
Reaction of N-Acetylcysteine with Cu2+: Appearance of Intermediates with High Free Radical Scavenging Activity: Implications for Anti-/Pro-Oxidant Properties of Thiols

 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Kinetics of ABTSr Scavenging: Cu2+ Catalysis and H+ Inhibition
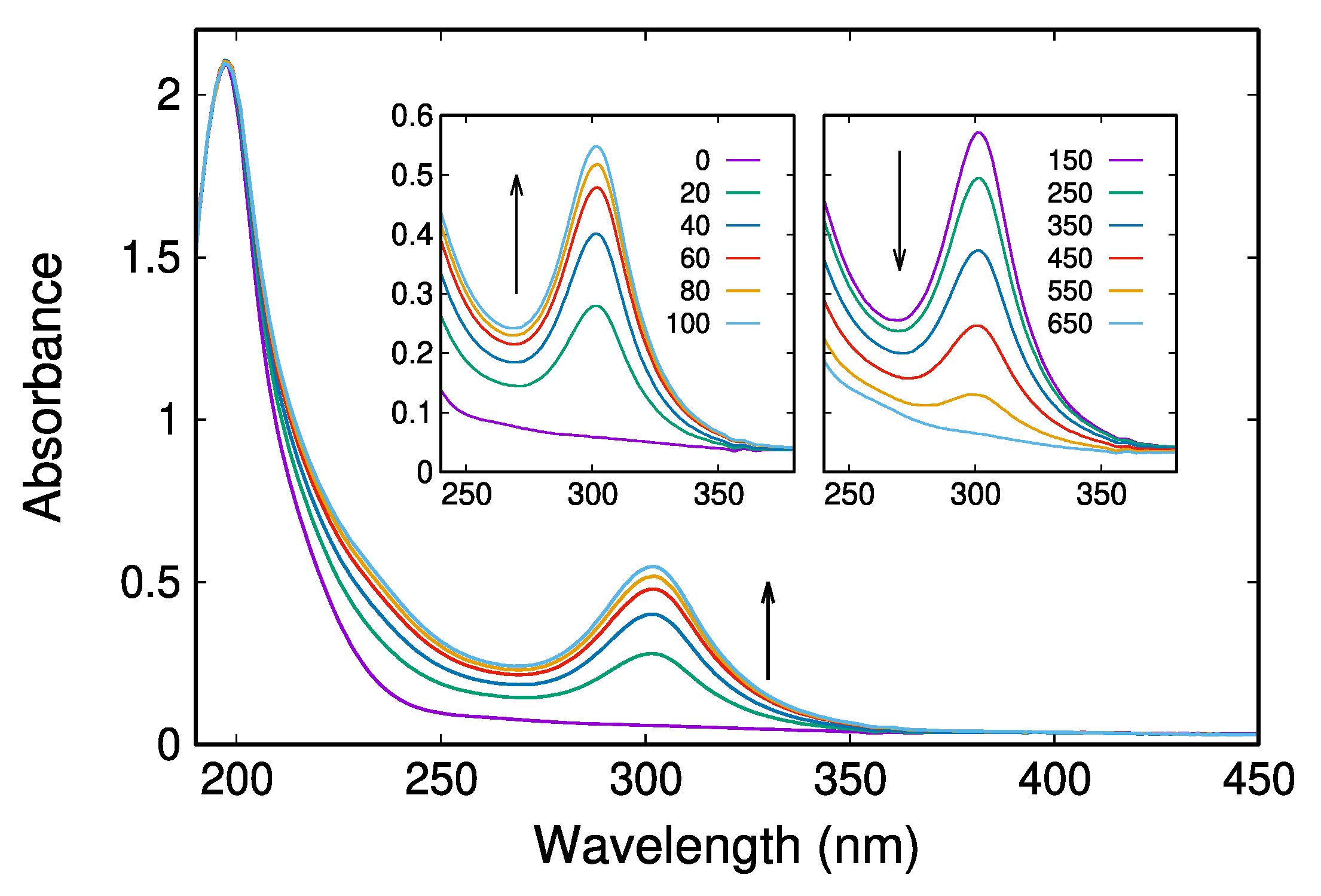
2.2. UV/Vis Absorption Spectra Reveal a Transient Characterized by an Absorption Maximum at 302 nm
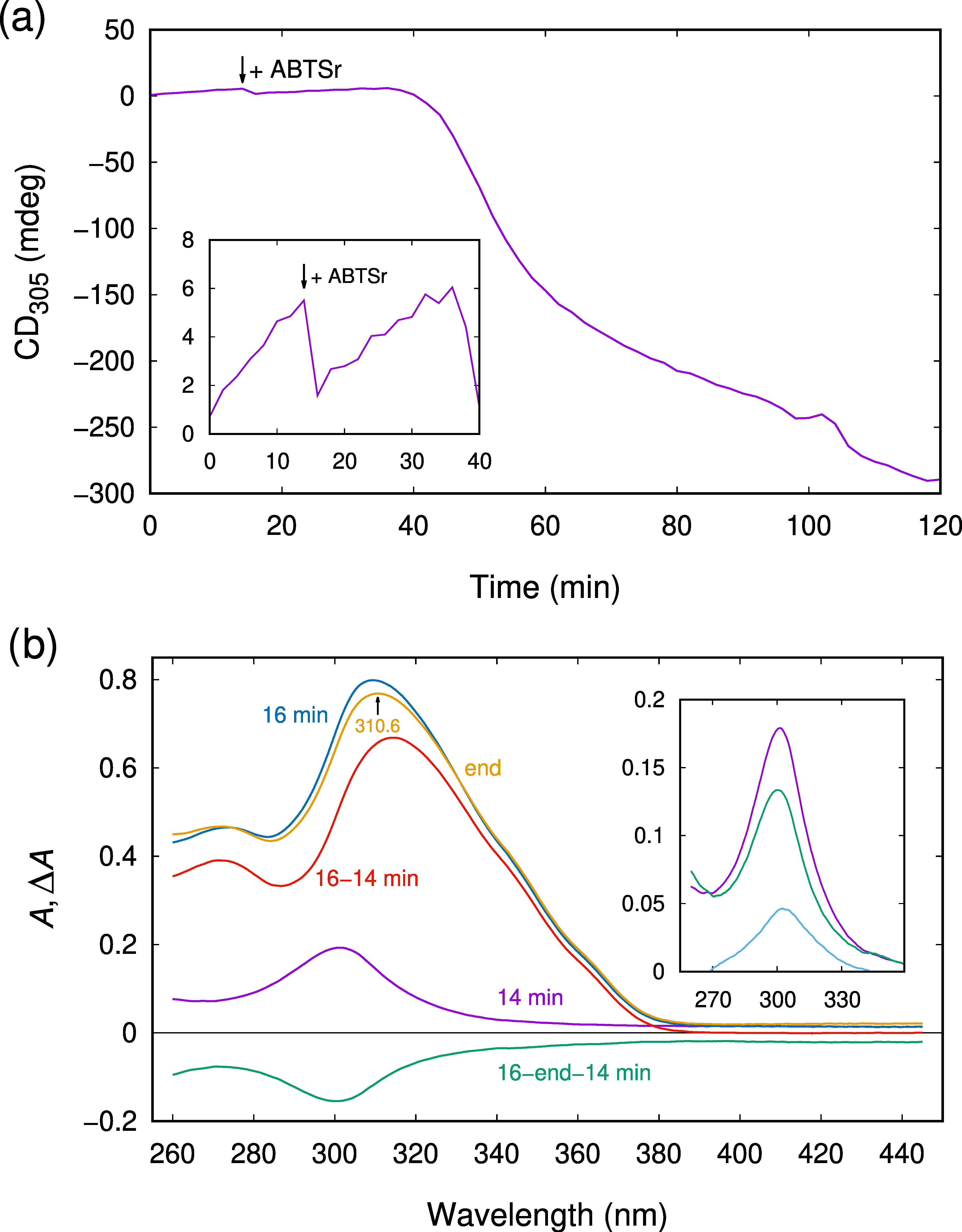
2.3. CD Spectroscopy Reveals Another Transient Showing Sigmoidal Production Kinetics
2.4. The Observed Intermediates React Rapidly with ABTSr
3. Discussion
3.1. Interpretation of the Absorption around 300 nm
3.2. Interpretation of the CD Spectra and Proposed Structures of the Intermediates
3.3. The Proposed Intermediates as Oxidation Catalysts
3.4. Mechanistic Considerations

3.5. An Alternative: Cu(I)–Thiolates as Catalytic Intermediates
4. Materials and Methods
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pfaff, A.R.; Beltz, J.; King, E.; Ercal, N. Medicinal thiols: Current status and new perspectives. Mini-Rev. Med. Chem. 2020, 20, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Dhouib, I.E.; Jallouli, M.; Annabi, A.; Gharbi, N.; Elfazaa, S.; Lasram, M.M. A minireview on N-acetylcysteine: An old drug with new approaches. Life Sci. 2016, 151, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lou, J.R.; Zhang, X.-X.; Benbrook, D.M.; Hanigan, M.H.; Lind, S.E.; Ding, W.-Q. N-Acetylcysteine interacts with copper to generate hydrogen peroxide and selectively induce cancer cell death. Cancer Lett. 2010, 298, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Valachova, K.; Juranek, I.; Rapta, P.; Valent, I.; Soltes, L. On infusion of high-dose ascorbate in treating cancer: Is it time for N-acetylcysteine pretreatment to enhance susceptibility and to lower side effects? Med. Hypotheses 2019, 122, 8–9. [Google Scholar] [CrossRef]
- Mlejnek, P.; Dolezel, P.; Maier, V.; Kikalova, K.; Skoupa, N. N-Acetylcysteine dual and antagonistic effect on cadmium cytotoxicity in human leukemia cells. Environ. Toxicol. Pharmacol. 2019, 71, 103213. [Google Scholar] [CrossRef]
- Amini, A.; Masoumi-Moghaddam, S.; Morris, D.L. Utility of Bromelain and N-Acetylcysteine in Treatment of Peritoneal Dissemination of Gastrointestinal Mucin-Producing Malignancies; Springer International Publishing: Cham, Switzerland, 2016; ISBN 9783319285689. [Google Scholar]
- Pedre, B.; Barayeu, U.; Ezerina, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Aruoma, O.; Halliwell, B.; Hoey, B.; Butler, J. The antioxidant action of N-acetylcysteine—Its reaction with hydrogen-peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Ezerina, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular HS and sulfane sulfur production. Cell Chem. Biol. 2018, 25, 447. [Google Scholar] [CrossRef]
- Sprong, R.; Winkelhuyzen-Janssen, A.; Aarsman, C.; van Oirschot, J.; van der Bruggen, T.; van Asbeck, B. Low-dose N-acetylcysteine protects rats against endotoxin-mediated oxidative stress, but high-dose increases mortality. Am. J. Respir. Crit. Care Med. 1998, 157, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Yamada, K.; Yamashita, N.; Tada-Oikawa, S.; Kawanishi, S. N-acetylcysteine, a cancer chemopreventive agent, causes oxidative damage to cellular and isolated DNA. Carcinogenesis 1999, 20, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.; Jacobs, C.; Kaminski, T.; Halliwell, B.; Leeuwenburgh, C. Supplementation with vitamin C and N-acetyl-cysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic. Biol. Med. 2001, 31, 745–753. [Google Scholar] [CrossRef]
- Sagrista, M.; Garcia, A.; De Madariaga, M.; Mora, M. Antioxidant and pro-oxidant effect of the thiolic compounds N-acetyl-l-cysteine and glutathione against free radical-induced lipid peroxidation. Free Radic. Res. 2002, 36, 329–340. [Google Scholar] [CrossRef]
- Saez, G.; Thornalley, P.; Hill, H.; Hems, R.; Bannister, J. The production of free-radicals during the autoxidation of cysteine and their effect on isolated rat hepatocytes. Biochim. Biophys. Acta 1982, 719, 24–31. [Google Scholar] [CrossRef]
- Vansteveninck, J.; Vanderzee, J.; Dubbelman, T. Site-specific and bulk-phase generation of hydroxyl radicals in the presence of cupric ions and thiol compounds. Biochem. J. 1985, 232, 309–311. [Google Scholar] [CrossRef]
- Kachur, A.; Koch, C.; Biaglow, J. Mechanism of copper-catalyzed autoxidation of cysteine. Free Radic. Res. 1999, 31, 23–34. [Google Scholar] [CrossRef]
- Burkitt, M. A critical overview of the chemistry of copper-dependent low density lipoprotein oxidation: Roles of lipid hydroperoxides, alpha-tocopherol, thiols, and ceruloplasmin. Arch. Biochem. Biophys. 2001, 394, 117–135. [Google Scholar] [CrossRef]
- Wu, M.-S.; Lien, G.-S.; Shen, S.-C.; Yang, L.-Y.; Chen, Y.-C. N-acetyl-l-cysteine enhances fisetin-induced cytotoxicity via induction of ROS-independent apoptosis in human colonic cancer cells. Mol. Carcinog. 2014, 53, E119–E129. [Google Scholar] [CrossRef]
- Qanungo, S.; Wang, M.; Nieminen, A. N-acetyl-L-cysteine enhances apoptosis through inhibition of nuclear factor-kappa B in hypoxic murine embryonic fibroblasts. J. Biol. Chem. 2004, 279, 50455–50464. [Google Scholar] [CrossRef]
- Rakshit, S.; Bagchi, J.; Mandal, L.; Paul, K.; Ganguly, D.; Bhattacharjee, S.; Ghosh, M.; Biswas, N.; Chaudhuri, U.; Bandyopadhyay, S. N-Acetyl cysteine enhances imatinib-induced apoptosis of Bcr-Abl(+) cells by endothelial nitric oxide synthase-mediated production of nitric oxide. Apoptosis 2009, 14, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Andreou, A.; Trantza, S.; Filippou, D.; Sipsas, N.; Tsiodras, S. COVID-19: The potential role of copper and N-acetylcysteine (NAC) in a combination of candidate antiviral treatments against SARS-COV-2. In Vivo 2020, 34, 1567–1588. [Google Scholar] [CrossRef] [PubMed]
- Mathews, A.; Walker, S. The action of metals and strong salt solutions on the spontaneous oxidation of cystein. J. Biol. Chem. 1909, 6, 299–312. [Google Scholar] [CrossRef]
- Harrison, D. The catalytic action of traces of iron and copper on the anaerobic oxidation of sulphydryl compounds. Biochem. J. 1927, 21, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Stricks, W.; Kolthoff, I. Polarographic investigations of reactions in aqueous solutions containing copper and cysteine (cystine). 1. Cuprous copper and cysteine in ammoniacal medium—The dissociation constant of cuprous cysteinate. J. Am. Chem. Soc. 1951, 73, 1723–1727. [Google Scholar] [CrossRef]
- Klotz, I.; Czerlinski, G.; Fiess, H. A mixed-valence copper complex with thiol compounds. J. Am. Chem. Soc. 1958, 80, 2920–2923. [Google Scholar] [CrossRef]
- Hemmerich, P.; Beinert, H.; Vanngard, T. Complex formation between copper and organic disulfides. Angew. Chem. Int. Ed. 1966, 5, 422–423. [Google Scholar] [CrossRef]
- Cavallini, D.; Demarco, C.; Dupre, S.; Rotilio, G. Copper catalyzed oxidation of cysteine to cystine. Arch. Biochem. Biophys. 1969, 130, 354–361. [Google Scholar] [CrossRef]
- Peisach, J.; Blumberg, W. A mechanism for action of penicillamine in treatment of Wilsons disease. Mol. Pharmacol. 1969, 5, 200–209. [Google Scholar]
- Hanaki, A.; Kamide, H. Autoxidation of cysteine catalyzed by copper in glycylglycine buffer. Chem. Pharm. Bull. 1975, 23, 1671–1676. [Google Scholar] [CrossRef]
- Vortisch, V.; Kroneck, P.; Hemmerich, P. Model studies on coordination of copper in enzymes. 4. Structure and stability of cuprous complexes with sulfur-containing ligands. J. Am. Chem. Soc. 1976, 98, 2821–2826. [Google Scholar] [CrossRef]
- Birker, P.; Freeman, H. Structure, properties, and function of a copper(I)-copper(II) complex of D-penicillamine–pentathallium(I) μ8-chloro-dodeca(D-penicillaminato)-octacuprate(I)hexacuprate(II) normal-hydrate. J. Am. Chem. Soc. 1977, 99, 6890–6899. [Google Scholar] [CrossRef]
- Lappin, A.; Mcauley, A. Reactions between copper(II) and 2-mercaptosuccinic acid in aqueous perchlorate solution. J. Chem. Soc.-Dalton Trans. 1978, 1606–1609. [Google Scholar] [CrossRef]
- Zwart, J.; Vanwolput, J.; Koningsberger, D. The mechanism of the copper-ion catalyzed autoxidation of cysteine in alkaline-medium. J. Mol. Catal. 1981, 12, 85–101. [Google Scholar] [CrossRef]
- Pecci, L.; Montefoschi, G.; Musci, G.; Cavallini, D. Novel findings on the copper catalysed oxidation of cysteine. Amino Acids 1997, 13, 355–367. [Google Scholar] [CrossRef]
- Gilbert, B.; Silvester, S.; Walton, P. Spectroscopic, kinetic and mechanistic studies of the influence of ligand and substrate concentration on the activation by peroxides of CuI-thiolate and other CuI complexes. J. Chem. Soc.-Perkin Trans. 2 1999, 1115–1121. [Google Scholar] [CrossRef]
- Itoh, S.; Nagagawa, M.; Fukuzumi, S. Fine tuning of the interaction between the copper(I) and disulfide bond. Formation of a bis(mu-thiolato)dicopper(II) complex by reductive cleavage of the disulfide bond with copper(I). J. Am. Chem. Soc. 2001, 123, 4087–4088. [Google Scholar] [CrossRef]
- Rigo, A.; Corazza, A.; di Paolo, M.; Rossetto, M.; Ugolini, R.; Scarpa, M. Interaction of copper with cysteine: Stability of cuprous complexes and catalytic role of cupric ions in anaerobic thiol oxidation. J. Inorg. Biochem. 2004, 98, 1495–1501. [Google Scholar] [CrossRef]
- Seko, H.; Tsuge, K.; Igashira-Kamiyama, A.; Kawamoto, T.; Konno, T. Autoxidation of thiol-containing amino acid to its disulfide derivative that links two copper(II) centers: The important role of auxiliary ligand. Chem. Commun. 2010, 46, 1962–1964. [Google Scholar] [CrossRef]
- Ording-Wenker, E.C.M.; Siegler, M.A.; Lutz, M.; Bouwman, E. Catalytic catechol oxidation by copper complexes: Development of a structure-activity relationship. Dalton Trans. 2015, 44, 12196–12209. [Google Scholar] [CrossRef]
- Buzuk, M.; Brinic, S.; Vladislavic, N.; Bralic, M.; Buljac, M.; Roncevic, I.S. Real-time monitoring of “self-oxidation” of cysteine in presence of Cu2+: Novel findings in the oxidation mechanism. Mon. Chem. 2016, 147, 359–367. [Google Scholar] [CrossRef]
- Ngamchuea, K.; Batchelor-McAuley, C.; Compton, R.G. The copper(II)-catalyzed oxidation of glutathione. Chem.-Eur. J. 2016, 22, 15937–15944. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Maia, L.B.; Moro, A.J.; Lima, J.C.; Cordas, C.M.; Moura, I.; Mourer, J.J.G. Unusual reduction mechanism of copper in cysteine-rich environment. Inorg. Chem. 2018, 57, 8078–8088. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.W.; Jones, Z.R.; Wu, G.; Teat, S.J.; Scott, S.L.; Hayton, T.W. Synthesis and characterization of “atlas-sphere” copper nanoclusters: New insights into the reaction of Cu2+ with thiols. Inorg. Chem. 2019, 58, 8739–8749. [Google Scholar] [CrossRef]
- Hu, Z.-E.; Li, J.; Wu, Z.-N.; Wei, Y.-J.; Liu, Y.-H.; Wang, N.; Yu, X.-Q. One-pot synthesis-biocompatible copper-tripeptide complex as a nanocatalytic medicine to enhance chemodynamic therapy. ACS Biomater. Sci. Eng. 2021, 7, 1394–1402. [Google Scholar] [CrossRef]
- Ehrenberg, L.; Harmsringdahl, M.; Fedorcsak, I.; Granath, F. Kinetics of the copper-catalyzed and iron-catalyzed oxidation of cysteine by dioxygen. Acta Chem. Scand. 1989, 43, 177–187. [Google Scholar] [CrossRef]
- Zanzen, U.; Bovenkamp-Langlois, L.; Klysubun, W.; Hormes, J.; Prange, A. The interaction of copper ions with Staphylococcus aureus, Pseudomonas aeruginosa, and Escherichia coli: An X-ray absorption near-edge structure (XANES) spectroscopy study. Arch. Microbiol. 2018, 200, 401–412. [Google Scholar] [CrossRef]
- Macomber, L.; Rensing, C.; Imlay, J.A. Intracellular copper does not catalyze the formation of oxidative DNA damage in Escherichia coli. J. Bacteriol. 2007, 189, 1616–1626. [Google Scholar] [CrossRef]
- Milne, L.; Nicotera, P.; Orrenius, S.; Burkitt, M. Effects of glutathione and chelating-agents on copper-mediated DNA oxidation—Prooxidant and antioxidant properties of glutathione. Arch. Biochem. Biophys. 1993, 304, 102–109. [Google Scholar] [CrossRef]
- Pham, A.N.; Xing, G.; Miller, C.J.; Waite, T.D. Fenton-like copper redox chemistry revisited: Hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J. Catal. 2013, 301, 54–64. [Google Scholar] [CrossRef]
- Valent, I.; Topolska, D.; Valachova, K.; Bujdak, J.; Soltes, L. Kinetics of ABTS derived radical cation scavenging by bucillamine, cysteine, and glutathione. Catalytic effect of Cu2+ ions. Biophys. Chem. 2016, 212, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y. Asymmetric photochemical-reactions in solution. Chem. Rev. 1992, 92, 741–770. [Google Scholar] [CrossRef]
- Harada, N.; Nakanishi, K. Circular Dichroic Spectroscopy: Exciton Coupling in Organic Stereochemistry; University Science Books: Mill Valley, CA, USA, 1983. [Google Scholar]
- Scott, S.; Chen, W.; Bakac, A.; Espenson, J. Spectroscopic parameters, electrode-potentials, acid ionization-constants, and electron-exchange rates of the 2,2’-azinobis(3-ethylbenzothiazoline-6-sulfonate) radicals and ions. J. Phys. Chem. 1993, 97, 6710–6714. [Google Scholar] [CrossRef]
- Packer, J. The radiation chemistry of thiols. In The Chemistry of the Thiol Group, Part 2; Patai, S., Ed.; Wiley: London, UK, 1974; pp. 481–517. [Google Scholar]
- Bagiyan, G.; Koroleva, I.; Soroka, N. Formation conditions of mononuclear copper(I) complexes with aminothiols. Zhurnal Neorg. Khimii 1978, 23, 416–424. [Google Scholar]
- Hanaki, A. Interaction of biologically relevant Cu(II)-peptide and cysteine—Transient complexes with Cu(II)N3S and Cu(II)N2S2 chromophores. Chem. Lett. 1980, 9, 629–630. [Google Scholar] [CrossRef]
- Mezyk, S.; Armstrong, D. Kinetics of oxidation of Cu(I) complexes of cysteine and penicillamine—Nature of intermediates and reactants at pH 10.0. Can. J. Chem.-Rev. Can. Chim. 1989, 67, 736–745. [Google Scholar] [CrossRef]
- Wilson, E.; Martin, R. Penicillamine deprotonations and interactions with copper ions. Arch. Biochem. Biophys. 1971, 142, 445–454. [Google Scholar] [CrossRef]
- Demarco, C.; Dupre, S.; Crifo, C.; Rotilio, G.; Cavallini, D. Copper-catalyzed oxidation of thiomalic acid. Arch. Biochem. Biophys. 1971, 144, 496–502. [Google Scholar] [CrossRef]
- Hanaki, A.; Funahashi, Y.; Odani, A. Ternary Cu(II) complexes, Cu(H-1L)(ACys(-)) and Cu(H-2L)(ACys(-)); L = peptides, ACys(-) = N-acetyl-cysteinate. Analogous complexes to the intermediates in the transport of Cu(II) from Cu(H-2L) to cysteine. J. Inorg. Biochem. 2006, 100, 305–315. [Google Scholar] [CrossRef]
- Sharma, R.; Pal, M.; Mishra, K.K. Entropy-controlled Cu(II)-catalyzed oxidation of N-acetyl-l-cysteine by methylene blue in acidic medium. Z. Phys. Chem. 2017, 231, 1093–1109. [Google Scholar] [CrossRef]
- Morgan, M.T.; Nguyen, L.A.H.; Hancock, H.L.; Fahrni, C.J. Glutathione limits aquacopper(I) to sub-femtomolar concentrations through cooperative assembly of a tetranuclear cluster. J. Biol. Chem. 2017, 292, 21558–21567. [Google Scholar] [CrossRef] [PubMed]
- Amundsen, A.; Whelan, J.; Bosnich, B. Biological analogs—Nature of binding-sites of copper-containing proteins. J. Am. Chem. Soc. 1977, 99, 6730–6739. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I. Spectroscopic methods in bioinorganic chemistry: Blue to green to red copper sites. Inorg. Chem. 2006, 45, 8012–8025. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Yokoyama, A.; Tanaka, H. Studies on the sulfur-containing chelating agents. XXIX. Reaction of cysteamine and its related compounds with copper. Chem. Pharm. Bull. 1970, 18, 2373–2385. [Google Scholar] [CrossRef][Green Version]
- Graf, L.; Fallab, S. Zum reaktionsmechanismus von oxydasen. Experientia 1964, 20, 46–47. [Google Scholar] [CrossRef]
- Blumberg, W.; Peisach, J. Bis(thiosemicarbazone) and other nitrogen and sulfur ligated complexes of copper(2). J. Chem. Phys. 1968, 49, 1793–1802. [Google Scholar] [CrossRef]
- Bagiyan, G.; Koroleva, I.; Soroka, N.; Ufimtsev, A. Kinetics of the catalytic oxidation reactions of thiol compounds in aqueous solutions in the presence of copper ions. Kinet. Catal. 2004, 45, 372–380. [Google Scholar] [CrossRef]
- Davis, F.; Gilbert, B.; Norman, R.; Symons, M. Electron-spin resonance studies. 66. Characterization of copper(II) complexes in the oxidation of D-penicillamine, L-cysteine, and related sulfur-containing-compounds. J. Chem. Soc.-Perkin Trans. 2 1983, 1763–1771. [Google Scholar] [CrossRef]
- Hanaki, A. Kinetic-studies on the role of dioxygen in the copper-catalyzed autoxidation of cysteine. Bull. Chem. Soc. Jpn. 1995, 68, 831–837. [Google Scholar] [CrossRef]
- Eigen, M.; Wilkins, R. Mechanisms of inorganic reactions. Adv. Chem. Ser. 1965, 49, 55. [Google Scholar]
- Pasquarello, A.; Petri, I.; Salmon, P.; Parisel, O.; Car, R.; Toth, E.; Powell, D.; Fischer, H.; Helm, L.; Merbach, A. First solvation shell of the Cu(II) aqua ion: Evidence for fivefold coordination. Science 2001, 291, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Ottersen, T.; Warner, L.; Seff, K. Synthesis and crystal-structure of a dimeric cyclic copper(I)-aliphatic disulfide complex—Cyclo-di-mu-bis[2-(N,N-dimethylamino)ethyl] disulfide-dicopper(I) tetrafluoroborate. Inorg. Chem. 1974, 13, 1904–1911. [Google Scholar] [CrossRef]
- Warner, L.; Ottersen, T.; Seff, K. Synthesis and crystal-structure of a polymeric copper(I) aliphatic disulfide complex—(bis[2-(2-pyridyl)ethyl] disulfide)copper(I) perchlorate. Inorg. Chem. 1974, 13, 2819–2826. [Google Scholar] [CrossRef]
- Ohta, T.; Tachiyama, T.; Yoshizawa, K.; Yamabe, T.; Uchida, T.; Kitagawa, T. Synthesis, structure, and H2O2-dependent catalytic functions of disulfide-bridged dicopper(I) and related thioether-copper(I) and thioether-copper(II) complexes. Inorg. Chem. 2000, 39, 4358–4369. [Google Scholar] [CrossRef] [PubMed]
- Neuba, A.; Haase, R.; Meyer-Klaucke, W.; Floerke, U.; Henkel, G. A halide-induced copper(I) disulfide/copper(II) thiolate interconversion. Angew. Chem. Int. Ed. 2012, 51, 1714–1718. [Google Scholar] [CrossRef] [PubMed]
- Ording-Wenker, E.C.M.; van der Plas, M.; Siegler, M.A.; Bonnet, S.; Bickelhaupt, F.M.; Guerra, C.F.; Bouwman, E. Thermodynamics of the CuII μ-thiolate and CuI disulfide equilibrium: A combined experimental and theoretical study. Inorg. Chem. 2014, 53, 8494–8504. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Hatcher, L.Q.; Vance, M.A.; Sarangi, R.; Milligan, A.E.; Sarjeant, A.A.N.; Incarvito, C.D.; Rheingold, A.L.; Hodgson, K.O.; Hedman, B.; et al. Copper(I) complex O2-reactivity with a N3S thioether ligand: A copper-dioxygen adduct including sulfur ligation, ligand oxygenation, and comparisons with all nitrogen ligand analogues. Inorg. Chem. 2007, 46, 6056–6068. [Google Scholar] [CrossRef]
- Eickman, N.; Himmelwright, R.; Solomon, E. Geometric and electronic-structure of oxyhemocyanin—Spectral and chemical correlations to met apo, half met, met, and dimer active-sites. Proc. Natl. Acad. Sci. USA 1979, 76, 2094–2098. [Google Scholar] [CrossRef]
- Panijpan, B. Chirality of disulfide bond in biomolecules. J. Chem. Educ. 1977, 54, 670–672. [Google Scholar] [CrossRef]
- Kuhn, W. The physical significance of optical rotatory power. Trans. Faraday Soc. 1930, 26, 0293–0307. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Yokojima, S.; Fukaminato, T.; Shiino, K.; Irie, M.; Nakamura, S. Anisotropic dissymmetry factor, g: Theoretical investigation on single molecule chiroptical spectroscopy. J. Phys. Chem. A 2014, 118, 5046–5057. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.; Tunnicliffe, H. The oxidation of reduced glutathione and other sulphydryl compounds. Proc. R. Soc. Lond. Ser. B 1923, 94, 266–297. [Google Scholar] [CrossRef]
- Cavallini, D.; Demarco, C.; Dupre, S. Luminol chemiluminescence studies of oxidation of cysteine and other thiols to disulfides. Arch. Biochem. Biophys. 1968, 124, 18–26. [Google Scholar] [CrossRef]
- Elwell, C.E.; Gagnon, N.L.; Neisen, B.D.; Dhar, D.; Spaeth, A.D.; Yee, G.M.; Tolman, W.B. Copper-oxygen complexes revisited: Structures, spectroscopy, and reactivity. Chem. Rev. 2017, 117, 2059–2107. [Google Scholar] [CrossRef]
- Koval, I.A.; Gamez, P.; Belle, C.; Selmeczi, K.; Reedijk, J. Synthetic models of the active site of catechol oxidase: Mechanistic studies. Chem. Soc. Rev. 2006, 35, 814–840. [Google Scholar] [CrossRef] [PubMed]
- Karlin, K.D.; Itoh, S.; Rokita, S. Copper-Oxygen Chemistry; Wiley Series of Reactive Intermediates in Chemistry and Biology; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper active sites in biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [PubMed]
- Kroneck, P.M.H. Walking the seven lines: Binuclear copper A in cytochrome c oxidase and nitrous oxide reductase. J. Biol. Inorg. Chem. 2018, 23, 27–39. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, D.-H.; Sarjeant, A.A.N.; Karlin, K.D. Thiol-copper(I) and disulfide-dicopper(I) complex O2-reactivity leading to sulfonate-copper(II) complex or the formation of a cross-linked thioether-phenol product with phenol addition. J. Inorg. Biochem. 2007, 101, 1845–1858. [Google Scholar] [CrossRef]
- Yamauchi, O.; Seki, H. Dioxygen absorption by a 2-1 copper(II)-disulfide system—Partial incorporation of oxygen-atoms from dioxygen upon copper(II)-catalyzed disulfide bond-cleavage to sulfinate. Chem. Lett. 1982, 11, 1241–1244. [Google Scholar] [CrossRef]
- Karlin, K.; Kaderli, S.; Zuberbuhler, A. Kinetics and thermodynamics of copper(I)/dioxygen interaction. Acc. Chem. Res. 1997, 30, 139–147. [Google Scholar] [CrossRef]
- Naumova, M.; Khakhulin, D.; Rebarz, M.; Rohrmueller, M.; Dicke, B.; Biednov, M.; Britz, A.; Espinoza, S.; Grimm-Lebsanft, B.; Kloz, M.; et al. Structural dynamics upon photoexcitation-induced charge transfer in a dicopper(I)-disulfide complex. Phys. Chem. Chem. Phys. 2018, 20, 6274–6286. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.; Sundaram, U.; Machonkin, T. Multicopper oxidases and oxygenases. Chem. Rev. 1996, 96, 2563–2605. [Google Scholar] [CrossRef]
- Solomon, E.I.; Augustine, A.J.; Yoon, J. O2 Reduction to H2O by the multicopper oxidases. Dalton Trans. 2008, 30, 3921–3932. [Google Scholar] [CrossRef]
- Casuso, P.; Carrasco, P.; Loinaz, I.; Grande, H.J.; Odriozola, I. Converting drugs into gelators: Supramolecular hydrogels from N-acetyl-l-cysteine and coinage-metal salts. Org. Biomol. Chem. 2010, 8, 5455–5458. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Tachi, Y.; Itoh, S. Interconversion between bis(μ-thiolato)dicopper(II) and disulfide-bridged dicopper(I) complexes mediated by chloride ion. J. Am. Chem. Soc. 2002, 124, 12428–12429. [Google Scholar] [CrossRef]
- Thomas, A.M.; Lin, B.-L.; Wasinger, E.C.; Stack, T.D.P. Ligand noninnocence of thiolate/disulfide in dinuclear copper complexes: Solvent-dependent redox isomerization and proton-coupled electron transfer. J. Am. Chem. Soc. 2013, 135, 18912–18919. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Notari, R. Kinetics and mechanism of captopril oxidation in aqueous-solution under controlled oxygen partial-pressure. Pharm. Res. 1987, 4, 98–103. [Google Scholar] [CrossRef]
- Bagiyan, G.; Koroleva, I.; Soroka, N.; Ufimtsev, A. Oxidation of thiol compounds by molecular oxygen in aqueous solutions. Russ. Chem. Bull. 2003, 52, 1135–1141. [Google Scholar] [CrossRef]
- Kachur, A.; Koch, C.; Biaglow, J. Mechanism of copper-catalyzed oxidation of glutathione. Free Radic. Res. 1998, 28, 259–269. [Google Scholar] [CrossRef]
- Soorkia, S.; Dehon, C.; Kumar, S.S.; Pedrazzani, M.; Frantzen, E.; Lucas, B.; Barat, M.; Fayeton, J.A.; Jouvet, C. UV photofragmentation dynamics of protonated cystine: Disulfide bond rupture. J. Phys. Chem. Lett. 2014, 5, 1110–1116. [Google Scholar] [CrossRef]
- Dance, I. Formation and X-ray structure of hexa(tert-butylthiolato)pentacuprate(I) monoanion. J. Chem. Soc.-Chem. Commun. 1976, 68–69. [Google Scholar] [CrossRef]
- Blower, P.; Dilworth, J. Thiolato-complexes of the transition-metals. Coord. Chem. Rev. 1987, 76, 121–185. [Google Scholar] [CrossRef]
- Fujisawa, K.; Imai, S.; Kitajima, N.; Moro-oka, Y. Preparation, spectroscopic, characterization, and molecular structure of copper(I) aliphatic thiolate complexes. Inorg. Chem. 1998, 37, 168–169. [Google Scholar] [CrossRef]
- Zeevi, S.; Tshuva, E.Y. Synthesis and X-ray characterization of mono- and polynuclear thiolatocopper(I) complexes: The effect of steric bulk on coordination number and nuclearity. Eur. J. Inorg. Chem. 2007, 2007, 5369–5376. [Google Scholar] [CrossRef]
- Ferrara, S.J.; Mague, J.T.; Donahue, J.P. Synthesis and structures of cuprous triptycylthiolate complexes. Inorg. Chem. 2012, 51, 6567–6576. [Google Scholar] [CrossRef] [PubMed]
- Yam, V.; Lam, C.; Fung, W.; Cheung, K. Syntheses, photophysics, and photochemistry of trinuclear copper(I) thiolate and hexanuclear copper(I) selenolate complexes: X-ray crystal structures of [Cu6(µ-dppm)4(µ3-SePh)4](BF4)2 and [Cu6{µ-(Ph2P)2NH}4(µ3-SePh)4](BF4)2. Inorg. Chem. 2001, 40, 3435–3442. [Google Scholar] [CrossRef]
- Butler, I.; Schoonen, M.; Rickard, D. Removal of dissolved-oxygen from water—A comparison of 4 common techniques. Talanta 1994, 41, 211–215. [Google Scholar] [CrossRef]
- Kizek, R.; Vacek, J.; Trnkova, L.; Jelen, F. Cyclic voltammetric study of the redox system of glutathione using the disulfide bond reductant tris(2-carboxyethyl)phosphine. Bioelectrochemistry 2004, 63, 19–24. [Google Scholar] [CrossRef]
- Pountney, D.; Schauwecker, I.; Zarn, J.; Vasak, M. Formation of mammalian Cu8-metallothionein in vitro: Evidence for the existence of 2 Cu(I)4-thiolate clusters. Biochemistry 1994, 33, 9699–9705. [Google Scholar] [CrossRef]
- Tarasava, K.; Loebus, J.; Freisinger, E. Localization and spectroscopic analysis of the Cu(I) binding site in wheat metallothionein Ec-1. Int. J. Mol. Sci. 2016, 17, 371. [Google Scholar] [CrossRef]
- Ford, P.; Vogler, A. Photochemical and photophysical properties of tetranuclear and hexanuclear clusters of metals with d10 and s2 electronic configurations. Acc. Chem. Res. 1993, 26, 220–226. [Google Scholar] [CrossRef]
- Artells, E.; Palacios, O.; Capdevila, M.; Atrian, S. In vivo-folded metal-metallothionein 3 complexes reveal the Cu-thionein rather than Zn-thionein character of this brain-specific mammalian metallothionein. FEBS J. 2014, 281, 1659–1678. [Google Scholar] [CrossRef]
- Scheller, J.S.; Irvine, G.W.; Wong, D.L.; Hartwig, A.; Stillman, M.J. Stepwise copper(I) binding to metallothionein: A mixed cooperative and non-cooperative mechanism for all 20 copper ions. Metallomics 2017, 9, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Casas-Finet, J.; Hu, S.; Hamer, D.; Karpel, R. Spectroscopic characterization of the copper(I)-thiolate cluster in the DNA-binding domain of yeast ACE1 transcription factor. FEBS Lett. 1991, 281, 205–208. [Google Scholar] [CrossRef]
- Subedi, P.; Paxman, J.J.; Wang, G.; Ukuwela, A.A.; Xiao, Z.; Heras, B. The Scs disulfide reductase system cooperates with the metallochaperone CueP in Salmonella copper resistance. J. Biol. Chem. 2019, 294, 15876–15888. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.-Y.; Yeom, J.-H.; Kim, J.-S.; Um, S.-H.; Jo, I.; Lee, K.; Kim, Y.-H.; Ha, N.-C. Direct ROS scavenging activity of CueP from Salmonella enterica serovar Typhimurium. Mol. Cells 2014, 37, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Goebl, C.; Morris, V.K.; van Dam, L.; Visscher, M.; Polderman, P.E.; Hartlmueller, C.; de Ruiter, H.; Hora, M.; Liesinger, L.; Birner-Gruenberger, R.; et al. Cysteine oxidation triggers amyloid fibril formation of the tumor suppressor p16INK4A. Redox Biol. 2020, 28, 101316. [Google Scholar] [CrossRef]
- Werner, T.E.R.; Bernson, D.; Esbjoerner, E.K.; Rocha, S.; Wittung-Stafshede, P. Amyloid formation of fish beta-parvalbumin involves primary nucleation triggered by disulfide-bridged protein dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 27997–28004. [Google Scholar] [CrossRef]
- Morgada, M.N.; Abriata, L.A.; Cefaro, C.; Gajda, K.; Banci, L.; Vila, A.J. Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of Cu-A in human cytochrome oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 11771–11776. [Google Scholar] [CrossRef]
- Osterberg, R.; Ligaarden, R.; Persson, D. Copper(I) complexes of penicillamine and glutathione. J. Inorg. Biochem. 1979, 10, 341–355. [Google Scholar] [CrossRef]
- Corazza, A.; Harvey, I.; Sadler, P. H-1,C-13-NMR and X-ray absorption studies of copper(I) glutathione complexes. Eur. J. Biochem. 1996, 236, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Koenigsberger, L.-C.; Koenigsberger, E.; Hefter, G.; May, P.M. Formation constants of copper(I) complexes with cysteine, penicillamine and glutathione: Implications for copper speciation in the human eye. Dalton Trans. 2015, 44, 20413–20425. [Google Scholar] [CrossRef] [PubMed]
- Kroneck, P. Models for electron-paramagnetic resonance nondetectable copper in blue oxidases—Binuclear copper(II) complex with oxidized glutathione. J. Am. Chem. Soc. 1975, 97, 3839–3841. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Sugiura, Y.; Ishizu, K.; Iitaka, Y.; Nakamura, H. Crystal-structure and spectroscopic properties of violet glutathione-copper(II) complex with axial sulfur coordination and 2 copper sites via a disulfide bridge. J. Am. Chem. Soc. 1980, 102, 6130–6136. [Google Scholar] [CrossRef]
- Huet, J.; Jouini, M.; Abello, L.; Lapluye, G. Structural study of copper oligopeptide complexes. 1. Oxidized glutathione Cu(II) system. J. Chim. Phys. - Chim. Biol. 1984, 81, 505–511. [Google Scholar] [CrossRef]
- Pedersen, J.; Steinkuhler, C.; Weser, U.; Rotilio, G. Copper-glutathione complexes under physiological conditions: Structures in solution different from the solid state coordination. Biometals 1996, 9, 3–9. [Google Scholar] [CrossRef]
- Shtyrlin, V.; Zyavkina, Y.; Ilakin, V.; Garipov, R.; Zakharov, A. Structure, stability, and ligand exchange of copper(II) complexes with oxidized glutathione. J. Inorg. Biochem. 2005, 99, 1335–1346. [Google Scholar] [CrossRef]
- Burkitt, M.; Duncan, J. Effects of trans-resveratrol on copper-dependent hydroxyl-radical formation and DNA damage: Evidence for hydroxyl-radical scavenging and a novel, glutathione-sparing mechanism of action. Arch. Biochem. Biophys. 2000, 381, 253–263. [Google Scholar] [CrossRef]
- Hrabarova, E.; Valachova, K.; Rychly, J.; Rapta, P.; Sasinkova, V.; Malikova, M.; Soltes, L. High-molar-mass hyaluronan degradation by Weissberger’s system: Pro- and anti-oxidative effects of some thiol compounds. Polym. Degrad. Stabil. 2009, 94, 1867–1875. [Google Scholar] [CrossRef]
- Aliaga, M.E.; Lopez-Alarcon, C.; Garcia-Rio, L.; Martin-Pastor, M.; Speisky, H. Redox-changes associated with the glutathione-dependent ability of the Cu(II)-GSSG complex to generate superoxide. Bioorg. Med. Chem. 2012, 20, 2869–2876. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valent, I.; Bednárová, L.; Schreiber, I.; Bujdák, J.; Valachová, K.; Šoltés, L. Reaction of N-Acetylcysteine with Cu2+: Appearance of Intermediates with High Free Radical Scavenging Activity: Implications for Anti-/Pro-Oxidant Properties of Thiols. Int. J. Mol. Sci. 2022, 23, 6199. https://doi.org/10.3390/ijms23116199
Valent I, Bednárová L, Schreiber I, Bujdák J, Valachová K, Šoltés L. Reaction of N-Acetylcysteine with Cu2+: Appearance of Intermediates with High Free Radical Scavenging Activity: Implications for Anti-/Pro-Oxidant Properties of Thiols. International Journal of Molecular Sciences. 2022; 23(11):6199. https://doi.org/10.3390/ijms23116199
Chicago/Turabian StyleValent, Ivan, Lucie Bednárová, Igor Schreiber, Juraj Bujdák, Katarína Valachová, and Ladislav Šoltés. 2022. "Reaction of N-Acetylcysteine with Cu2+: Appearance of Intermediates with High Free Radical Scavenging Activity: Implications for Anti-/Pro-Oxidant Properties of Thiols" International Journal of Molecular Sciences 23, no. 11: 6199. https://doi.org/10.3390/ijms23116199
APA StyleValent, I., Bednárová, L., Schreiber, I., Bujdák, J., Valachová, K., & Šoltés, L. (2022). Reaction of N-Acetylcysteine with Cu2+: Appearance of Intermediates with High Free Radical Scavenging Activity: Implications for Anti-/Pro-Oxidant Properties of Thiols. International Journal of Molecular Sciences, 23(11), 6199. https://doi.org/10.3390/ijms23116199