
Picrasidine I Triggers Heme Oxygenase-1-Induced Apoptosis in Nasopharyngeal Carcinoma Cells via ERK and Akt Signaling Pathways

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
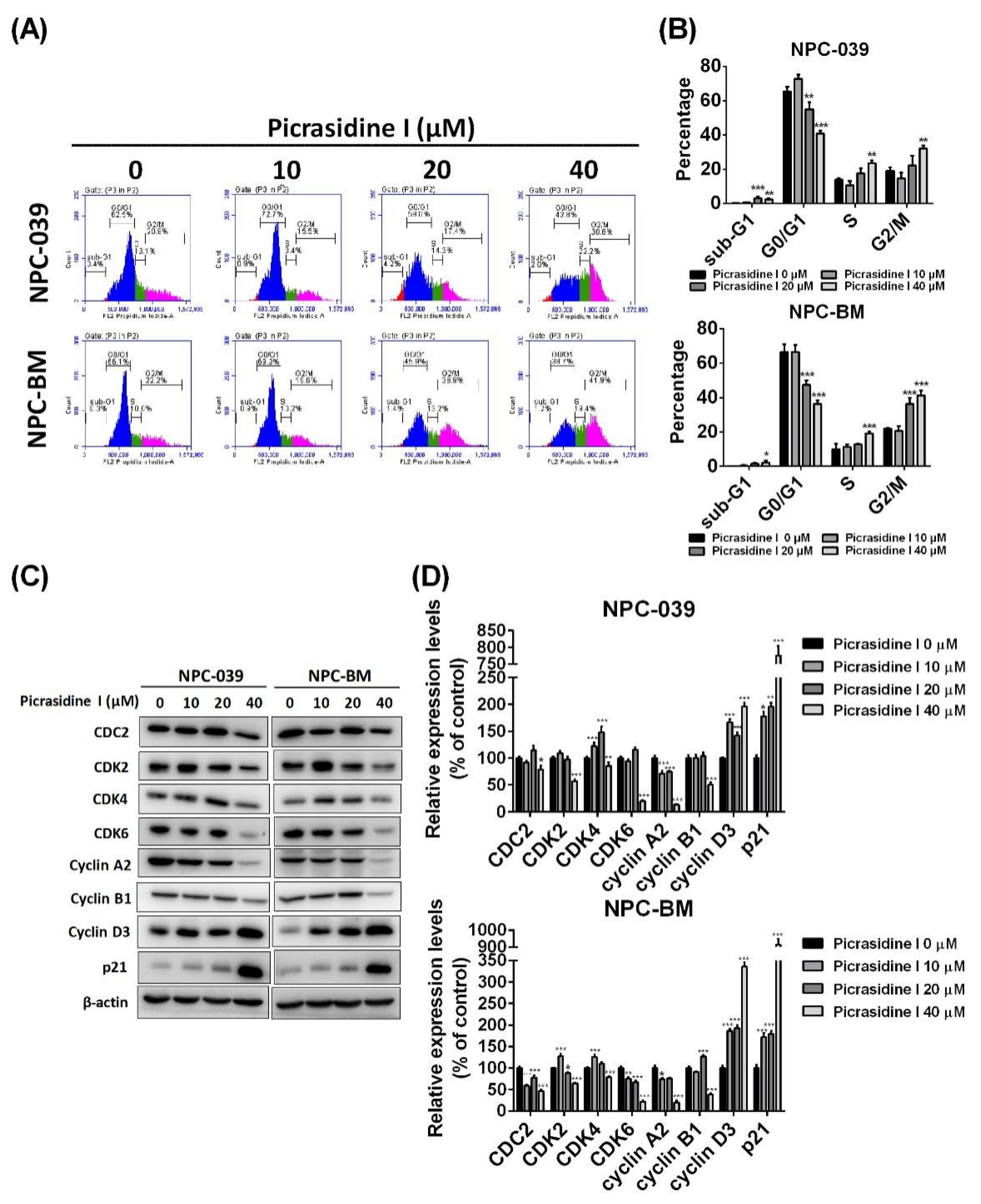
2.1. Picrasidine I Decreased the Cell Viability of NPC Cell Lines and Caused Cell Accumulation in Sub-G1, S, and G2/M Phases
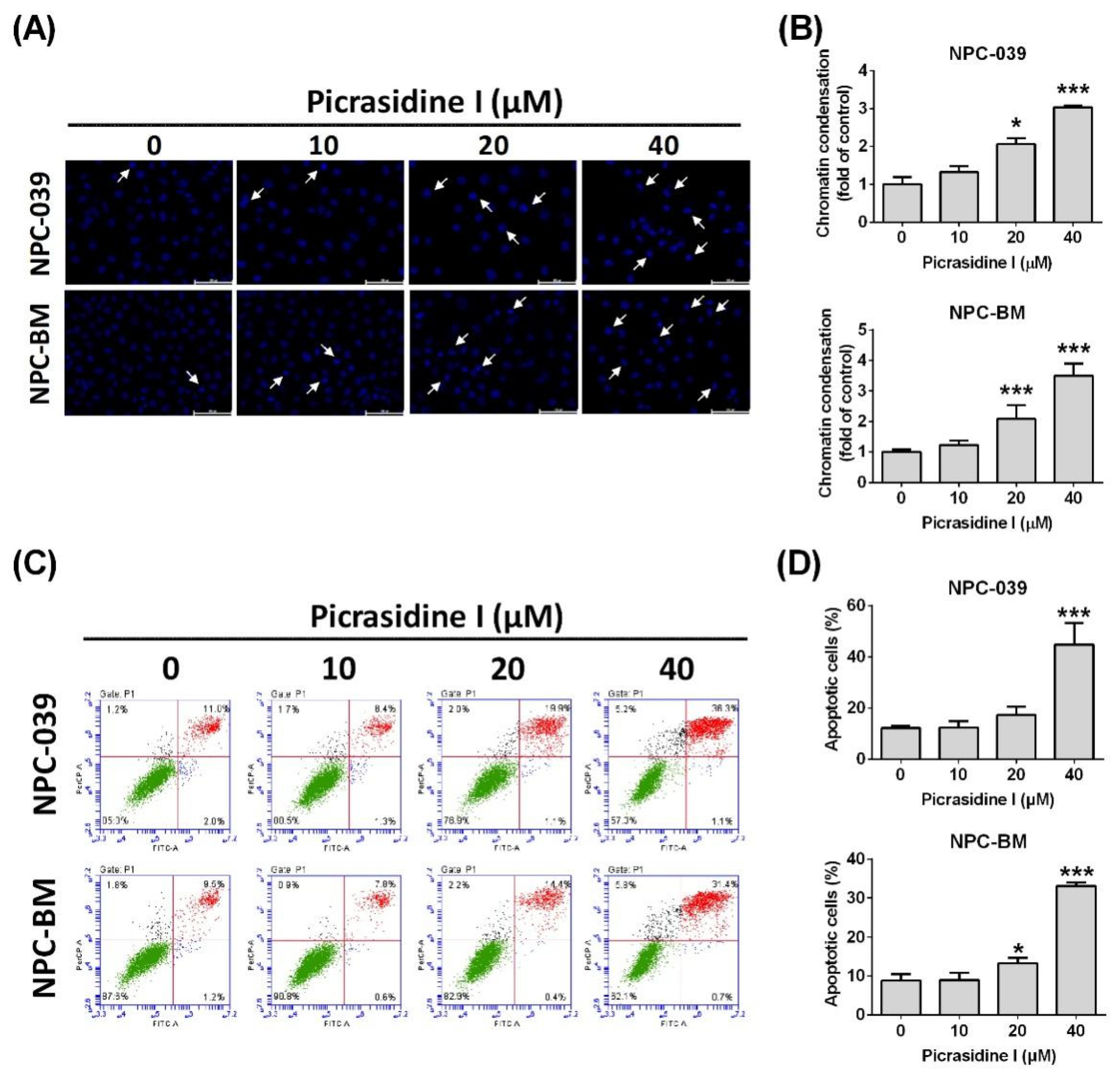
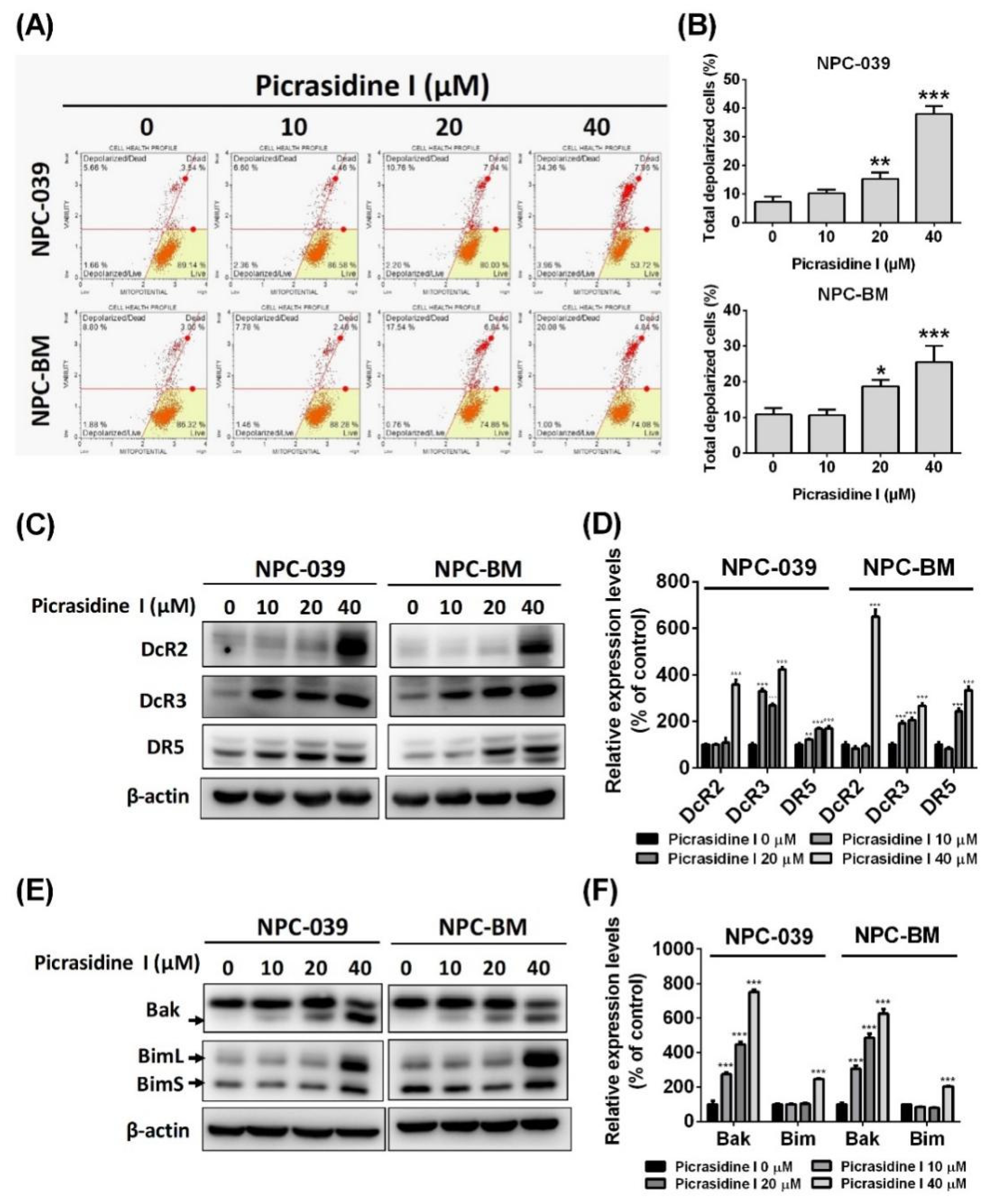
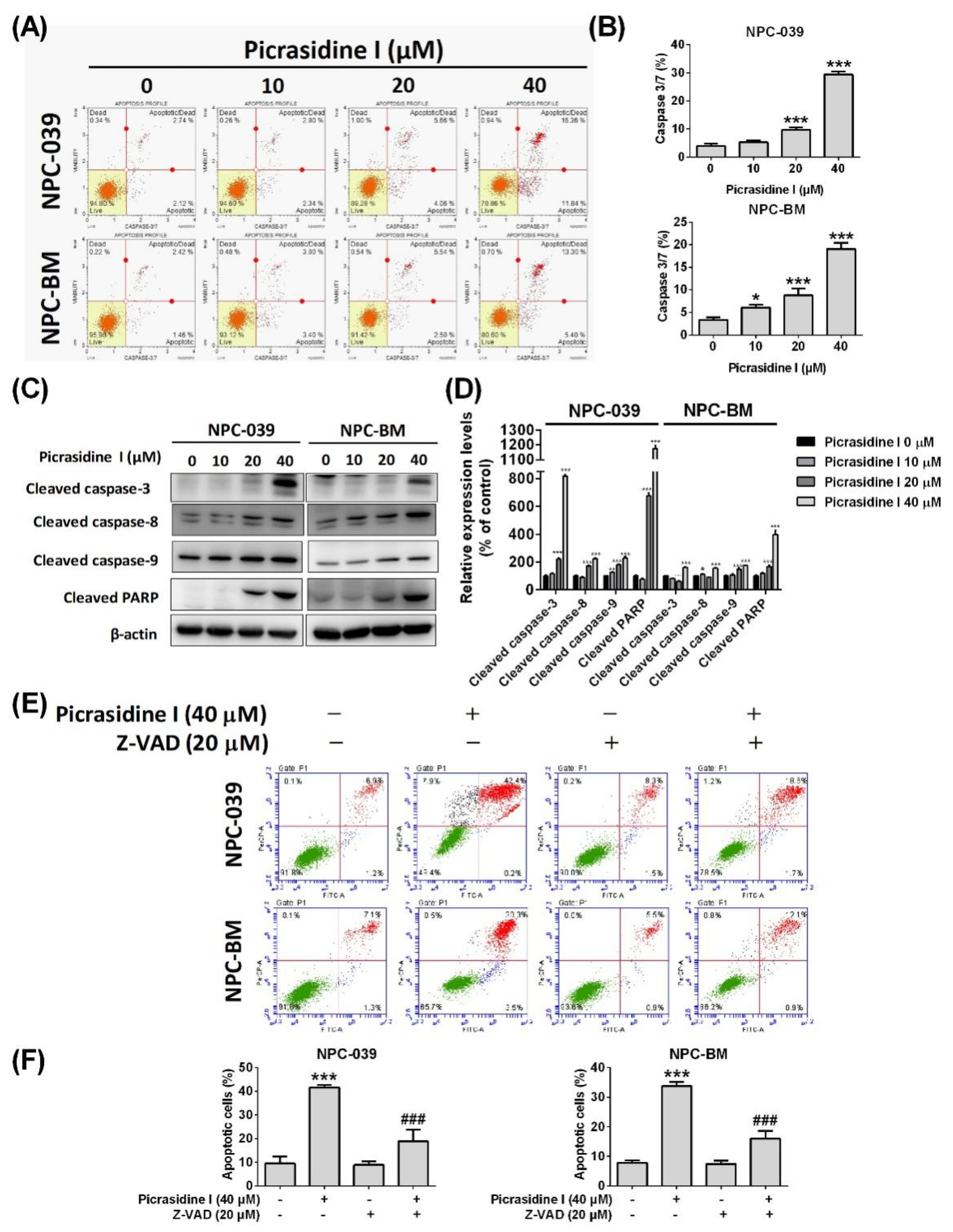
2.2. Picrasidine I Induced Apoptosis in NPC Cells via Extrinsic and Intrinsic Pathways
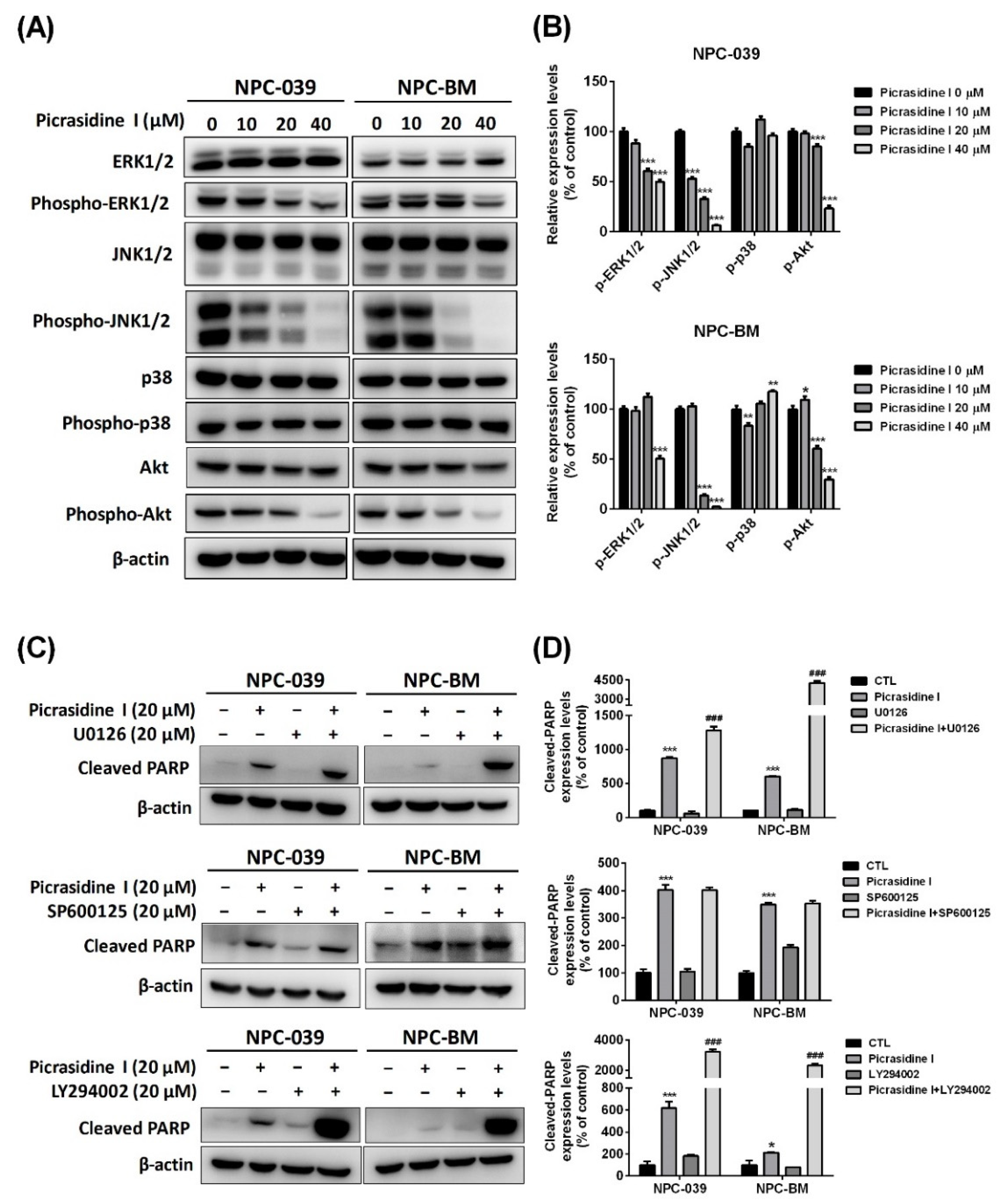
2.3. The Activation of Mitogen-Activated Protein Kinases (MAPKs) and Akt Signaling Pathways Is Involved in Picrasidine I-Induced Apoptosis
2.4. HO-1 Participates in Picrasidine I-Induced Apoptosis via Activating the ERK1/2 and Akt Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Cell Viability Assay
4.3. Colony Formation Assay
4.4. Cell Cycle Analysis
4.5. Western Blot Analysis
4.6. Nuclear Morphology Analysis
4.7. Apoptosis Analysis
4.8. Mitochondrial Membrane Potential Analysis
4.9. Caspase-3/7 Activity Assay
4.10. Proteome Profiler Analysis
4.11. RNA Interference Experiments
4.12. Bioinformatic Analysis
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Q.; Li, C.F.; Li, J.; Chen, W.H.; Chen, Q.Y.; Yuan, L.X.; Lai, X.P.; He, Y.; Xu, Y.X.; Hu, D.P.; et al. Establishment and validation of prognostic nomograms for endemic nasopharyngeal carcinoma. J. Natl. Cancer Inst. 2016, 108, djv291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, D.T.; Sham, J.S.; Kwong, D.L.; Au, G.K. Treatment outcome after radiotherapy alone for patients with stage i-ii nasopharyngeal carcinoma. Cancer 2003, 98, 74–80. [Google Scholar] [CrossRef]
- Al-Sarraf, M.; LeBlanc, M.; Giri, P.G.; Fu, K.K.; Cooper, J.; Vuong, T.; Forastiere, A.A.; Adams, G.; Sakr, W.A.; Schuller, D.E.; et al. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: Phase iii randomized intergroup study 0099. J. Clin. Oncol. 1998, 16, 1310–1317. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. Targeting apoptosis pathways in cancer therapy. Curr. Cancer. Drug. Targets 2004, 4, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef] [Green Version]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar]
- Niedzwiecki, A.; Roomi, M.W.; Kalinovsky, T.; Rath, M. Anticancer efficacy of polyphenols and their combinations. Nutrients 2016, 8, 552. [Google Scholar] [CrossRef] [Green Version]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.J.; Bao, J.L.; Chen, X.P.; Huang, M.; Wang, Y.T. Alkaloids isolated from natural herbs as the anticancer agents. Evid. Based Complement Altern. Med. 2012, 2012, 485042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levinson, H.Z. The defensive role of alkaloids in insects and plants. Experientia 1976, 32, 408–411. [Google Scholar] [CrossRef]
- Isah, T. Anticancer alkaloids from trees: Development into drugs. Pharm. Rev. 2016, 10, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Wang, B.; Yang, X.; Guo, H.; Zhang, K.; Zhu, Z.; Liu, J.; Hao, D. Picrasidine i from picrasma quassioides suppresses osteoclastogenesis via inhibition of rankl induced signaling pathways and attenuation of ros production. Cell Physiol. Biochem. 2017, 43, 1425–1435. [Google Scholar] [CrossRef]
- Yamashita, N.; Kondo, M.; Zhao, S.; Li, W.; Koike, K.; Nemoto, K.; Kanno, Y. Picrasidine g decreases viability of mda-mb 468 egfr-overexpressing triple-negative breast cancer cells through inhibition of egfr/stat3 signaling pathway. Bioorg. Med. Chem. Lett. 2017, 27, 2608–2612. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, X.; Fredimoses, M.; Song, M.; Chen, H.; Liu, K.; Lee, M.H.; Dong, Z. Fgfr2 regulation by picrasidine q inhibits the cell growth and induces apoptosis in esophageal squamous cell carcinoma. J. Cell Biochem. 2018, 119, 2231–2239. [Google Scholar] [CrossRef]
- Zhao, S.; Kanno, Y.; Li, W.; Wakatabi, H.; Sasaki, T.; Koike, K.; Nemoto, K.; Li, H. Picrasidine n is a subtype-selective pparbeta/delta agonist. J. Nat. Prod. 2016, 79, 879–885. [Google Scholar] [CrossRef]
- Zhao, S.; Kanno, Y.; Li, W.; Sasaki, T.; Zhang, X.; Wang, J.; Cheng, M.; Koike, K.; Nemoto, K.; Li, H. Identification of picrasidine c as a subtype-selective pparalpha agonist. J. Nat. Prod. 2016, 79, 3127–3133. [Google Scholar] [CrossRef]
- Yang, Y.T.; Hsieh, M.J.; Chuang, Y.C.; Lin, C.C.; Lo, Y.S.; Ho, H.Y.; Kumar, V.B.; Ko, J.L. Anticancer effects of picrasidine i on oral squamous cell carcinoma. Env. Toxicol. 2022, 37, 627–636. [Google Scholar] [CrossRef]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.; Zhu, F.; Zhang, J.; Keum, Y.S.; Zykova, T.; Yao, K.; Peng, C.; Zheng, D.; Cho, Y.Y.; Ma, W.Y.; et al. Mst1 promotes apoptosis through phosphorylation of histone h2ax. J. Biol. Chem. 2010, 285, 39108–39116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Madhunapantula, S.V.; Mosca, P.J.; Robertson, G.P. The akt signaling pathway: An emerging therapeutic target in malignant melanoma. Cancer Biol. 2011, 12, 1032–1049. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. Mapk signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharm. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- So, K.Y.; Kim, S.H.; Jung, K.T.; Lee, H.Y.; Oh, S.H. Mapk/jnk1 activation protects cells against cadmium-induced autophagic cell death via differential regulation of catalase and heme oxygenase-1 in oral cancer cells. Toxicol. Appl. Pharm. 2017, 332, 81–91. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Lin, C.C.; Lo, Y.S.; Chuang, Y.C.; Ho, H.Y.; Chen, M.K. Chrysosplenol d triggers apoptosis through heme oxygenase-1 and mitogen-activated protein kinase signaling in oral squamous cell carcinoma. Cancers 2021, 13, 4327. [Google Scholar] [CrossRef]
- Skrzypek, K.; Tertil, M.; Golda, S.; Ciesla, M.; Weglarczyk, K.; Collet, G.; Guichard, A.; Kozakowska, M.; Boczkowski, J.; Was, H.; et al. Interplay between heme oxygenase-1 and mir-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid Redox Signal. 2013, 19, 644–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.H.; Chung, M.J.; et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Lin, C.C.; Chuang, Y.C.; Lo, Y.S.; Hsieh, M.J.; Chen, M.K. Apoptotic effects of dehydrocrenatidine via jnk and erk pathway regulation in oral squamous cell carcinoma. Biomed Pharm. 2021, 137, 111362. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Lin, M.L.; Meng, M.; Chen, S.S. Galangin induces p53-independent s-phase arrest and apoptosis in human nasopharyngeal carcinoma cells through inhibiting pi3k-akt signaling pathway. Anticancer Res. 2018, 38, 1377–1389. [Google Scholar]
- Feng, S.; Wang, Y.; Zhang, R.; Yang, G.; Liang, Z.; Wang, Z.; Zhang, G. Curcumin exerts its antitumor activity through regulation of mir-7/skp2/p21 in nasopharyngeal carcinoma cells. Onco Targets 2017, 10, 2377–2388. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Krueger, A.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. Flice-inhibitory proteins: Regulators of death receptor-mediated apoptosis. Mol. Cell Biol. 2001, 21, 8247–8254. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Qu, Z.; Lin, Y.; Lee, C.S.; Tai, W.C.; Chen, S. Brevilin a induces cell cycle arrest and apoptosis in nasopharyngeal carcinoma. Front. Pharm. 2019, 10, 594. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. Map kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.Y.; Chen, P.J.; Lo, Y.S.; Lin, C.C.; Chuang, Y.C.; Hsieh, M.J.; Chen, M.K. Luteolin-7-o-glucoside inhibits cell proliferation and modulates apoptosis through the akt signaling pathway in human nasopharyngeal carcinoma. Env. Toxicol. 2021, 36, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.J.; Wang, C.W.; Lin, J.T.; Chuang, Y.C.; Hsi, Y.T.; Lo, Y.S.; Lin, C.C.; Chen, M.K. Celastrol, a plant-derived triterpene, induces cisplatin-resistance nasopharyngeal carcinoma cancer cell apoptosis though erk1/2 and p38 mapk signaling pathway. Phytomedicine 2019, 58, 152805. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am. J. Kidney Dis. 2017, 69, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme oxygenase 1 in the nervous system: Does it favor neuronal cell survival or induce neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, M.; Sitasawad, S. Mir-140-5p attenuates hypoxia-induced breast cancer progression by targeting nrf2/ho-1 axis in a keap1-independent mechanism. Cells 2021, 11, 12. [Google Scholar] [CrossRef]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes rsl3-induced ferroptosis by suppressing the nrf2/ho-1 signalling pathway in kras mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. Ho-1 induction in cancer progression: A matter of cell adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of nrf2/ho-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Jozkowicz, A.; Dulak, J. Ho-1/co system in tumor growth, angiogenesis and metabolism—targeting ho-1 as an anti-tumor therapy. Vasc. Pharm. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Tsuji, M.H.; Yanagawa, T.; Iwasa, S.; Tabuchi, K.; Onizawa, K.; Bannai, S.; Toyooka, H.; Yoshida, H. Heme oxygenase-1 expression in oral squamous cell carcinoma as involved in lymph node metastasis. Cancer Lett. 1999, 138, 53–59. [Google Scholar] [CrossRef]
- Tien, H.F.; Lee, F.Y.; Chuang, S.M.; Lin, C.T. Cytogenetic characterization of a nasopharyngeal carcinoma cell line and its subline. Cancer Genet. Cytogenet. 1990, 49, 31–36. [Google Scholar] [CrossRef]
- Liao, S.K.; Perng, Y.P.; Shen, Y.C.; Chung, P.J.; Chang, Y.S.; Wang, C.H. Chromosomal abnormalities of a new nasopharyngeal carcinoma cell line (npc-bm1) derived from a bone marrow metastatic lesion. Cancer Genet. Cytogenet. 1998, 103, 52–58. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Mahmood, T.; Yang, P.C. Western blot: Technique, theory, and trouble shooting. N. Am. J. Med. Sci. 2012, 4, 429–434. [Google Scholar]
- Mandelkow, R.; Gumbel, D.; Ahrend, H.; Kaul, A.; Zimmermann, U.; Burchardt, M.; Stope, M.B. Detection and quantification of nuclear morphology changes in apoptotic cells by fluorescence microscopy and subsequent analysis of visualized fluorescent signals. Anticancer Res. 2017, 37, 2239–2244. [Google Scholar] [CrossRef] [Green Version]
- Sakamuru, S.; Attene-Ramos, M.S.; Xia, M. Mitochondrial membrane potential assay. Methods Mol. Biol. 2016, 1473, 17–22. [Google Scholar] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, H.-Y.; Chen, P.-J.; Chuang, Y.-C.; Lo, Y.-S.; Lin, C.-C.; Hsieh, M.-J.; Chen, M.-K. Picrasidine I Triggers Heme Oxygenase-1-Induced Apoptosis in Nasopharyngeal Carcinoma Cells via ERK and Akt Signaling Pathways. Int. J. Mol. Sci. 2022, 23, 6103. https://doi.org/10.3390/ijms23116103
Ho H-Y, Chen P-J, Chuang Y-C, Lo Y-S, Lin C-C, Hsieh M-J, Chen M-K. Picrasidine I Triggers Heme Oxygenase-1-Induced Apoptosis in Nasopharyngeal Carcinoma Cells via ERK and Akt Signaling Pathways. International Journal of Molecular Sciences. 2022; 23(11):6103. https://doi.org/10.3390/ijms23116103
Chicago/Turabian StyleHo, Hsin-Yu, Ping-Ju Chen, Yi-Ching Chuang, Yu-Sheng Lo, Chia-Chieh Lin, Ming-Ju Hsieh, and Mu-Kuan Chen. 2022. "Picrasidine I Triggers Heme Oxygenase-1-Induced Apoptosis in Nasopharyngeal Carcinoma Cells via ERK and Akt Signaling Pathways" International Journal of Molecular Sciences 23, no. 11: 6103. https://doi.org/10.3390/ijms23116103
APA StyleHo, H.-Y., Chen, P.-J., Chuang, Y.-C., Lo, Y.-S., Lin, C.-C., Hsieh, M.-J., & Chen, M.-K. (2022). Picrasidine I Triggers Heme Oxygenase-1-Induced Apoptosis in Nasopharyngeal Carcinoma Cells via ERK and Akt Signaling Pathways. International Journal of Molecular Sciences, 23(11), 6103. https://doi.org/10.3390/ijms23116103