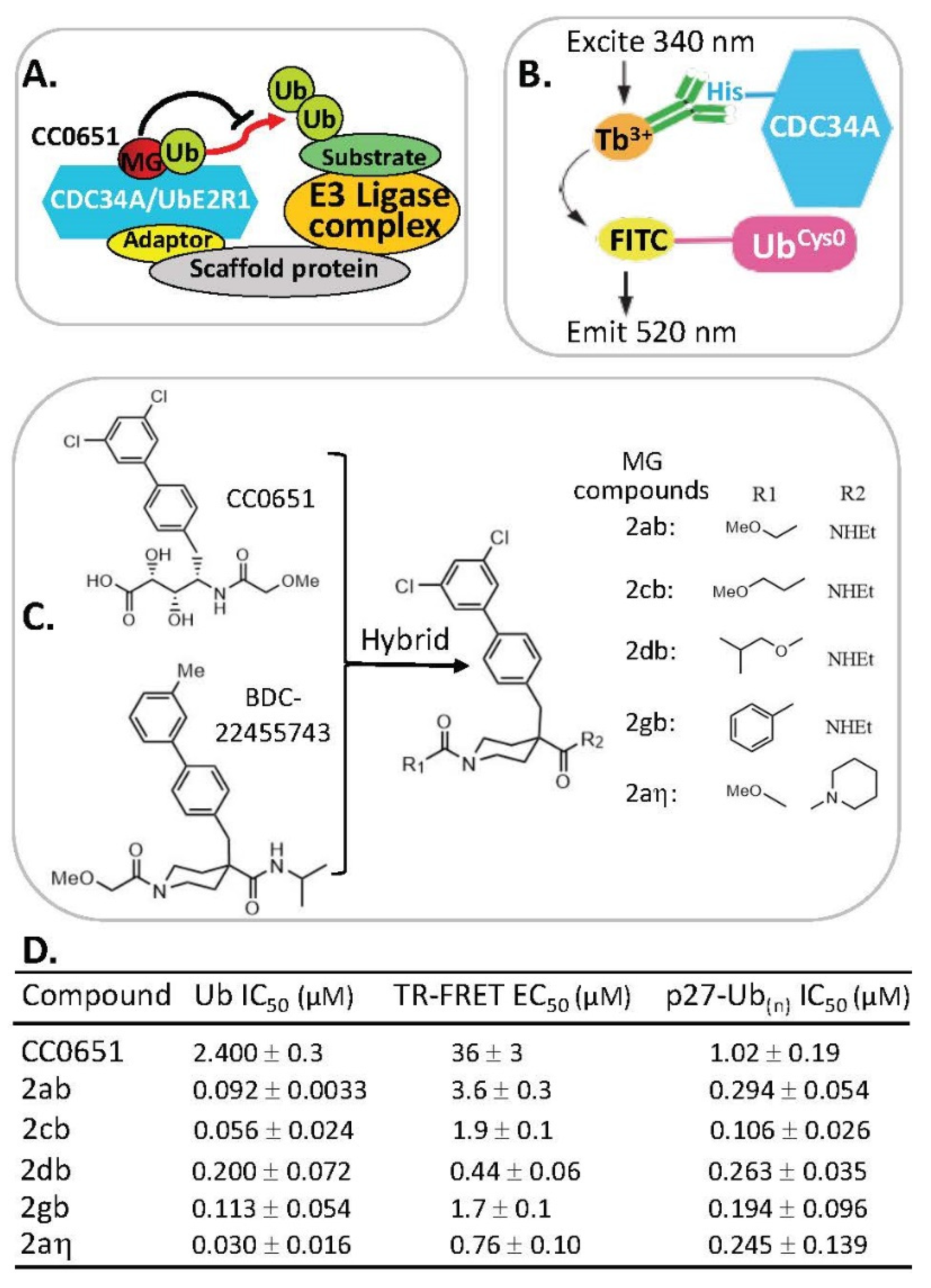
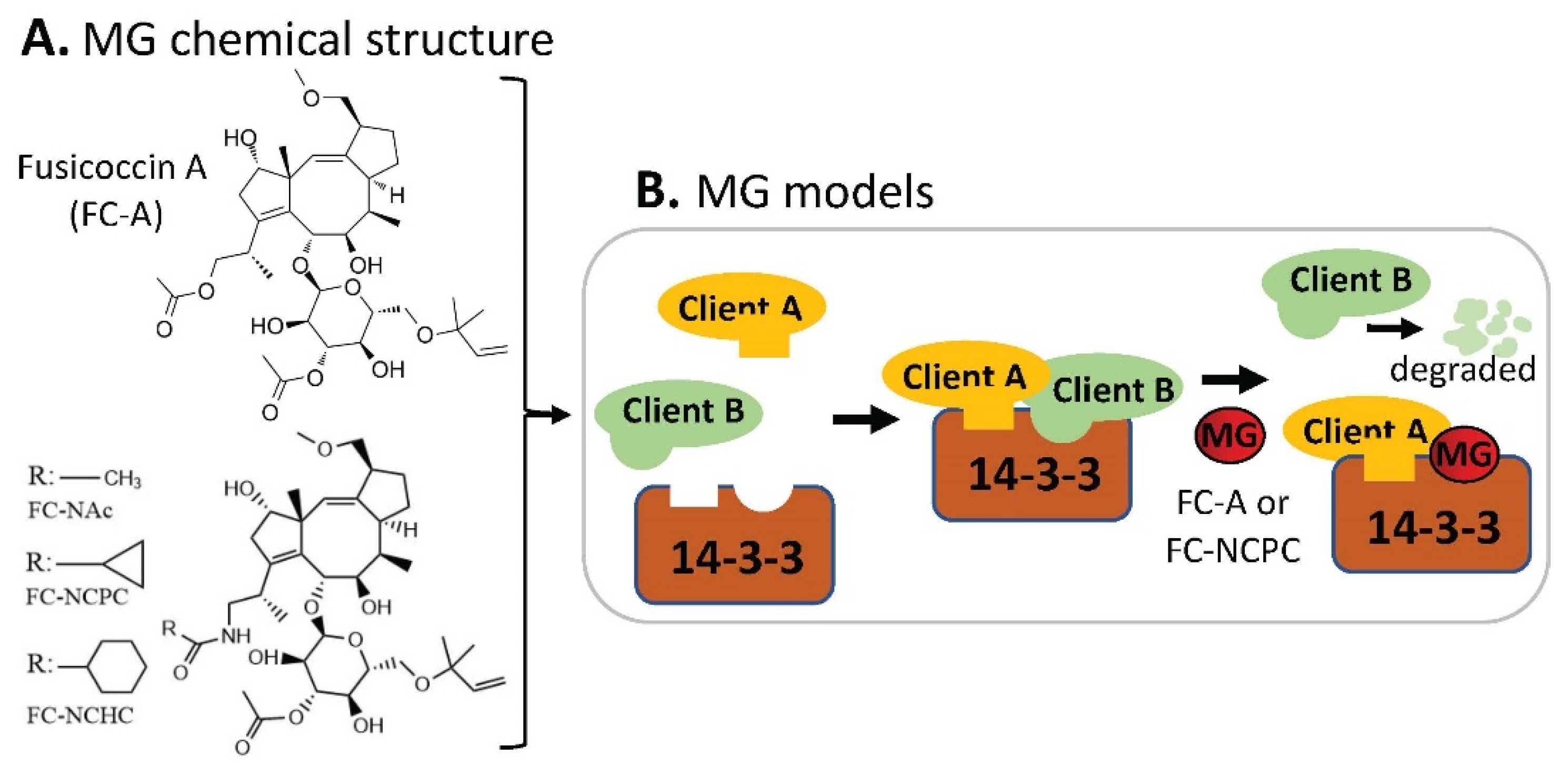
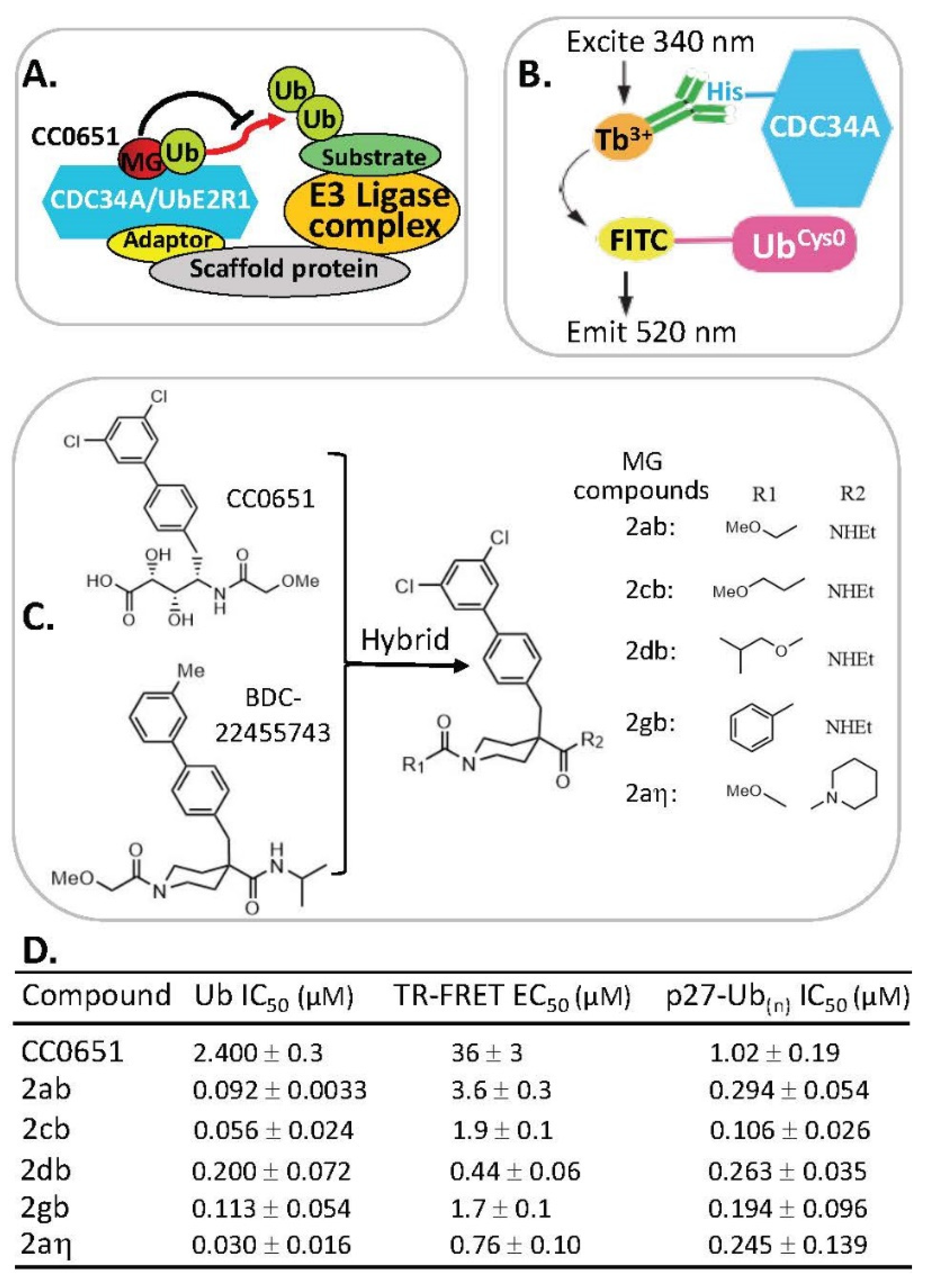
2.7. MG Stabilization of Weak Protein–Ubiquitin Interactions for Function Inhibition or Activation
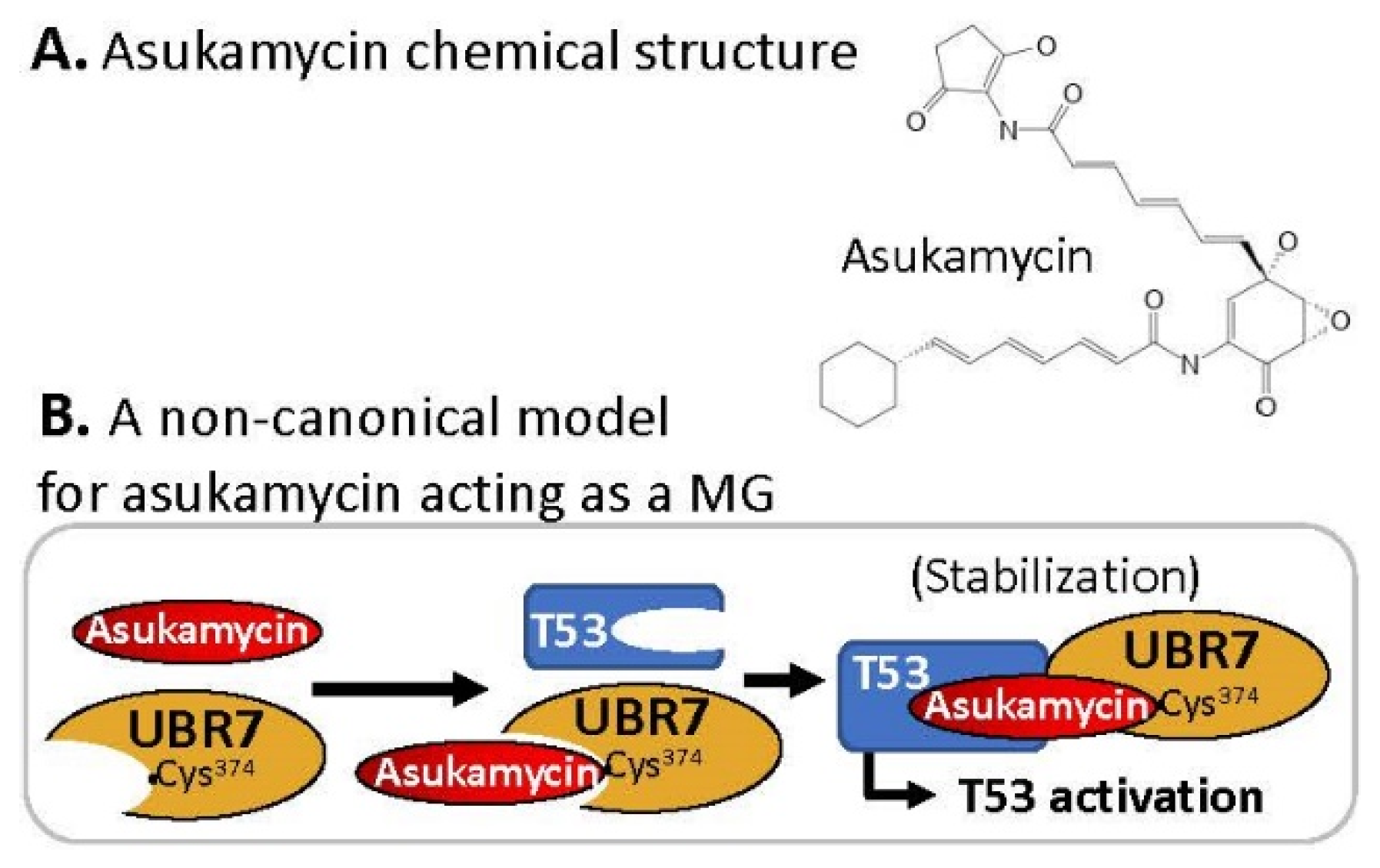
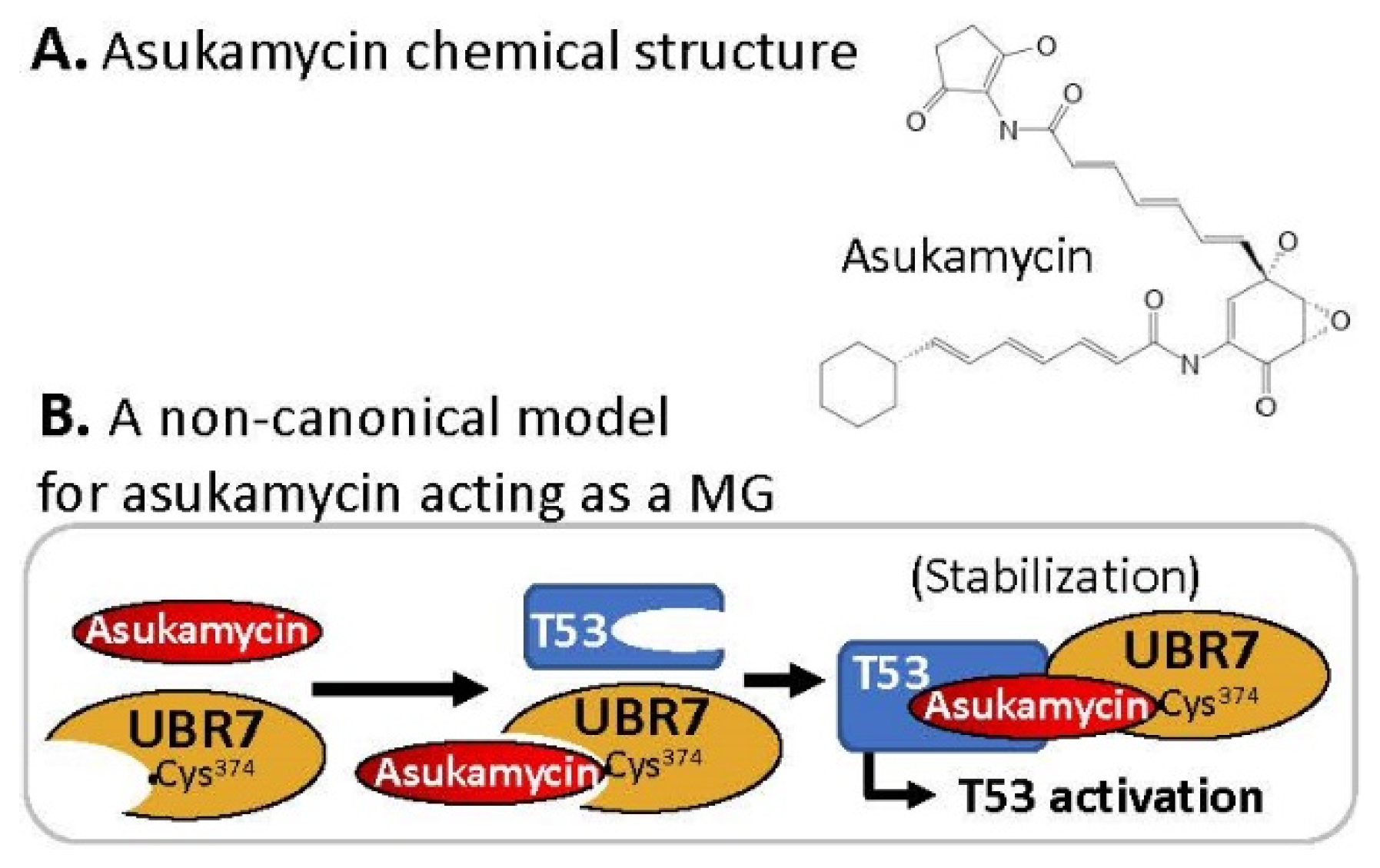
2.8. MG Covalently Engaged/Bound to E3 Ligase and Recruited TP53/p53 for p53 Activation
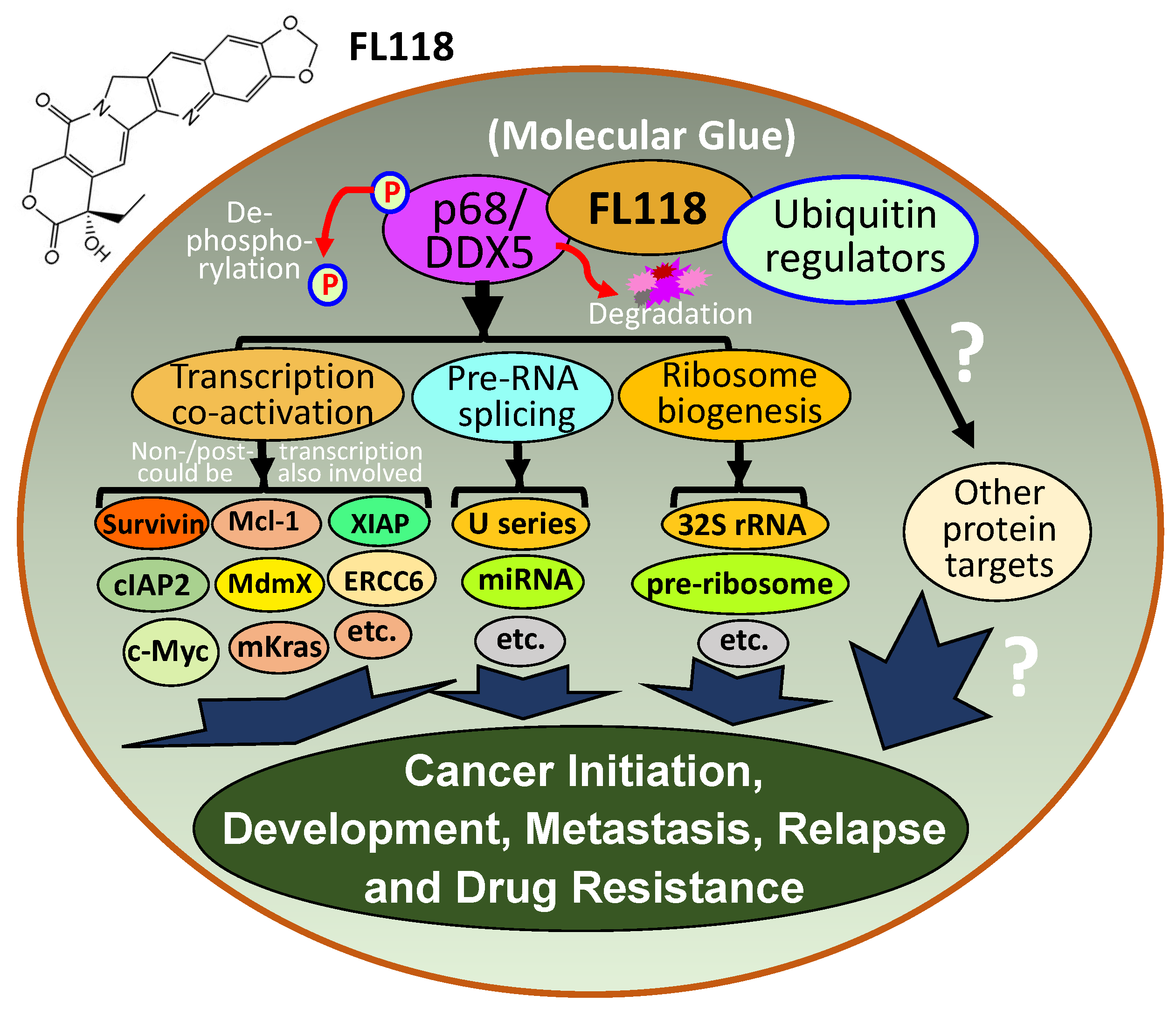
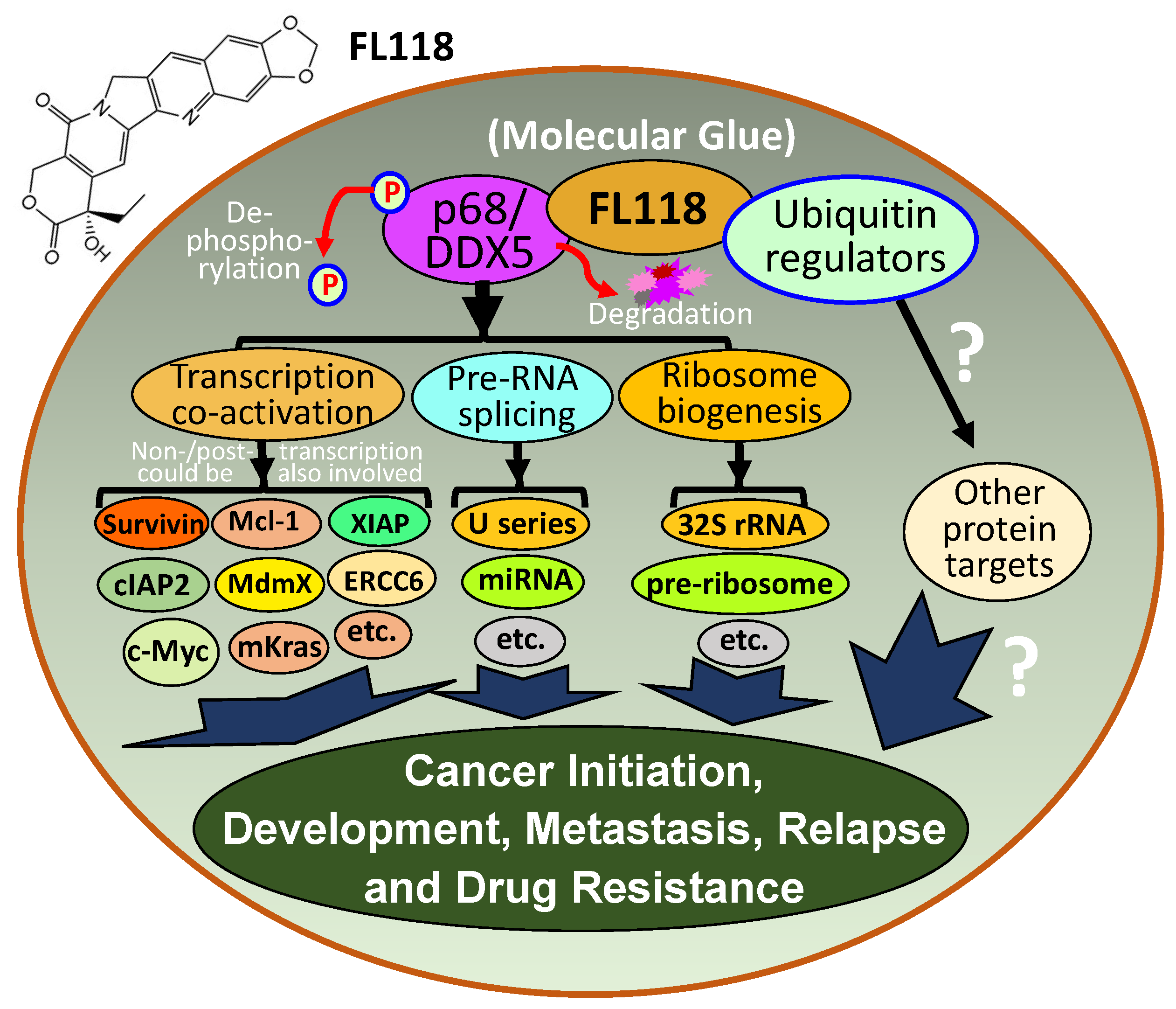
2.9. FL118, as an MG, Binds to Dephosphorylates and Degrades Oncogenic Protein DDX5
3. Molecular Glues (MGs) for Treating Neurodegenerative Diseases
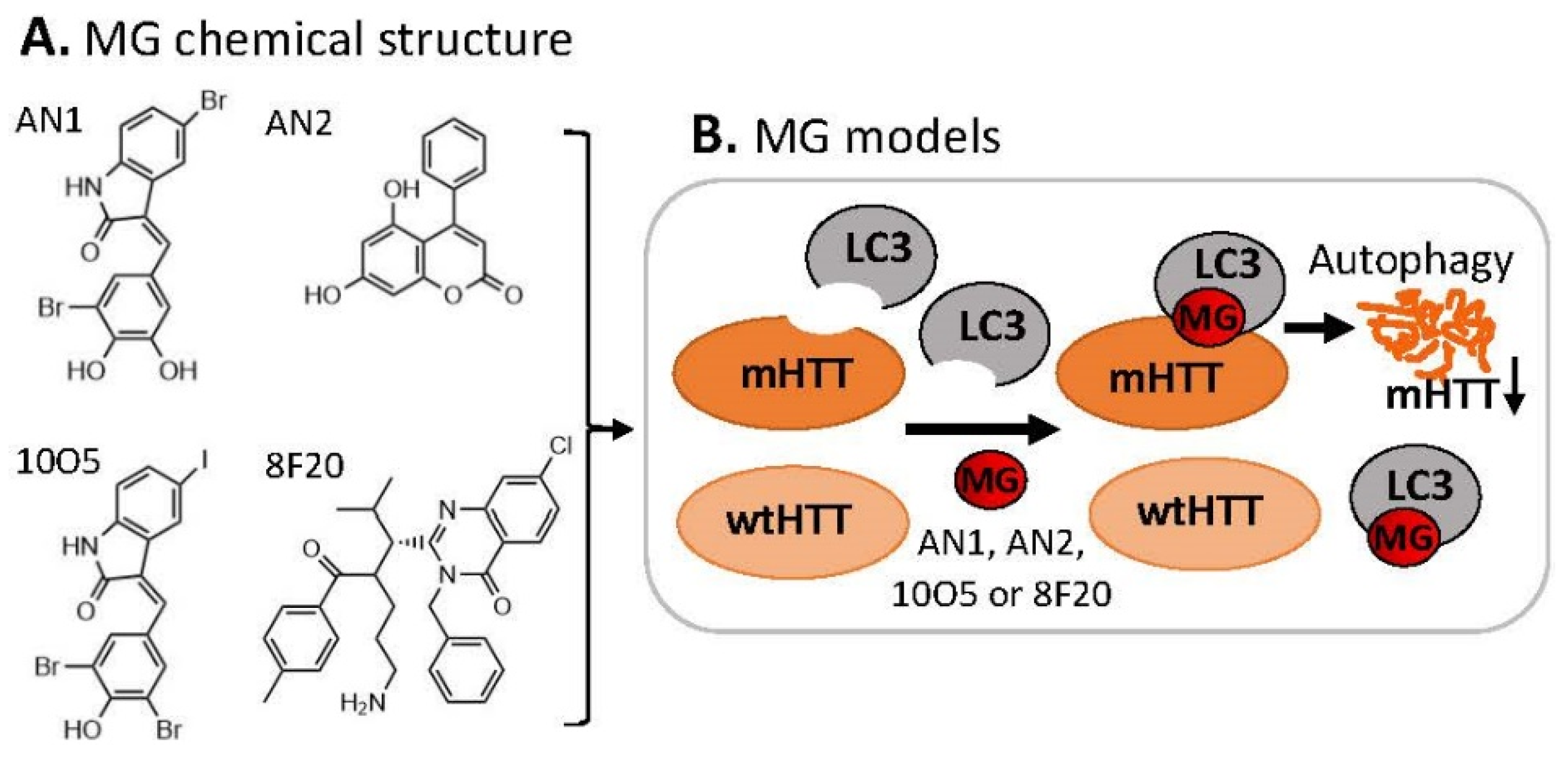
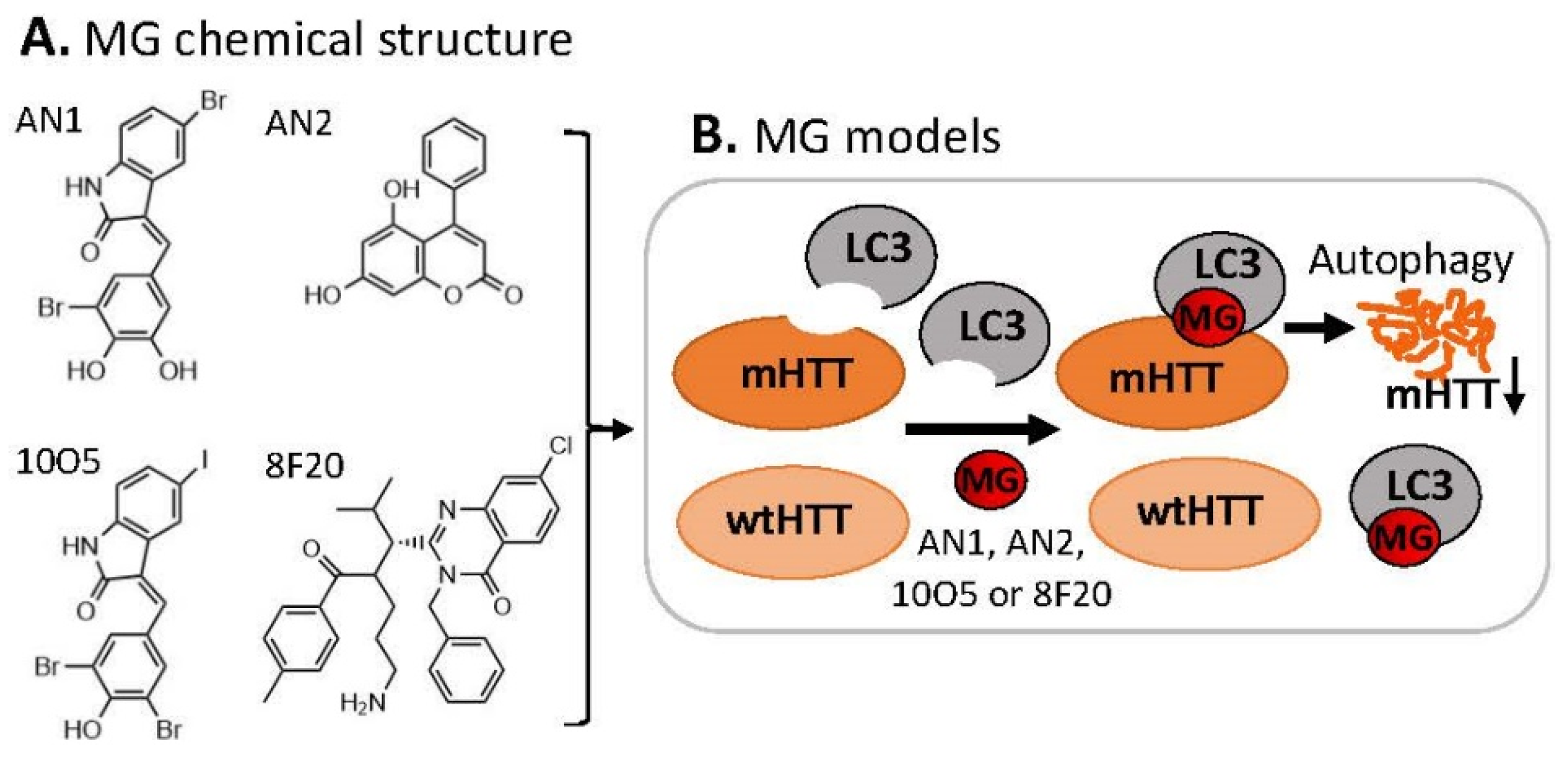
3.1. MG for Treating Huntington’s Disease
3.2. Other Neurodegenerative Diseases
4. Perspectives and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faux, M.C.; Scott, J.D. Molecular glue: Kinase anchoring and scaffold proteins. Cell 1996, 85, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.L. The Rise of Molecular Glues. Cell 2021, 184, 3–9. [Google Scholar] [CrossRef]
- Tan, X.; Calderon-Villalobos, L.I.; Sharon, M.; Zheng, C.; Robinson, C.V.; Estelle, M.; Zheng, N. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 2007, 446, 640–645. [Google Scholar] [CrossRef]
- Sheard, L.B.; Tan, X.; Mao, H.; Withers, J.; Ben-Nissan, G.; Hinds, T.R.; Kobayashi, Y.; Hsu, F.F.; Sharon, M.; Browse, J.; et al. Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature 2010, 468, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Kronke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Dong, G.; Ding, Y.; He, S.; Sheng, C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64, 10606–10620. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Kozicka, Z.; Thoma, N.H. Haven’t got a glue: Protein surface variation for the design of molecular glue degraders. Cell Chem. Biol. 2021, 28, 1032–1047. [Google Scholar] [CrossRef]
- Teng, M.; Lu, W.; Donovan, K.A.; Sun, J.; Krupnick, N.M.; Nowak, R.P.; Li, Y.D.; Sperling, A.S.; Zhang, T.; Ebert, B.L.; et al. Development of PDE6D and CK1alpha Degraders through Chemical Derivatization of FPFT-2216. J. Med. Chem. 2022, 65, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, J.; Kaniskan, H.U.; Wei, W.; Jin, J. Folate-Guided Protein Degradation by Immunomodulatory Imide Drug-Based Molecular Glues and Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 12273–12285. [Google Scholar] [CrossRef] [PubMed]
- Pech, M.F.; Fong, L.E.; Villalta, J.E.; Chan, L.J.; Kharbanda, S.; O’Brien, J.J.; McAllister, F.E.; Firestone, A.J.; Jan, C.H.; Settleman, J. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. Elife 2019, 8, e47362. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Quarni, W.; Goralski, M.; Wan, S.; Jin, H.; Van de Velde, L.A.; Fang, J.; Wu, Q.; Abu-Zaid, A.; Wang, T.; et al. Targeting the spliceosome through RBM39 degradation results in exceptional responses in high-risk neuroblastoma models. Sci. Adv. 2021, 7, eabj5405. [Google Scholar] [CrossRef]
- Nijhuis, A.; Sikka, A.; Yogev, O.; Herendi, L.; Balcells, C.; Ma, Y.; Poon, E.; Eckold, C.; Valbuena, G.N.; Xu, Y.; et al. Indisulam targets RNA splicing and metabolism to serve as a therapeutic strategy for high-risk neuroblastoma. Nat. Commun. 2022, 13, 1380. [Google Scholar] [CrossRef]
- Gosavi, P.M.; Ngan, K.C.; Yeo, M.J.R.; Su, C.; Li, J.; Lue, N.Z.; Hoenig, S.M.; Liau, B.B. Profiling the Landscape of Drug Resistance Mutations in Neosubstrates to Molecular Glue Degraders. ACS Cent. Sci. 2022, 8, 417–429. [Google Scholar] [CrossRef]
- Benson, C.; White, J.; De Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Oumata, N.; Bettayeb, K.; Ferandin, Y.; Demange, L.; Lopez-Giral, A.; Goddard, M.L.; Myrianthopoulos, V.; Mikros, E.; Flajolet, M.; Greengard, P.; et al. Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J. Med. Chem. 2008, 51, 5229–5242. [Google Scholar] [CrossRef]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Slabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207. [Google Scholar] [CrossRef]
- Lv, L.; Chen, P.; Cao, L.; Li, Y.; Zeng, Z.; Cui, Y.; Wu, Q.; Li, J.; Wang, J.H.; Dong, M.Q.; et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. eLife 2020, 9, e59994. [Google Scholar] [CrossRef]
- Dieter, S.M.; Siegl, C.; Codo, P.L.; Huerta, M.; Ostermann-Parucha, A.L.; Schulz, E.; Zowada, M.K.; Martin, S.; Laaber, K.; Nowrouzi, A.; et al. Degradation of CCNK/CDK12 is a druggable vulnerability of colorectal cancer. Cell Rep. 2021, 36, 109394. [Google Scholar] [CrossRef]
- Kerres, N.; Steurer, S.; Schlager, S.; Bader, G.; Berger, H.; Caligiuri, M.; Dank, C.; Engen, J.R.; Ettmayer, P.; Fischerauer, B.; et al. Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep. 2017, 20, 2860–2875. [Google Scholar] [CrossRef] [Green Version]
- Slabicki, M.; Yoon, H.; Koeppel, J.; Nitsch, L.; Roy Burman, S.S.; Di Genua, C.; Donovan, K.A.; Sperling, A.S.; Hunkeler, M.; Tsai, J.M.; et al. Small-molecule-induced polymerization triggers degradation of BCL6. Nature 2020, 588, 164–168. [Google Scholar] [CrossRef]
- Garvie, C.W.; Wu, X.; Papanastasiou, M.; Lee, S.; Fuller, J.; Schnitzler, G.R.; Horner, S.W.; Baker, A.; Zhang, T.; Mullahoo, J.P.; et al. Structure of PDE3A-SLFN12 complex reveals requirements for activation of SLFN12 RNase. Nat. Commun. 2021, 12, 4375. [Google Scholar] [CrossRef]
- Yan, B.; Ding, Z.; Zhang, W.; Cai, G.; Han, H.; Ma, Y.; Cao, Y.; Wang, J.; Chen, S.; Ai, Y. Multiple PDE3A modulators act as molecular glues promoting PDE3A-SLFN12 interaction and induce SLFN12 dephosphorylation and cell death. Cell Chem. Biol. 2022, 29, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, N.; Huang, Y.; Wang, Y.; Sun, Y.; Wu, Q.; Li, D.; Gao, S.; Wang, H.W.; Huang, N.; et al. Structure of PDE3A-SLFN12 complex and structure-based design for a potent apoptosis inducer of tumor cells. Nat. Commun. 2021, 12, 6204. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, Y.; Ni, Z.; Yang, C.; Tong, Y.; Liu, Y.; Li, H.; Rao, Y. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res. 2021, 31, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.F.; Tang, X.; Pelletier, B.; Orlicky, S.; Xie, W.; Plantevin, V.; Neculai, D.; Chou, Y.C.; Ogunjimi, A.; Al-Hakim, A.; et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 2011, 145, 1075–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Ceccarelli, D.F.; Orlicky, S.; St-Cyr, D.J.; Ziemba, A.; Garg, P.; Plamondon, S.; Auer, M.; Sidhu, S.; Marinier, A.; et al. E2 enzyme inhibition by stabilization of a low-affinity interface with ubiquitin. Nat. Chem. Biol. 2014, 10, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Cyr, D.; Ceccarelli, D.F.; Orlicky, S.; van der Sloot, A.M.; Tang, X.; Kelso, S.; Moore, S.; James, C.; Posternak, G.; Coulombe-Huntington, J.; et al. Identification and optimization of molecular glue compounds that inhibit a noncovalent E2 enzyme-ubiquitin complex. Sci. Adv. 2021, 7, eabi5797. [Google Scholar] [CrossRef]
- Simonetta, K.R.; Taygerly, J.; Boyle, K.; Basham, S.E.; Padovani, C.; Lou, Y.; Cummins, T.J.; Yung, S.L.; von Soly, S.K.; Kayser, F.; et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat. Commun. 2019, 10, 1402. [Google Scholar] [CrossRef]
- Sijbesma, E.; Visser, E.; Plitzko, K.; Thiel, P.; Milroy, L.G.; Kaiser, M.; Brunsveld, L.; Ottmann, C. Structure-based evolution of a promiscuous inhibitor to a selective stabilizer of protein-protein interactions. Nat. Commun. 2020, 11, 3954. [Google Scholar] [CrossRef]
- Isobe, Y.; Okumura, M.; McGregor, L.M.; Brittain, S.M.; Jones, M.D.; Liang, X.; White, R.; Forrester, W.; McKenna, J.M.; Tallarico, J.A.; et al. Manumycin polyketides act as molecular glues between UBR7 and P53. Nat. Chem. Biol. 2020, 16, 1189–1198. [Google Scholar] [CrossRef]
- Li, F.; Fountzilas, C.; Puzanov, I.; Attwood, K.M.; Morrison, C.; Ling, X. Multiple functions of the DEAD-box RNA helicase, DDX5 (p68), make DDX5 a superior oncogenic biomarker and target for targeted cancer therapy. Am. J. Cancer Res. 2021, 11, 5190–5213. [Google Scholar] [PubMed]
- Ling, X.; Cao, S.; Cheng, Q.; Keefe, J.T.; Rustum, Y.M.; Li, F. A Novel Small Molecule FL118 That Selectively Inhibits Survivin, Mcl-1, XIAP and cIAP2 in a p53-Independent Manner, Shows Superior Antitumor Activity. PLoS ONE 2012, 7, e45571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, X.; Li, F. An intravenous (i.v.) route-compatible formulation of FL118, a survivin, Mcl-1, XIAP, and cIAP2 selective inhibitor, improves FL118 antitumor efficacy and therapeutic index (TI). Am. J. Transl. Res. 2013, 5, 139–154. [Google Scholar] [PubMed]
- Ling, X.; Xu, C.; Fan, C.; Zhong, K.; Li, F.; Wang, X. FL118 Induces p53-Dependent Senescence in Colorectal Cancer Cells by Promoting Degradation of MdmX. Cancer Res. 2014, 74, 7487–7497. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Ling, X.; Cao, S.; Liu, X.; Wan, S.; Jiang, T.; Li, F. Antitumor activity of FL118, a survivin, Mcl-1, XIAP, cIAP2 selective inhibitor, is highly dependent on its primary structure and steric configuration. Mol. Pharm. 2014, 11, 457–467. [Google Scholar] [CrossRef]
- Westover, D.; Ling, X.; Lam, H.; Welch, J.; Jin, C.; Gongora, C.; Del Rio, M.; Wani, M.; Li, F. FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol. Cancer 2015, 14, 92. [Google Scholar] [CrossRef] [Green Version]
- Ling, X.; Wu, W.; Fan, C.; Xu, C.; Liao, J.; Rich, L.J.; Huang, R.Y.; Repasky, E.A.; Wang, X.; Li, F. An ABCG2 non-substrate anticancer agent FL118 targets drug-resistant cancer stem-like cells and overcomes treatment resistance of human pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 240. [Google Scholar] [CrossRef] [Green Version]
- Holthof, L.C.; van der Horst, H.J.; van Hal-van Veen, S.E.; Ruiter, R.W.J.; Li, F.; Buijze, M.; Andersen, M.N.; Yuan, H.; de Bruijn, J.; van de Donk, N.; et al. Preclinical evidence for an effective therapeutic activity of FL118, a novel survivin inhibitor, in patients with relapsed/refractory multiple myeloma. Haematologica 2020, 105, e80–e83. [Google Scholar] [CrossRef]
- Ling, X.; Wu, W.; Aljahdali, I.A.M.; Liao, J.; Santha, S.; Fountzilas, C.; Boland, P.M.; Li, F. FL118, acting as a ‘molecular glue degrader’, binds to, dephosphorylates and degrades the oncoprotein DDX5 (p68) to control c-Myc, survivin and mutant Kras against colorectal and pancreatic cancer with high efficacy. Clin. Transl. Med. 2022, 12, e881. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, C.; Wang, Z.; Zhu, C.; Li, J.; Sha, T.; Ma, L.; Gao, C.; Yang, Y.; Sun, Y.; et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 2019, 575, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.; Andrei, S.A.; van Regteren Altena, A.; Simas, T.; Banerjee, S.L.; Kato, N.; Bisson, N.; Higuchi, Y.; Ottmann, C.; Fournier, A.E. Polypharmacological Perturbation of the 14-3-3 Adaptor Protein Interactome Stimulates Neurite Outgrowth. Cell Chem. Biol. 2020, 27, 657–667. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MG Compounds | Neosubstrates/ Protein Targets | Neosubstrates/ Protein Targets | Molecular Glue (MG) Compounds | |
|---|---|---|---|---|
| Thalidomide | IKZF1 #, IKZF3, ZNF692, ZNF276, SALL4, RNF166, ZBTB16, FAM83F, p63 | IKZF1 | thalidomide, lenalidomide, pomalidomide, avadomide/CC-122, FPFT-2216, iberdomide/CC-220, CC-3060, CC-92480, CC-885 | |
| Lenalidomide (Revlimid) | IKZF1, IKZF3, ZFP91, ZFP692, ZNF276, ZNF653, ZNF827, SALL4, RNF166, WIZ1, CK1α, FAM83F, RAB28 | IKZF3 | thalidomide, lenalidomide, pomalidomide, avadomide/CC-122, iberdomide/CC-220, 92480, CC-885 | |
| Pomalidomide | IKZF1, IKZF3, ZFP91, ZFP692, ZNF276, ZNF653, ZNF827, SALL4, RNF166, GZF1, ZBTB39, ZNF98, WIZ1, ZBTB16, FAM83F, RAB28, DTWD1 | ZNF692 | thalidomide, lenalidomide, pomalidomide, | |
| Avadomide (CC-122) | IKZF1, IKZF3, ZFP91 | ZNF276 | thalidomide, lenalidomide, pomalidomide, | |
| 5-hydroxy-thalidomide | SALL4, ZBTB16 | SALL4 | thalidomide, lenalidomide, pomalidomide, FPFT-2216, | |
| FPFT-2216 | IKZF1, CK1α | RNF166 | thalidomide, lenalidomide, pomalidomide, | |
| Iberdomide (CC-220) | IKZF1, IKZF3, ZFP91, ZNF98 | ZBTB16 | thalidomide, pomalidomide, 5-hydroxythalidomide, CC-647, CC-3060 | |
| CC-647 | ZBTB16 | FAM83F | thalidomide, lenalidomide, pomalidomide, | |
| CC-3060 | ZBTB16, IKZF1, ZFP91, ZNF276 | p63 | thalidomide | |
| CC-92480 | IKZF1, IKZF3 | ZFP91 | lenalidomide, pomalidomide, avadomide/CC-122, iberdomide/CC-220, CC-3060 | |
| CC-885 | IKZF1, IKZF3, GSPT1, CK1α, PLK1, HBS1L | ZNF653 | lenalidomide, pomalidomide, | |
| CC-90009 | GSPT1 | ZNF827 | lenalidomide, pomalidomide, | |
| ZXH-1-161 | GSPT1, GSPT2 | WIZ1 | thalidomide, lenalidomide, | |
| CK1α | lenalidomide, FPFT-2216, CC-885 | |||
| RAB28 | lenalidomide, pomalidomide, | |||
| GZF1 | pomalidomide, | |||
| ZBTB39 | pomalidomide, | |||
| ZNF98 | pomalidomide, iberdomide/CC-220, | |||
| DTWD1 | pomalidomide, | |||
| ZNF276 | CC-3060 | |||
| GSPT1 | CC-885, CC-90009, ZXH-1-161 | |||
| PLK1 | CC-885 | |||
| HBS1L | CC-885 | |||
| GSPT2 | ZXH-1-161 |
| Compounds | Protein Targets/Neosubstrates | Protein Targets | MG Compounds | |
|---|---|---|---|---|
| Indisulam | # RBM39, RBM23 | RBM39 | Indisulam, E7820, Tasisulam, CQS, dCeMM1 | |
| E7820 | RBM39, RBM23 | RBM23 | Indisulam, E7820, Tasisulam, CQS, | |
| Tasisulam | RBM39, RBM23 | |||
| CQS | RBM39, RBM23 | |||
| dCeMM1 | RBM39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Aljahdali, I.A.M.; Ling, X. Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein–Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 6206. https://doi.org/10.3390/ijms23116206
Li F, Aljahdali IAM, Ling X. Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein–Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(11):6206. https://doi.org/10.3390/ijms23116206
Chicago/Turabian StyleLi, Fengzhi, Ieman A. M. Aljahdali, and Xiang Ling. 2022. "Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein–Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 11: 6206. https://doi.org/10.3390/ijms23116206
APA StyleLi, F., Aljahdali, I. A. M., & Ling, X. (2022). Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein–Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases. International Journal of Molecular Sciences, 23(11), 6206. https://doi.org/10.3390/ijms23116206