
Generation and Selection of Specific Aptamers Targeting Brucella Species through an Enhanced Cell-SELEX Methodology

Abstract
:1. Introduction
2. Results
2.1. PCR Amplification and ssDNA Generation
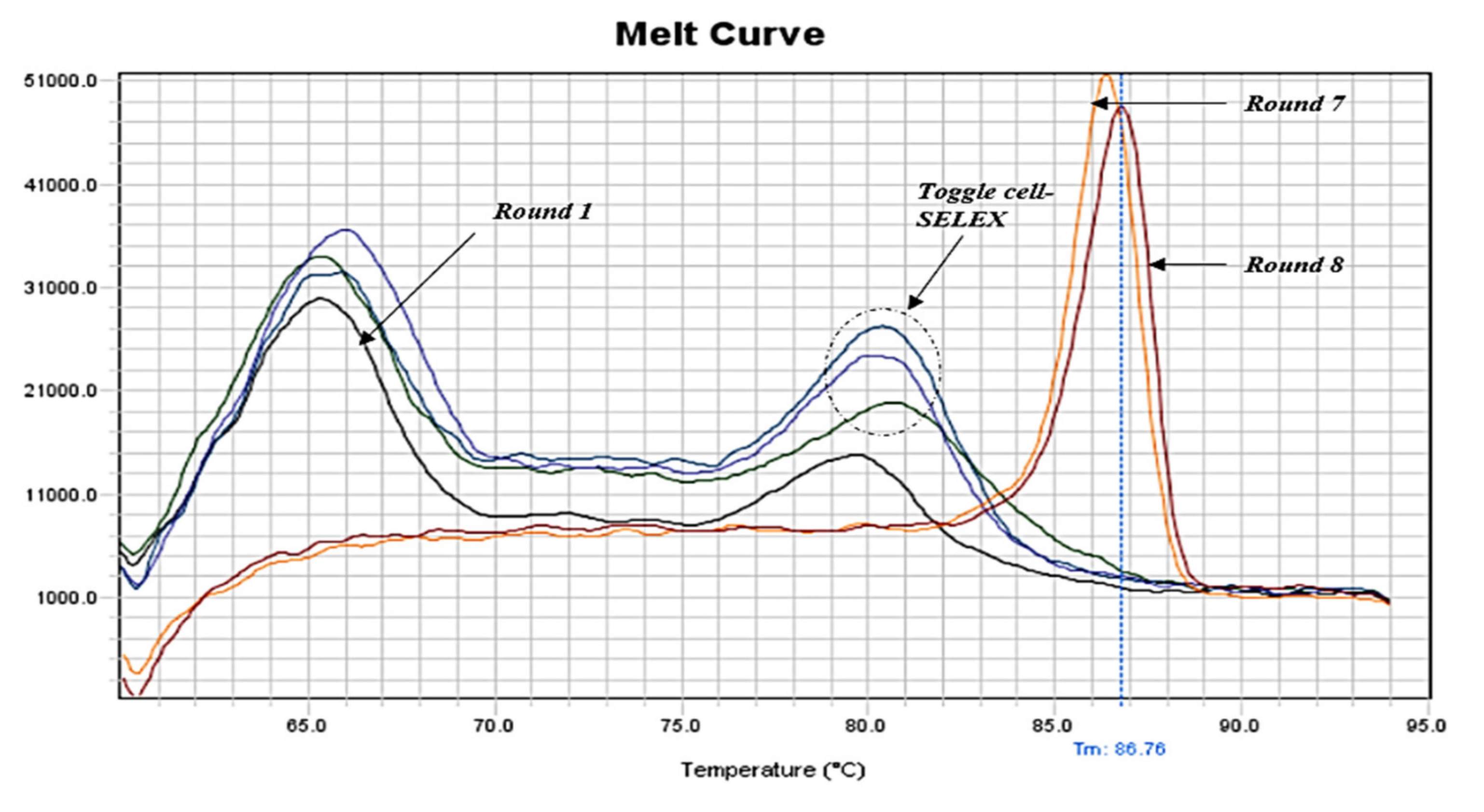
2.2. qPCR Monitoring of Cell-SELEX and Enrichment Determination
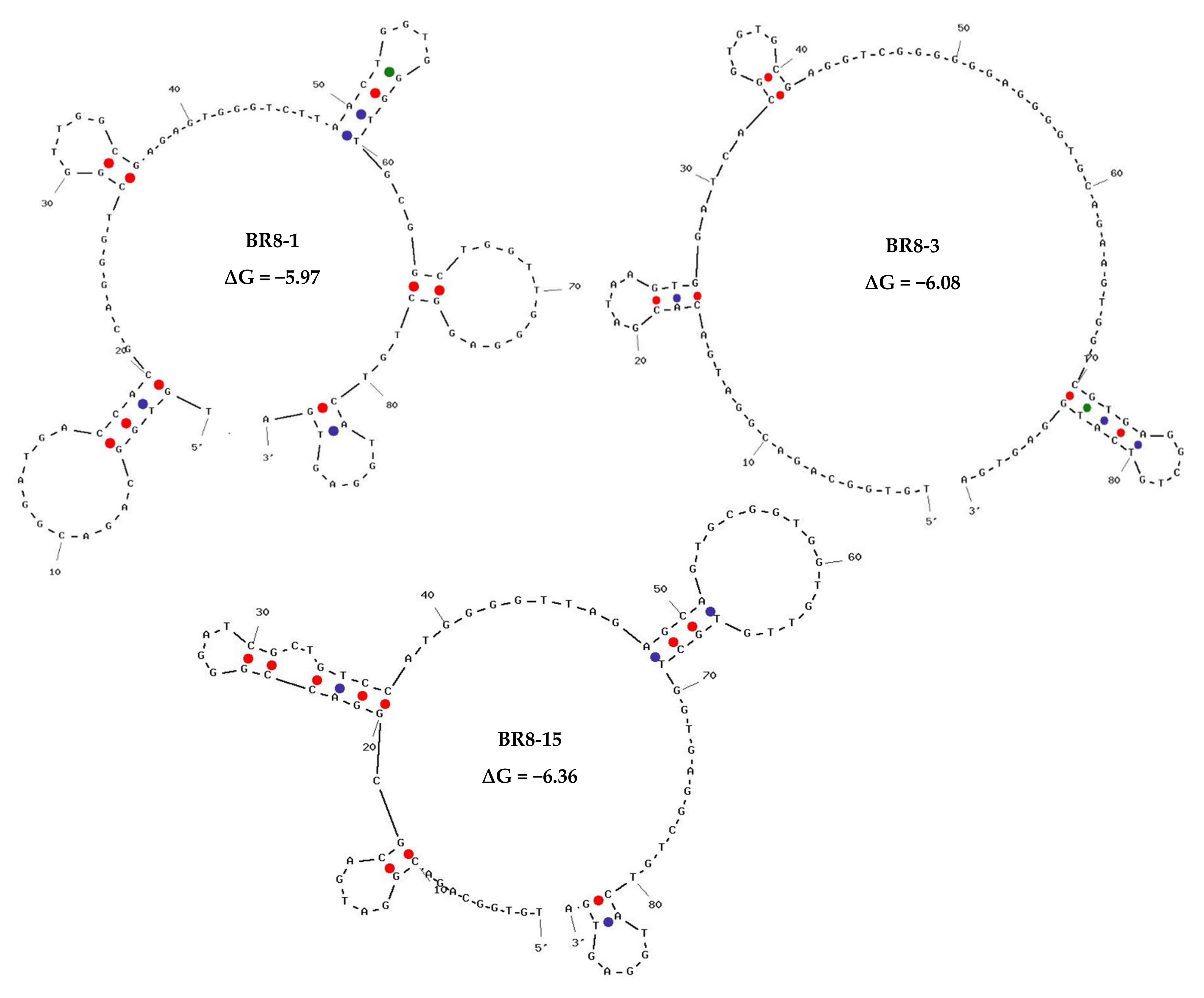
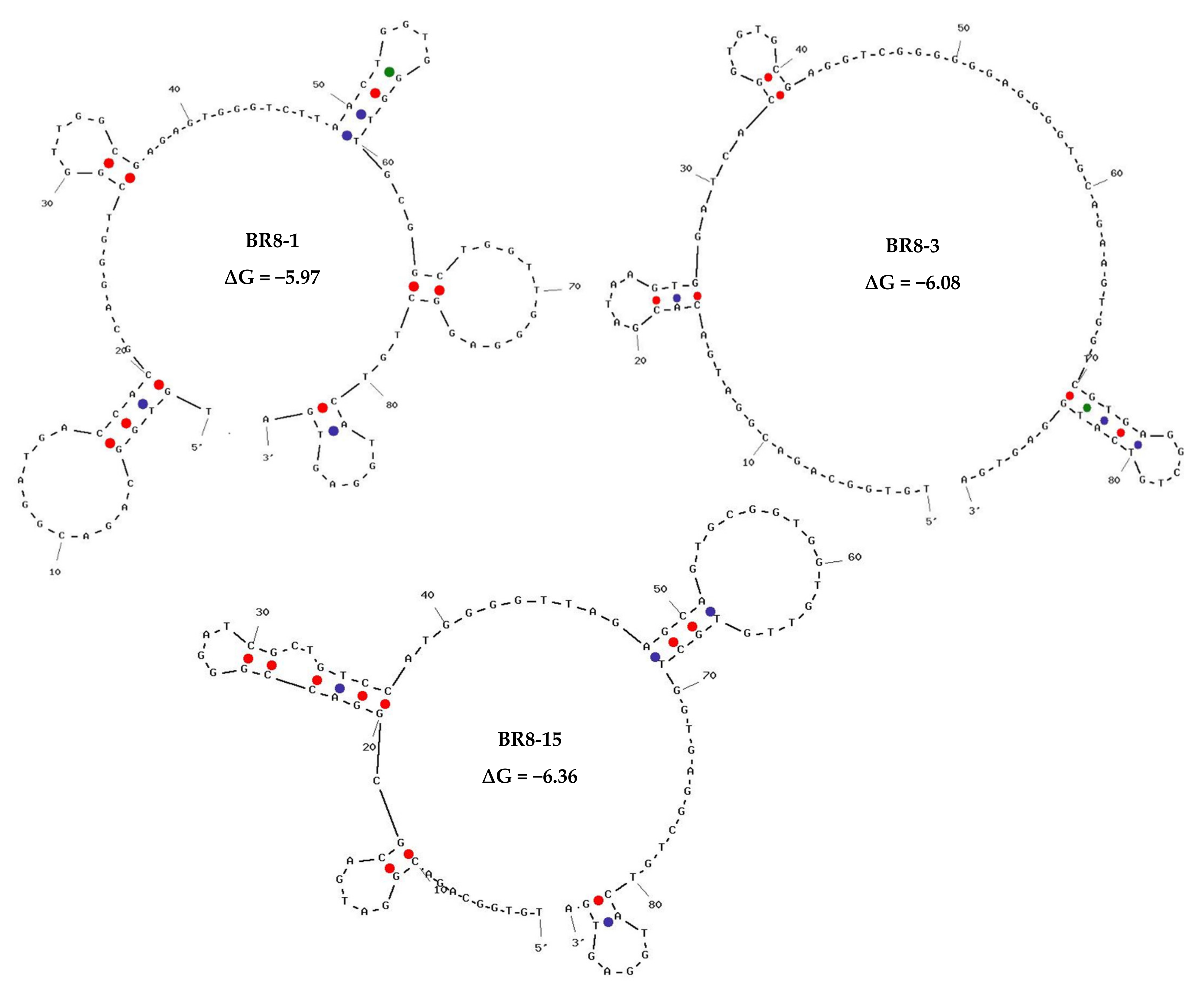
2.3. High-Throughput Sequencing Results, Bioinformatics Analysis, and Aptamer Selection
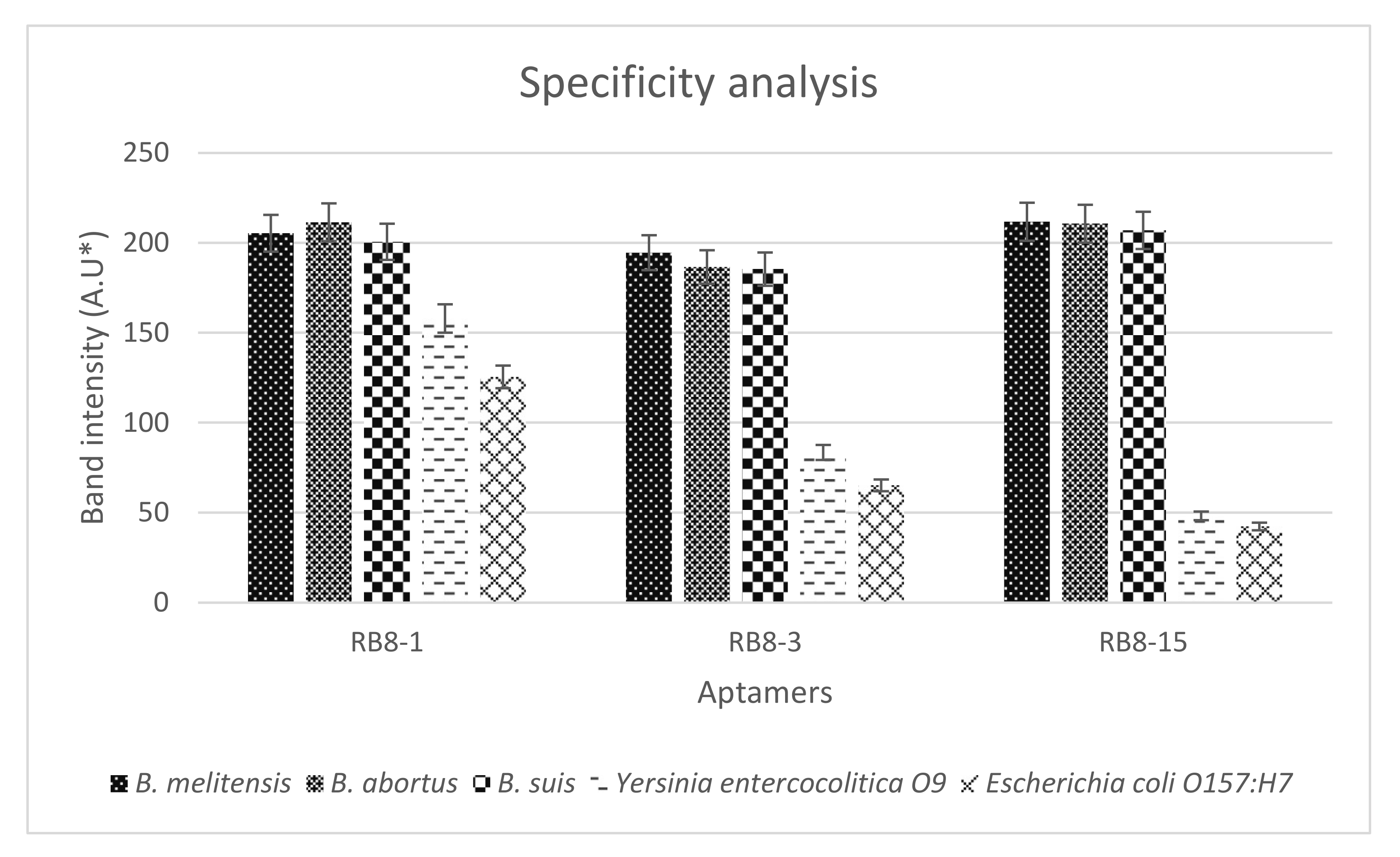
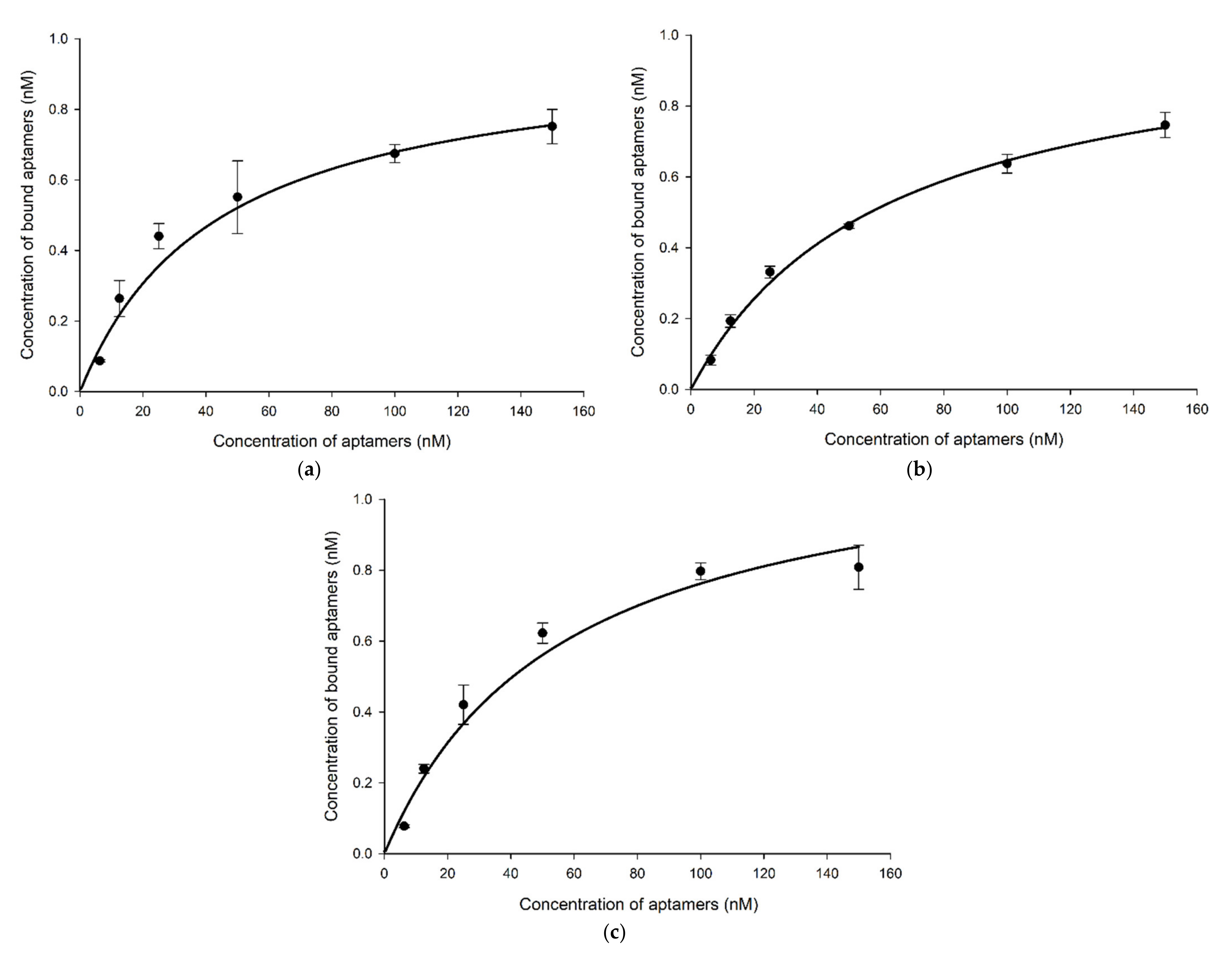
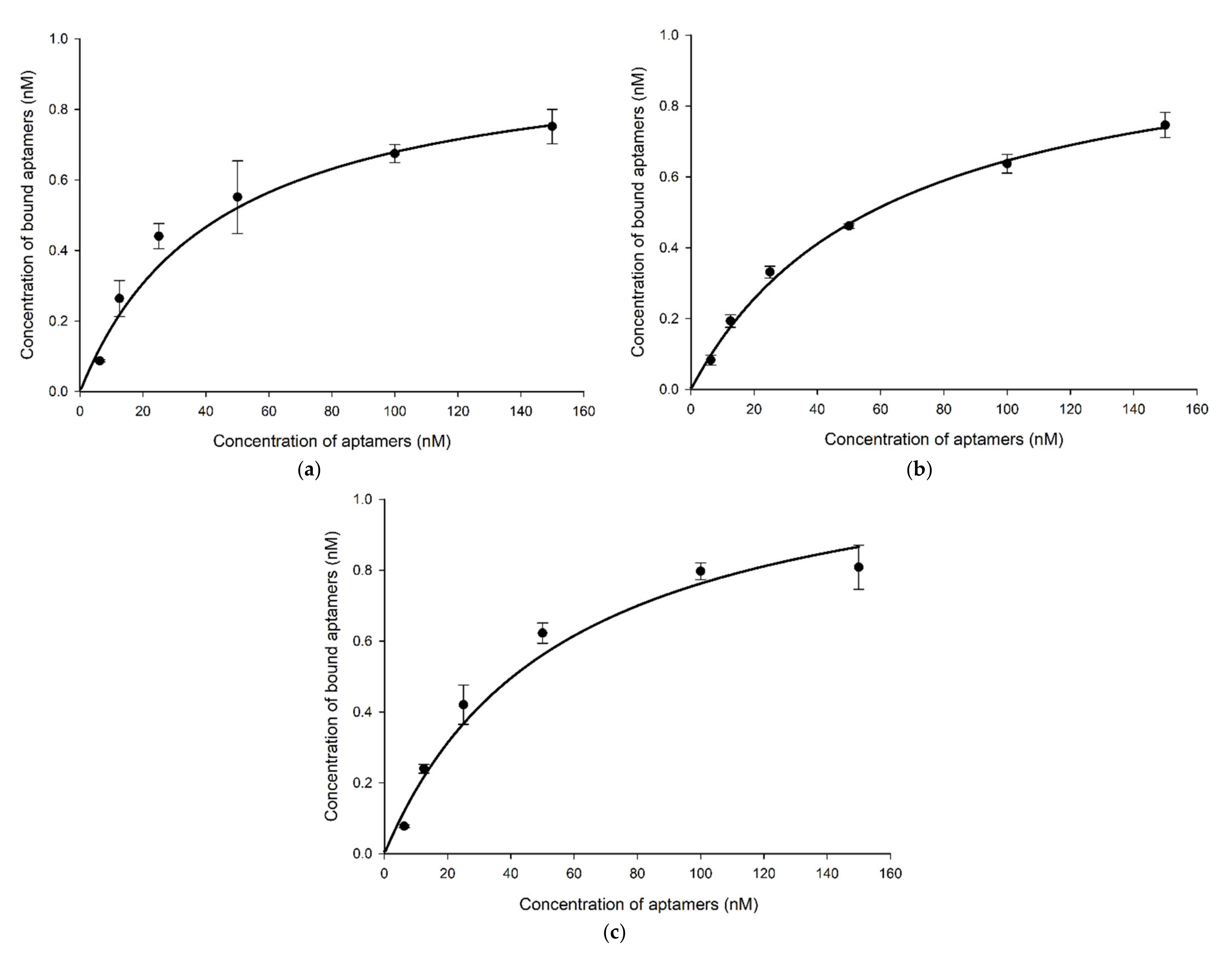
2.4. Specificity, Binding Affinity, and Sensitivity of Selected Aptamers
3. Discussion
4. Materials and Methods
4.1. Bacterial Targets, Culturing Conditions, and Dilution
4.2. ssDNA Library and Primers Design
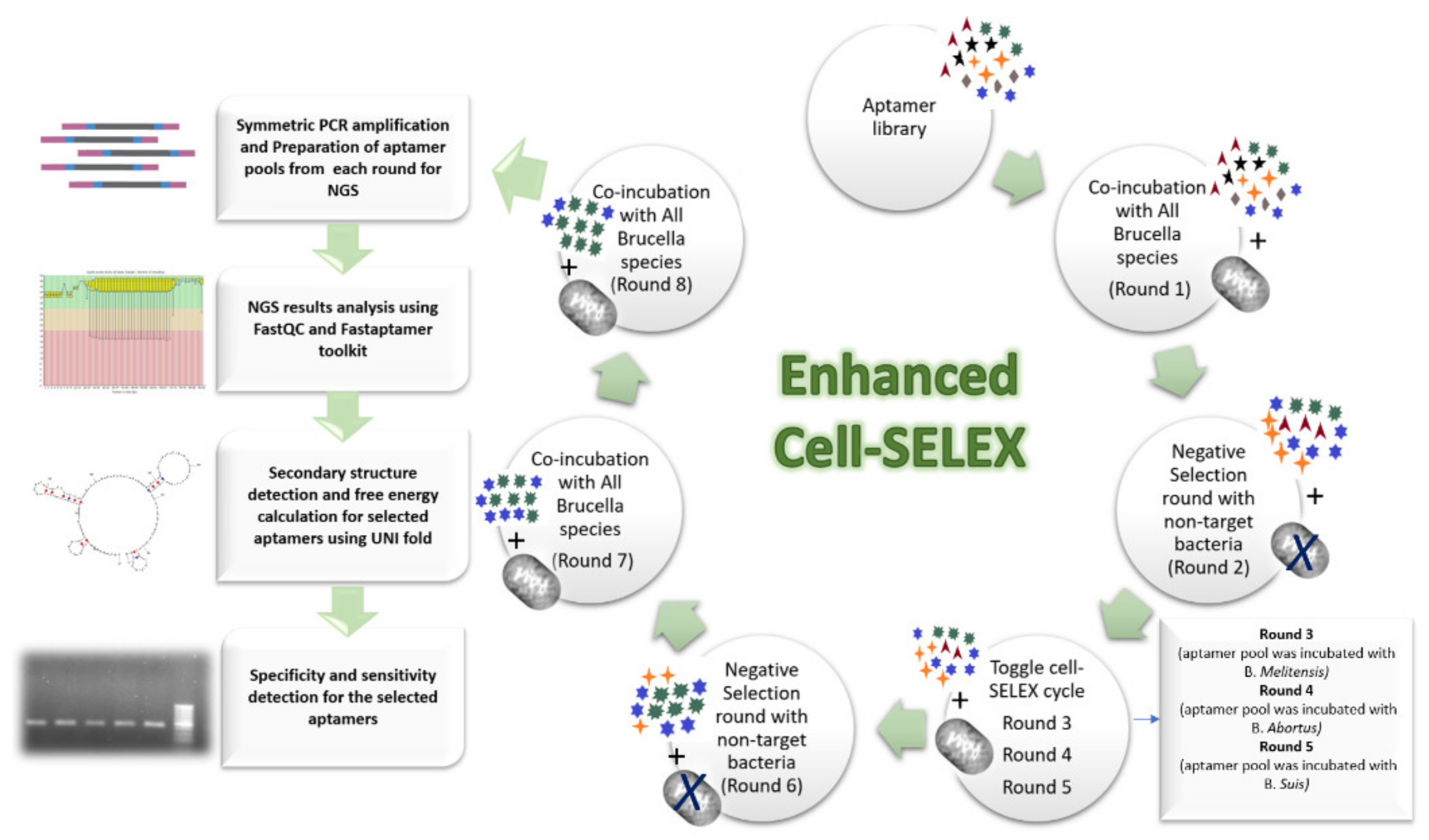
4.3. Cell-SELEX Workflow
4.4. Cell-SELEX Rounds
4.5. PCR Amplification and ssDNA Generation
4.5.1. Asymmetric PCR Amplification
4.5.2. Symmetric Followed by Asymmetric PCR Amplification
4.6. Gel Electrophoresis and ImageJ Analysis
4.7. Monitoring of Cell-SELEX Progress and Aptamer Enrichment Using qPCR
4.8. High-Throughput Sequencing (HTS) by Illumina Technology
4.9. Bioinformatics Analysis and Aptamer Selection
4.10. Specificity and Sensitivity Analysis of Selected Aptamers
4.11. Binding Affinity Analysis of the Selected Aptamer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Young, E.J. An overview of human brucellosis. Clin. Infect. Dis. 1995, 21, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Yagupsky, P. Detection of brucellae in blood cultures. J. Clin. Microbiol. 1999, 37, 3437–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttigieg, S.C.; Savic, S.; Cauchi, D.; Lautier, E.; Canali, M.; Aragrande, M. Brucellosis control in Malta and Serbia: A one health evaluation. Front. Vet. Sci. 2018, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.C.; Palmer, M.V. Advancement of Knowledge of Brucella Over the Past 50 Years. Vet. Pathol. 2014, 51, 1076–1089. [Google Scholar] [CrossRef]
- Crawford, R.P.; Huber, J.D.; Adams, B.S. Epidemiology and Surveillance. In Animal Brucellosis; Nielsen, K., Duncan, J.R., Eds.; CRC Press: Boca Raton, FL, USA, 1990; pp. 131–151. [Google Scholar]
- Meltzer, E.; Sidi, Y.; Smolen, G.; Banai, M.; Bardenstein, S.; Schwartz, E. Sexually Transmitted brucellosis in humans. Clin. Infect. Dis. 2010, 51, 12–15. [Google Scholar] [CrossRef]
- Tuon, F.F.; Gondolfo, R.B.; Cerchiari, N. Human-to-Human transmission of brucella-a systematic review. Trop. Med. Int. Health 2017, 22, 539–546. [Google Scholar] [CrossRef] [Green Version]
- McDermott, J.; Grace, D.; Zinsstag, J. Economics of brucellosis impact and control in low-income countries. Rev. Sci. Tech. 2013, 32, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Khurana, S.K.; Sehrawat, A.; Tiwari, R.; Prasad, M.; Gulati, B.; Shabbir, M.Z.; Chhabra, R.; Karthik, K.; Patel, S.K.; Pathak, M.; et al. Bovine Brucellosis–A comprehensive review. Vet. Q. 2021, 41, 61–88. [Google Scholar] [CrossRef]
- Goodwin, Z.I.; Pascual, D.W. Brucellosis vaccines for livestock. Vet. Immunol. Immunopathol. 2016, 181, 51–58. [Google Scholar] [CrossRef] [Green Version]
- González-Espinoza, G.; Arce-Gorvel, V.; Mémet, S.; Gorvel, J.-P. Brucella: Reservoirs and niches in animals and humans. Pathogens 2021, 10, 186. [Google Scholar] [CrossRef]
- Moreno, E. Retrospective and prospective perspectives on zoonotic brucellosis. Front. Microbiol. 2014, 5, e213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliendo, A.M.; Gilbert, D.N.; Ginocchio, C.C.; Hanson, K.E.; May, L.; Quinn, T.C.; Tenover, F.C.; Alland, D.; Blaschke, A.J.; Bonomo, R.A.; et al. Better tests, better care: Improved diagnostics for infectious diseases. Clin. Infect. Dis. 2013, 57, 139–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miguel, M.J.; Marín, C.M.; Muñoz, P.M.; Dieste, L.; Grilló, M.J.; Blasco, J.M. Development of a selective culture medium for primary isolation of the main brucella species. J. Clin. Microbiol. 2011, 49, 1458–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, K. Bovine brucellosis. In Manual of Standards for Diagnostic Tests and Vaccines; World Organisation for Animal Health (OIE): Paris, France, 2012; pp. 616–650. [Google Scholar]
- Nielsen, K.; Yu, W.L. Serological diagnosis of brucellosis. Prilozi 2010, 31, 65–89. [Google Scholar] [PubMed]
- Boschiroli, M.L.; Foulongne, V.; O’Callaghan, D. Brucellosis: A Worldwide Zoonosis. Curr. Opin. Microbiol. 2001, 4, 58–64. [Google Scholar] [CrossRef]
- Khan, M.Z.; Zahoor, M. An Overview of Brucellosis in Cattle and Humans, and Its Serological and Molecular Diagnosis in Control Strategies. Trop. Med. Infect. Dis. 2018, 3, 65. [Google Scholar] [CrossRef] [Green Version]
- Di Bonaventura, G.; Angeletti, S.; Ianni, A.; Petitti, T.; Gherardi, G. Microbiological laboratory diagnosis of human brucellosis: An overview. Pathogens 2021, 10, 1623. [Google Scholar] [CrossRef]
- Mohammadi, E.; Golchin, M. Detection of brucella abortus by immunofluorescence assay using anti outer membrane protein of 19 KDa antibody. Adv. Clin. Exp. Med. 2018, 27, 643–648. [Google Scholar] [CrossRef]
- Hans, R.; Yadav, P.K.; Sharma, P.K.; Boopathi, M.; Thavaselvam, D. Development and validation of immunoassay for whole cell detection of brucella abortus and brucella Melitensis. Sci. Rep. 2020, 10, 8543. [Google Scholar] [CrossRef]
- Mei, J.-J.; Wang, X.-L.; Li, X.-Y. Development and verification of double antibody sandwich ELISA for B. Melitensis. Chin. J. Biol. 2009, 22, 823–825. [Google Scholar]
- Kulabhusan, P.K.; Hussain, B.; Yüce, M. Current perspectives on aptamers as diagnostic tools and therapeutic agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Jia, W.N.; Li, X.Y.; Zhang, L.; Liu, C.; Wu, J. Advances in detection of infectious agents by aptamer-based technologies. Emerg. Microbes Infect. 2020, 9, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, A.; Tombelli, S. Aptamer optical switches: From biosensing to intracellular sensing. Sens. Actuators Rep. 2021, 3, 100030. [Google Scholar] [CrossRef]
- Villalonga, A.; Pérez-Calabuig, A.M.; Villalonga, R. Electrochemical biosensors based on nucleic acid aptamers. Anal. Bioanal. Chem. 2020, 412, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.I.; Song, E. Lab-on-a-Chip systems for aptamer-based biosensing. Micromachines 2020, 11, 220. [Google Scholar] [CrossRef] [Green Version]
- Majdinasab, M.; Badea, M.; Marty, J.L. Aptamer-Based lateral flow assays: Current trends in clinical diagnostic rapid tests. Pharmaceuticals 2022, 15, 90. [Google Scholar] [CrossRef]
- Hong, L.; Pan, M.; Xie, X.; Liu, K.; Yang, J.; Wang, S.; Wang, S. Aptamer-Based fluorescent biosensor for the rapid and sensitive detection of allergens in food matrices. Foods 2021, 10, 2598. [Google Scholar] [CrossRef]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef]
- Li, L.; Li, B.; Qi, Y.; Jin, Y. Label-Free aptamer-based colorimetric detection of mercury ions in aqueous media using unmodified gold nanoparticles as colorimetric probe. Anal. Bioanal. Chem. 2009, 393, 2051–2057. [Google Scholar] [CrossRef]
- Chen, J.; Zhuang, X.; Zheng, J.; Yang, R.; Wu, F.; Zhang, A.; Fang, B. Aptamer-Based cell-free detection system to detect target protein. Synth. Syst. Biotechnol. 2021, 6, 209–215. [Google Scholar] [CrossRef]
- Teng, J.; Yuan, F.; Ye, Y.; Zheng, L.; Yao, L.; Xue, F.; Chen, W.; Li, B. Aptamer-Based technologies in foodborne pathogen detection. Front. Microbiol. 2016, 7, 1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bognár, Z.; Gyurcsányi, R.E. Aptamers against Immunoglobulins: Design, selection and bioanalytical applications. Int. J. Mol. Sci. 2020, 21, 5748. [Google Scholar] [CrossRef] [PubMed]
- Elskens, J.P.; Elskens, J.M.; Madder, A. Chemical modification of aptamers for increased binding affinity in diagnostic applications: Current status and future prospects. Int. J. Mol. Sci. 2020, 21, 4522. [Google Scholar] [CrossRef] [PubMed]
- Dollins, C.M.; Nair, S.; Sullenger, B.A. Aptamers in immunotherapy. Gene Ther. 2008, 19, 443–450. [Google Scholar] [CrossRef]
- Song, S.; Wang, L.; Li, J.; Zhao, J.; Fan, C. Aptamer-Based biosensors. Trends Anal. Chem. 2008, 27, 108–117. [Google Scholar] [CrossRef]
- Que-Gewirth, N.S.; Sullenger, B.A. Gene therapy progress and prospects: RNA aptamers. Gene Ther. 2007, 14, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Liang, Z.; Zhou, N. Design strategies for aptamer-based biosensors. Sensors 2010, 10, 4541–4557. [Google Scholar] [CrossRef] [Green Version]
- Byun, J. Recent progress and opportunities for nucleic acid aptamers. Life 2021, 11, 193. [Google Scholar] [CrossRef]
- Souza, A.G.; Marangoni, K.; Patrícia, T.; Alves, P.T.; Silva, M.J.; Goulart, R.; Goulart, V.A. 3D Cell SELEX: Development of RNA aptamers as molecular probes for PC-3 tumor cell line. Exp. Cell Res. 2016, 341, 147–156. [Google Scholar] [CrossRef]
- Liu, J.; Lu, Y. Fast colorimetric sensing of adenosine and cocaine based on a general sensor design involving aptamers and nanoparticles. Angew. Chem. Int. Ed. 2006, 45, 90–94. [Google Scholar] [CrossRef]
- Han, K.; Chen, L.; Lin, Z.; Li, G. Target Induced Dissociation (TID) strategy for the development of electrochemical aptamer-based biosensor. Electrochem. Commun. 2009, 11, 157–160. [Google Scholar] [CrossRef]
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z.C.; Chen, H.W.; Mallikaratchy, P.; Sefah, K.; Yang, C.J.; Tan, W. Aptamers Evolved from Live Cells as Effective Molecular Probes for Cancer Study. Proc. Natl. Acad. Sci. USA 2006, 103, 11838–11843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Ding, J.; Deng, L.; Xiang, Y.; Liu, D.; Zhang, Y.; Chen, X.; Yang, Q. Selection of DNA aptamers recognizing epcam-positive prostate cancer by cell-selex for in vitro and in vivo Mr. Imaging. Drug Des. Devel. Ther. 2021, 15, 3985–3996. [Google Scholar] [CrossRef] [PubMed]
- Afrasiabi, S.; Pourhajibagher, M.; Raoofian, R.; Tabarzad, M.; Bahador, A. Therapeutic applications of nucleic acid aptamers in microbial infections. J. Biomed. Sci. 2020, 27, 6. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, X.; Zhou, Y.; Ma, M.; Wang, M.; Ying, B. Systematic evolution of ligands by exponential enrichment technologies and aptamer-based applications: Recent progress and challenges in precision medicine of infectious diseases. Front. Bioeng. Biotechnol. 2021, 9, 704077. [Google Scholar] [CrossRef]
- Wang, T.; Chen, C.; Larcher, L.M.; Barrero, R.A.; Veedu, R.N. Three decades of nucleic acid aptamer technologies: Lessons learned, progress and opportunities on aptamer development. Biotechnol. Adv. 2019, 37, 28–50. [Google Scholar] [CrossRef]
- Kumar, T.; Bruno, J.G.; Dhiman, A. ABCs of DNA aptamer and related assay development. Biotechnol. Adv. 2017, 35, 275–301. [Google Scholar] [CrossRef]
- Moon, J.; Kim, G.; Park, S.; Lim, J.; Mo, C. Comparison of Whole-Cell SELEX methods for the identification of staphylococcus aureus-specific DNA aptamers. Sensors 2015, 15, 8884–8897. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Wilhelm, N.; Perle, N.; Stoll, H.; Schlensak, C.; Wendel, H.P. Absolute quantification of cell-bound DNA aptamers during SELEX. Nucleic Acid Ther. 2013, 23, 125–130. [Google Scholar] [CrossRef]
- Kolm, C.; Cervenka, I.; Aschl, U.J.; Baumann, N.; Jakwerth, S.; Krska, R.; Mach, R.L.; Sommer, R.; DeRosa, M.C.; Kirschner, A.K.T.; et al. DNA aptamers against bacterial cells can be efficiently selected by a SELEX process using state-of-the art QPCR and ultra-deep sequencing. Sci. Rep. 2020, 10, 20917. [Google Scholar] [CrossRef]
- Mohammadinezhad, R.; Jalali, S.A.H.; Farahmand, H. Evaluation of different direct and indirect SELEX monitoring methods and implementation of melt-curve analysis for rapid discrimination of variant aptamer sequences. Anal. Methods 2020, 12, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Aquino-Jarquin, G.; Toscano-Garibay, J.D. RNA aptamer evolution: Two decades of selection. Int. J. Mol. Sci. 2011, 12, 9155–9171. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Z.; Yu, Y.; Wang, M.; Li, J.; Zhang, Z.; Liu, J.; Wu, X.; Lu, A.; Zhang, G.; Zhang, B. Recent advances in SELEX technology and aptamer applications in biomedicine. Int. J. Mol. Sci. 2017, 18, 2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Yu, Y.; Jiang, F.; Zhou, J.; Li, Y.; Liang, C.; Dang, L.; Lu, A.; Zhang, G. Development of Cell-SELEX technology and its application in cancer diagnosis and therapy. Int. J. Mol. Sci. 2016, 17, 2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using Cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef]
- Dwivedi, H.P.; Smiley, R.D.; Jaykus, L.-A. Selection and characterization of DNA aptamers with binding selectivity to campylobacter Jejuni using whole-Cell SELEX. Appl. Microbiol. Biotechnol. 2010, 87, 2323–2334. [Google Scholar] [CrossRef]
- Duan, N.; Wu, S.; Chen, X.; Huang, Y.; Wang, Z. Selection and identification of a DNA aptamer targeted to vibrio parahemolyticus. J. Agric. Food Chem. 2012, 60, 4034–4038. [Google Scholar] [CrossRef]
- Moon, J.; Kim, G.; Lee, S.; Park, S. Identification of salmonella typhimurium-specific DNA aptamers developed using whole-cell SELEX and FACS analysis. J. Microbiol. Methods 2013, 95, 162–166. [Google Scholar] [CrossRef]
- Hamula, C.L.A.; Peng, H.; Wang, Z.; Tyrrell, G.J.; Li, X.F.; Le, X.C. An improved SELEX technique for selection of DNA aptamers binding to M-Type 11 of streptococcus pyogenes. Methods 2016, 97, 51–57. [Google Scholar] [CrossRef]
- Marton, S.; Cleto, F.; Krieger, M.A.; Cardoso, J. Isolation of an aptamer that binds specifically to E. coli. PLoS ONE 2016, 11, e0153637. [Google Scholar] [CrossRef] [Green Version]
- Mozioglu, E.; Gokmen, O.; Tamerler, C.; Kocagoz, Z.T.; Akgoz, M. Selection of nucleic acid aptamers specific for mycobacterium tuberculosis. Appl. Biochem. Biotechnol. 2016, 178, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Yahyaabadi, M.Y.; Dorraj, G.S.; Heiat, M.; Latifi, A.M. Utilizing Cell-SELEX, as a Promising Strategy to Isolate SsDNA aptamer probes for detection of staphylococcus aureus. J. Appl. Biotechnol. Rep. 2017, 4, 633–638. [Google Scholar]
- Saad, M.; Chinerman, D.; Tabrizian, M.; Faucher, S.P. Identification of two aptamers binding to legionella pneumophila with high affinity and specificity. Sci. Rep. 2020, 10, 9145. [Google Scholar] [CrossRef] [PubMed]
- Gopinathan, P.; Hung, L.Y.; Wang, C.H.; Chiang, N.J.; Wang, Y.C.; Shan, Y.; Lee, G.B. Automated selection of aptamers against cholangiocarcinoma cells on an integrated microfluidic platform. Biomicrofluidics 2017, 11, e044101. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, S.K.; Bowser, M.T. Microfluidic methods for aptamer selection and characterization. Analyst 2018, 143, 21–32. [Google Scholar] [CrossRef]
- Komarova, N.; Kuznetsov, A. Inside the black box: What makes selex better? Molecules 2019, 24, 3598. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yu, J. Challenges of SELEX and Demerits of Aptamer-Based Methods. In Aptamers for Analytical Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; pp. 345–364. [Google Scholar]
- Ohuchi, S. Cell-SELEX Technology. Biores. Open Access 2012, 1, 265–272. [Google Scholar] [CrossRef]
- Song, M.Y.; Nguyen, D.; Hong, S.W.; Kim, B.C. Broadly Reactive aptamers targeting bacteria belonging to different genera using a sequential toggle Cell-SELEX. Sci. Rep. 2017, 7, e43641. [Google Scholar] [CrossRef] [Green Version]
- White, R.; Rusconi, C.; Scardino, E.; Wolberg, A.; Lawson, J.; Hoffman, M.; Sullenger, B. Generation of Species Cross-Reactive Aptamers Using “Toggle” SELEX. Mol. Ther. 2001, 4, 567–573. [Google Scholar] [CrossRef]
- Kohlberger, M.; Gadermaier, G. SELEX: Critical factors and optimization strategies for successful aptamer selection. Biotechnol. Appl. Biochem. 2021, 1, 22. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhao, J.; Li, J.; Liao, X.; Chen, F. Advances of aptamers screened by Cell-Selex in selection procedure, cancer diagnostics and therapeutics. Anal. Biochem. 2020, 598, 113620. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, J.; Chen, K.; Xu, Y.; Liu, B.; Liao, J.; Zhu, L.; Hu, X.; Li, J.; Pu, Y.; et al. Selection and Characterization of DNA aptamer against glucagon receptor by Cell-SELEX. Sci. Rep. 2017, 7, 7179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SAITO, S. SELEX-Based DNA Aptamer Selection: A perspective from the advancement of separation techniques. Anal. Sci. 2021, 37, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Tolle, F.; Wilke, J.; Wengel, J.; Mayer, G. By-Product formation in repetitive PCR amplification of DNA libraries during SELEX. PLoS ONE 2014, 9, e114693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosch, J.C.; Balikov, D.A.; Gong, F.; Lippmann, E.S. A systematic evolution of ligands by exponential enrichment workflow with consolidated counter selection to efficiently isolate high-affinity aptamers. Eng. Rep. 2020, 2, e12089. [Google Scholar] [CrossRef]
- Citartan, M.; Tang, T.; Tan, S.; Hoe, C.; Saini, R. Asymmetric PCR for Good Quality SsDNA Generation towards DNA Aptamer Production Asymmetric PCR for Good Quality SsDNA Generation towards DNA Aptamer Production. Songklanakarin J. Sci. Technol. 2012, 34, 125–131. [Google Scholar]
- Tabarzad, M.; Kazemi, B.; Vahidi, H.; Aboofazeli, R.; Shahhosseini, S.; Nafissi-Varcheh, N. Challenges to design and develop of DNA aptamers for protein targets. I. optimization of asymmetric PCR for generation of a single stranded DNA library. Iran. J. Pharm. Res. 2014, 13, 133–141. [Google Scholar]
- Heiat, M.; Ranjbar, R.; Latifi, A.M.; Rasaee, M.J.; Farnoosh, G. Essential strategies to optimize asymmetric PCR conditions as a reliable method to generate large amount of SsDNA aptamers. Biotechnol. Appl. Biochem. 2017, 64, 541–548. [Google Scholar] [CrossRef]
- Paul, A.; Avci-Adali, M.; Ziemer, G.; Wendel, H.P. Streptavidin-Coated magnetic beads for DNA strand separation implicate a multitude of problems during Cell-SELEX. Oligonucleotides 2009, 19, 243–254. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Paul, A.; Wilhelm, N.; Ziemer, G.; Wendel, H.P. Upgrading SELEX technology by using lambda exonuclease digestion for single-stranded DNA generation. Molecules 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Svobodová, M.; Pinto, A.; Nadal, P.; O’ Sullivan, C.K. Comparison of different methods for generation of single-stranded DNA for SELEX processes. Anal. Bioanal. Chem. 2012, 404, 835–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Li, D.; Zhang, G.; Li, H.; Shao, N.; Liang, Z.; Zhang, L.; Lu, A.; Zhang, G. Comparison of the methods for generating single- stranded DNA in SELEX. Analyst 2015, 140, 3439–3444. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; He, L.; Wang, J.; Fang, X.; Zhang, L. Developing a combined strategy for monitoring the progress of aptamer selection. Analst 2017, 142, 3136–3139. [Google Scholar] [CrossRef] [PubMed]
- Mencin, N.; Šmuc, T.; Vraničar, M.; Mavri, J.; Hren, M.; Galeša, K.; Krkoč, P.; Ulrich, H.; Šolar, B. Optimization of SELEX: Comparison of different methods for monitoring the progress of in vitro selection of aptamers. J. Pharm. Biomed. Anal. 2014, 91, 151–159. [Google Scholar] [CrossRef]
- Komarova, N.; Barkova, D.; Kuznetsov, A. Implementation of high-throughput sequencing (Hts) in aptamer selection technology. Int. J. Mol. Sci. 2020, 21, 8774. [Google Scholar] [CrossRef]
- Cho, M.; Xiao, Y.; Nie, J.; Stewart, R.; Csordas, A.T.; Oh, S.S.; Thomson, J.A.; Soh, H.T. Quantitative Selection of DNA Aptamers through Microfluidic Selection and High-Throughput Sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 15373–15378. [Google Scholar] [CrossRef] [Green Version]
- Schütze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Mörl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M.; et al. Probing the SELEX Process with Next-Generation Sequencing. PLoS ONE 2011, 6, e29604. [Google Scholar] [CrossRef] [Green Version]
- Hoinka, J.; Berezhnoy, A.; Dao, P.; Sauna, Z.E.; Gilboa, E.; Przytycka, T.M. Large scale analysis of the mutational landscape in HT-SELEX improves aptamer discovery. Nucleic Acids Res. 2015, 43, 5699–5707. [Google Scholar] [CrossRef]
- Alton, G.G.; Jones, L.M.; Angus, R.D.; Verger, J.M. Techniques for the Brucellosis Laboratory; Institut National de la Recherche Agronomique: Paris, France, 1988.
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Meth. 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 October 2019).
- Alam, K.K.; Chang, J.L.; Burke, D.H. FASTAptamer: A Bioinformatic toolkit for high-throughput sequence analysis of combinatorial selections. Mol. Ther.-Nucleic Acids 2015, 4, e230. [Google Scholar] [CrossRef]
- Siddiqui, S.; Jie, Y. Binding characteristics study of DNA based aptamers for E. Coli O157:H7. Molecules 2021, 26, 204. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Fernández-Algar, M.; Fernández-Chamorro, J.; Ramajo, J.; Martínez-Salas, E.; Briones, C. A combined ELONA-(RT)QPCR approach for characterizing DNA and RNA aptamers selected against PCBP-2. Molecules 2019, 24, 1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layman, B.; Mandella, B.; Carter, J.; Breen, H.; Rinehart, J.; Cavinato, A. Isolation and characterization of a SsDNA aptamer against major soluble antigen of Renibacterium salmoninarum. Molecules 2022, 27, 1853. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank in BR8 | Normalized Frequency | Name | Sequence | Rank in BR6 (Aptamers Bound to Non-Target Cells)/Normalized Frequency |
|---|---|---|---|---|
| #1 | 27371.16 | BR8-1 | TGTGGCAGACGGATGACCACGCAGGGTCGGTTGGCGAGAGTGGGTCTTAACTGGTGGGTTGCGGCTGGTTGGGAGGCTGTCATGGAGTGA | 3/1446.38 |
| #3 | 8497.90 | BR8-3 | TGTGGCAGACGGATGACACGATAAGTGGATCACGGTGTGCGAGGTCGGGGGGAGGGGTGCAGAAGTGGTCGTGAGGCTGTCATGGAGTGA | 12/619.03 |
| #15 | 3282.00 | BR8-15 | TGTGGCAGACGGATGACGCGGACCGGGATCGCTGTCCATGGGGTTAGAGCAGTGCGGTGGTGTTGTGCTGGTGAGGCTGTCATGGAGTGA | 5145/11.90 |
| Round No. | Cell-SELEX Type | Selection Pressure | |||||||
|---|---|---|---|---|---|---|---|---|---|
| BSA (mg/mL) | tRNA (mg/mL) | Bacterial Cell Concentration (CFU/mL) | Co-Incubation Time (min) | Shaking (rpm) | Washing Volume (µL) | Washing Frequency | Washing Incubation Time (min) | ||
| 1 | Conventional | 1 | 0.1 | 108 | 45 | 200 | 250 | 1 | 1 |
| 2 | Negative | 1 | ---------- | 108 | 45 | 200 | 250 | 1 | 1 |
| 3 | Toggle | 20 | 0.2 | 107 | 30 | 400 | 500 | 2 | 3 |
| 4 | Negative | 1 | --------- | 107 | 30 | 200 | 500 | 2 | 1 |
| 5 | Conventional | 40 | 0.3 | 106 | 20 | 600 | 750 | 3 | 5 |
| 6 | Conventional | 60 | 0.4 | 105 | 15 | 800 | 1000 | 3 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Husseini, D.M.; Sayour, A.E.; Melzer, F.; Mohamed, M.F.; Neubauer, H.; Tammam, R.H. Generation and Selection of Specific Aptamers Targeting Brucella Species through an Enhanced Cell-SELEX Methodology. Int. J. Mol. Sci. 2022, 23, 6131. https://doi.org/10.3390/ijms23116131
El-Husseini DM, Sayour AE, Melzer F, Mohamed MF, Neubauer H, Tammam RH. Generation and Selection of Specific Aptamers Targeting Brucella Species through an Enhanced Cell-SELEX Methodology. International Journal of Molecular Sciences. 2022; 23(11):6131. https://doi.org/10.3390/ijms23116131
Chicago/Turabian StyleEl-Husseini, Dalia M., Ashraf E. Sayour, Falk Melzer, Magda F. Mohamed, Heinrich Neubauer, and Reham H. Tammam. 2022. "Generation and Selection of Specific Aptamers Targeting Brucella Species through an Enhanced Cell-SELEX Methodology" International Journal of Molecular Sciences 23, no. 11: 6131. https://doi.org/10.3390/ijms23116131
APA StyleEl-Husseini, D. M., Sayour, A. E., Melzer, F., Mohamed, M. F., Neubauer, H., & Tammam, R. H. (2022). Generation and Selection of Specific Aptamers Targeting Brucella Species through an Enhanced Cell-SELEX Methodology. International Journal of Molecular Sciences, 23(11), 6131. https://doi.org/10.3390/ijms23116131