
Effects of Linkers and Substitutions on Multitarget Directed Ligands for Alzheimer’s Diseases: Emerging Paradigms and Strategies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
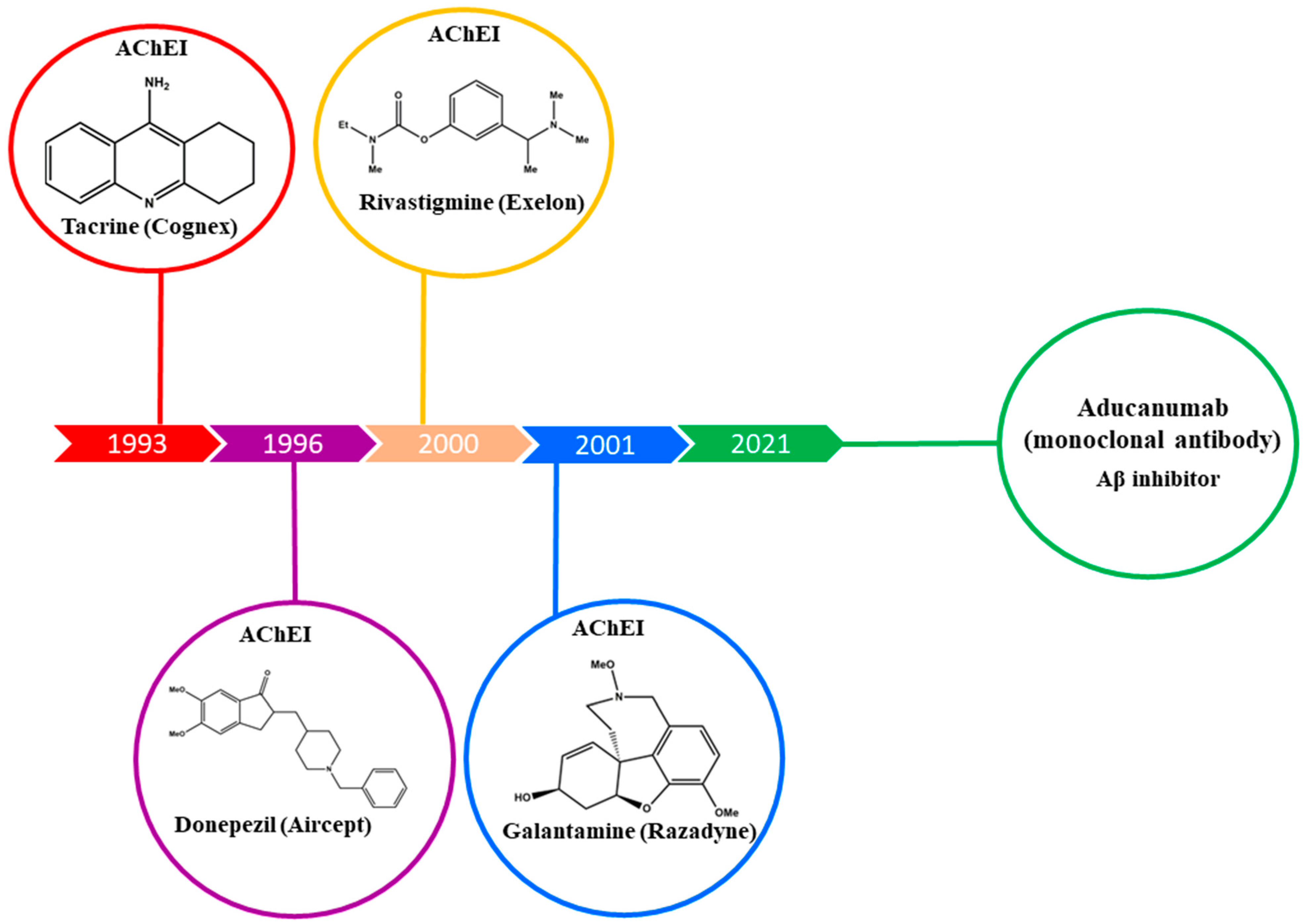
:1. Introduction
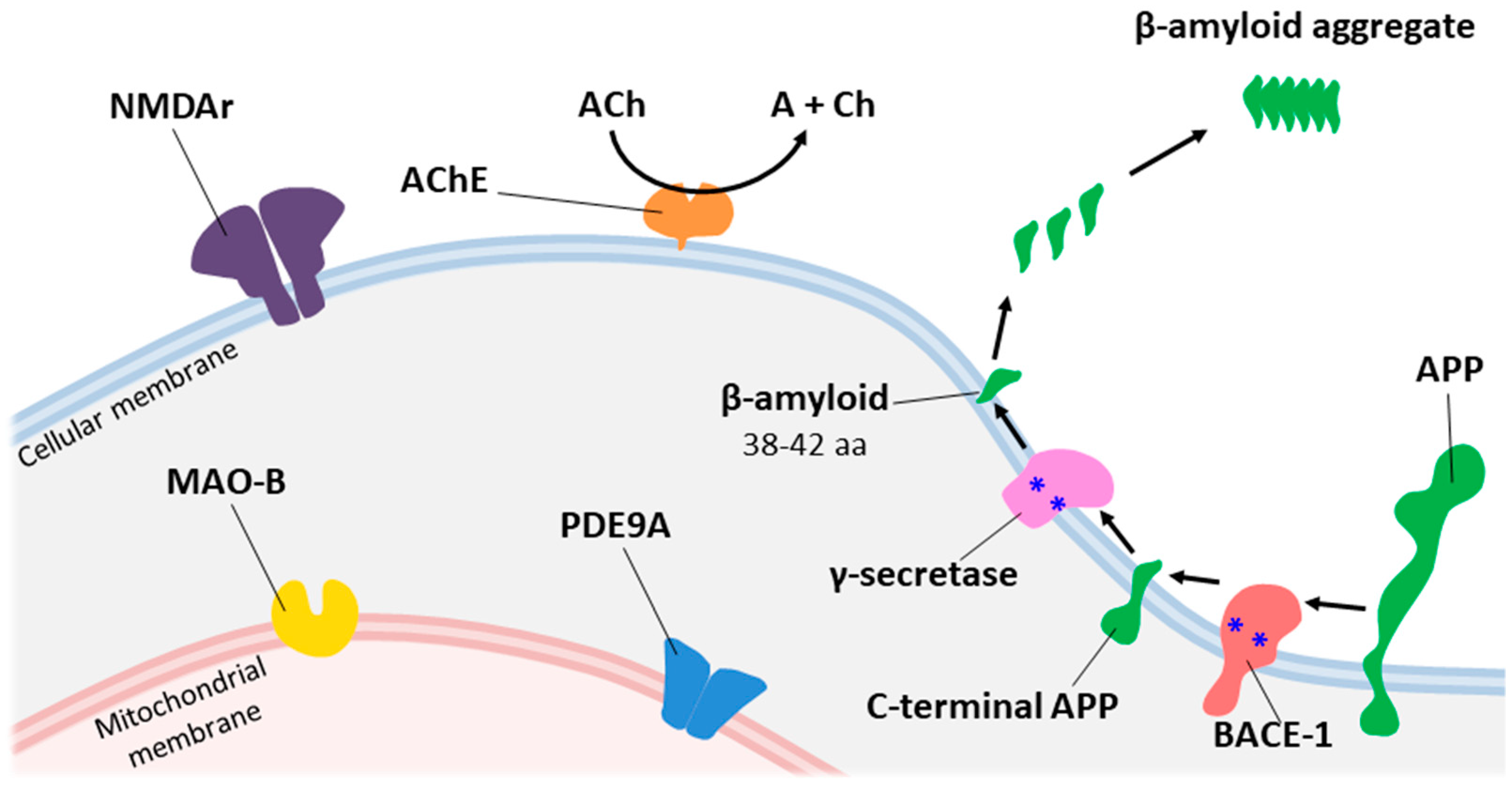
1.1. The Cholinergic Hypothesis
1.2. The Amyloid Cascade Hypothesis
1.3. The Oxidative Stress Hypothesis
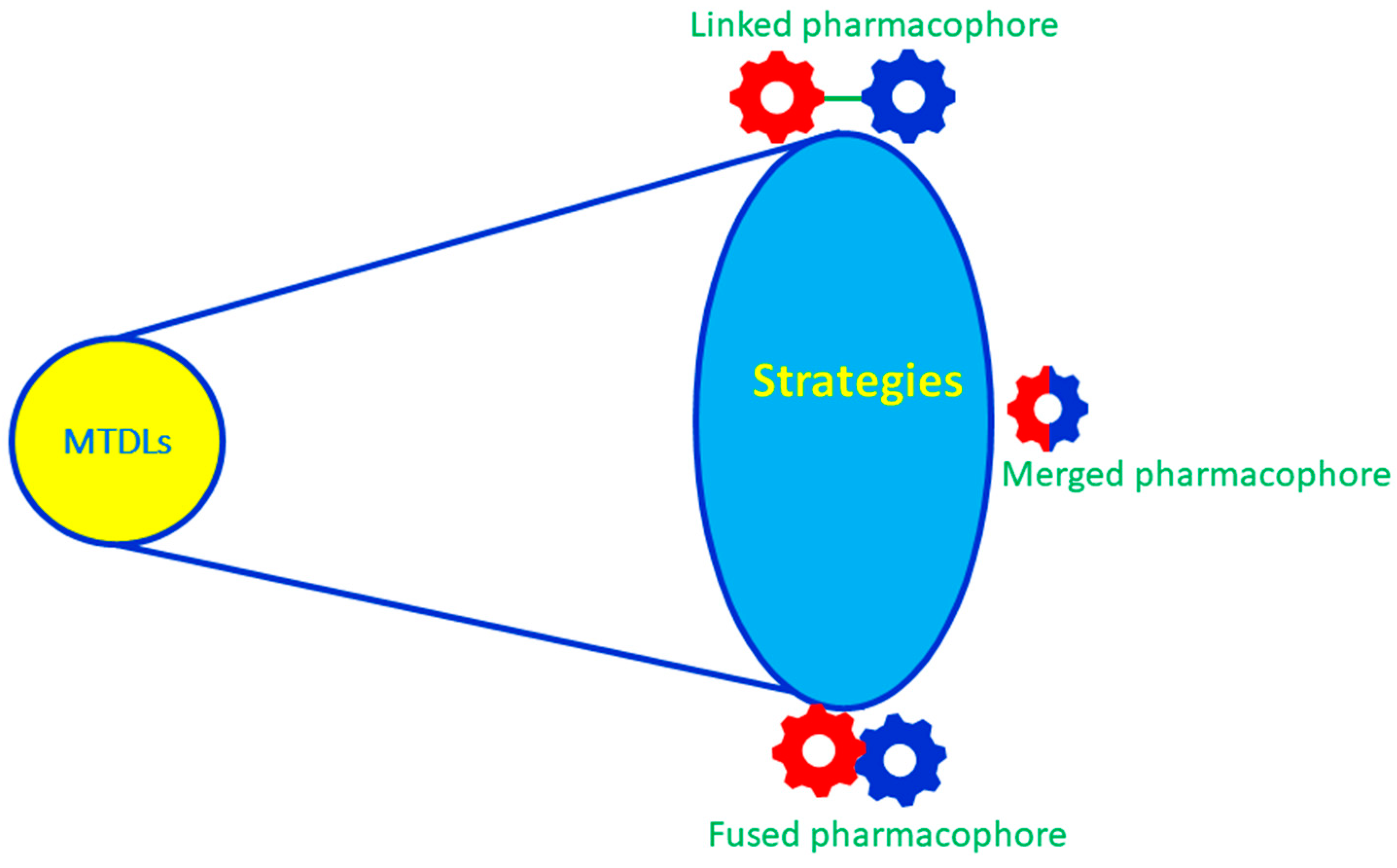
2. Design and Development of Multitargeted Directed Ligands (MTDLs)
2.1. Rational Design
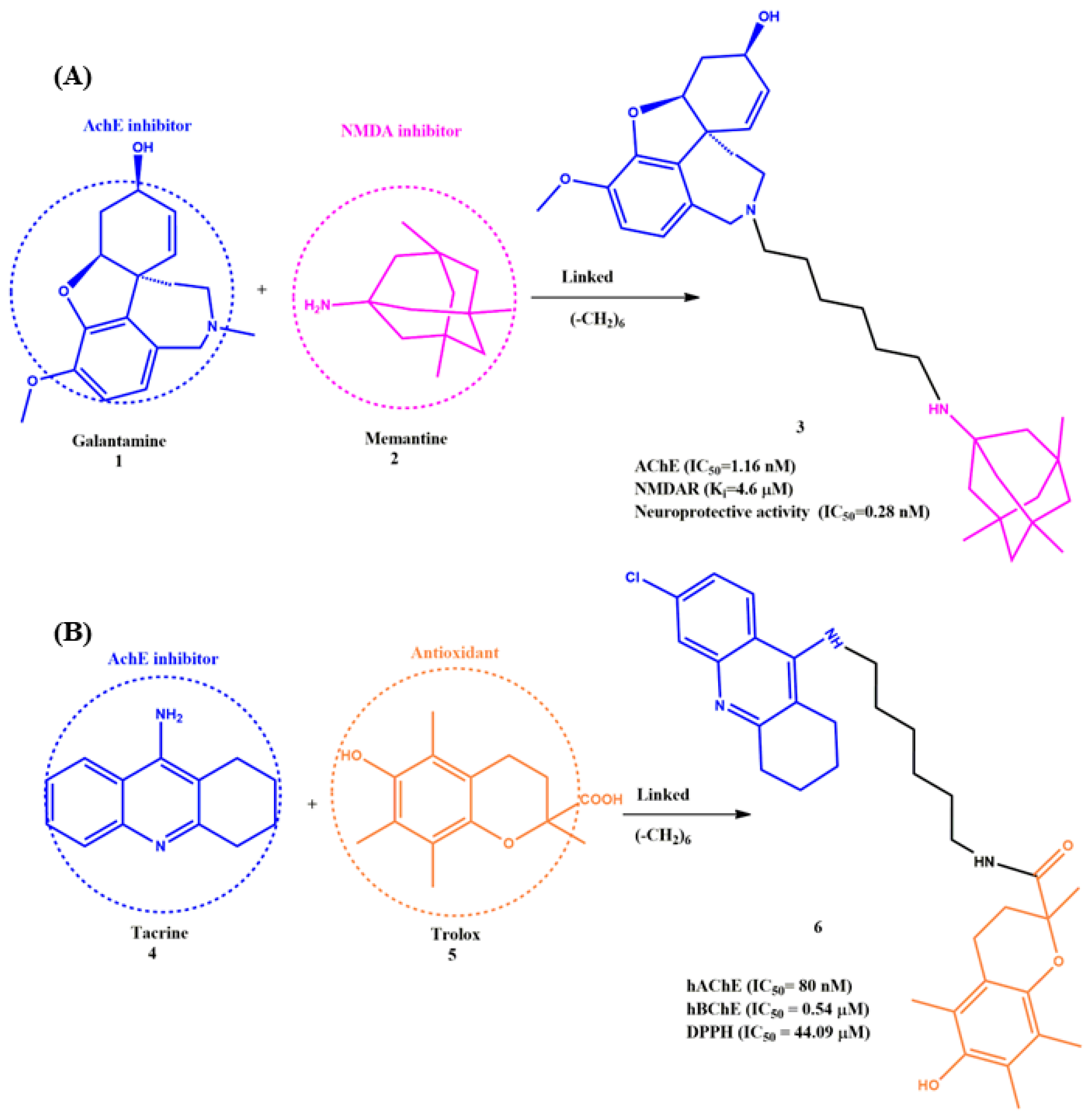
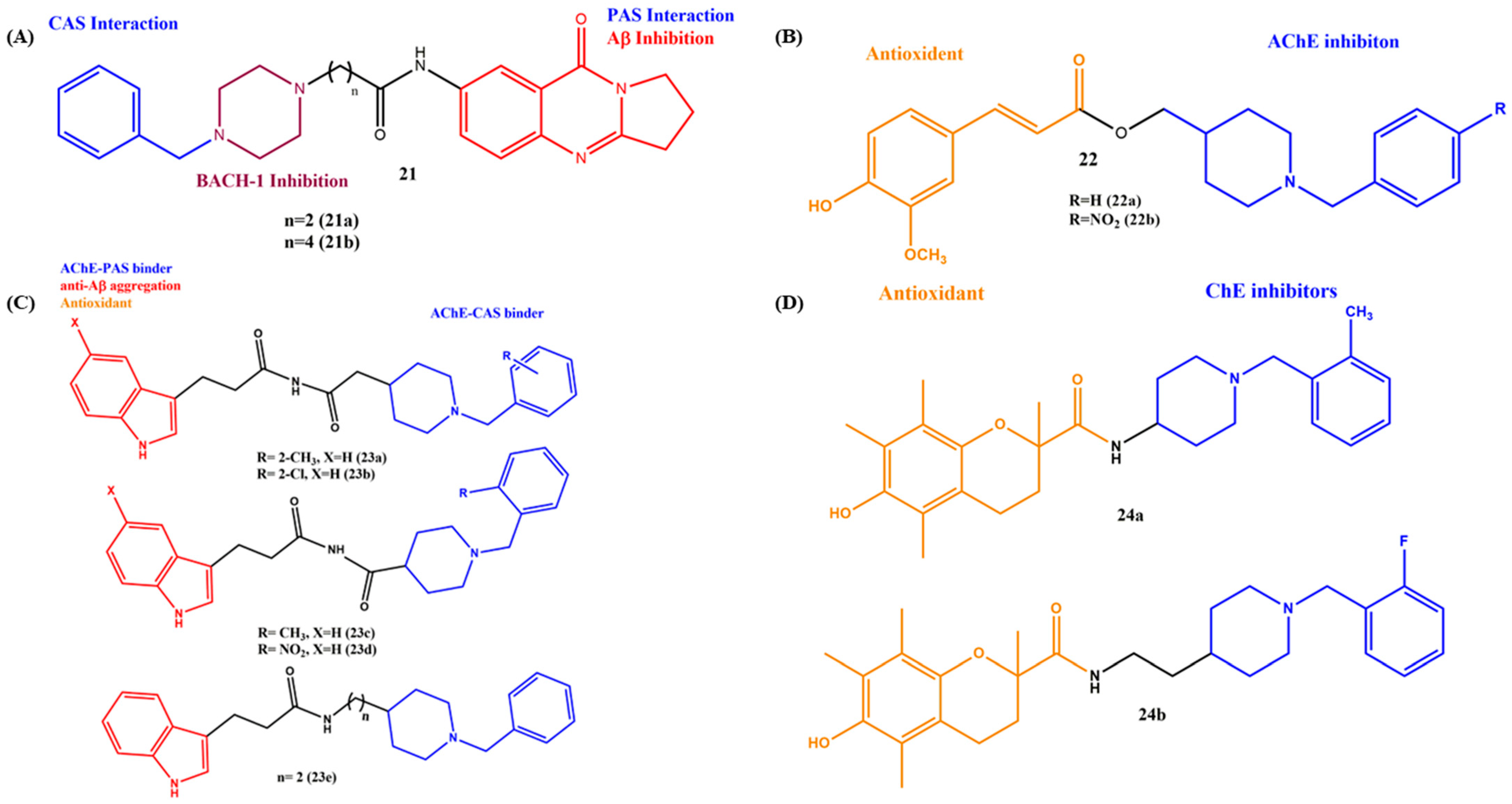
2.2. Linked MTDLs for AD Targets
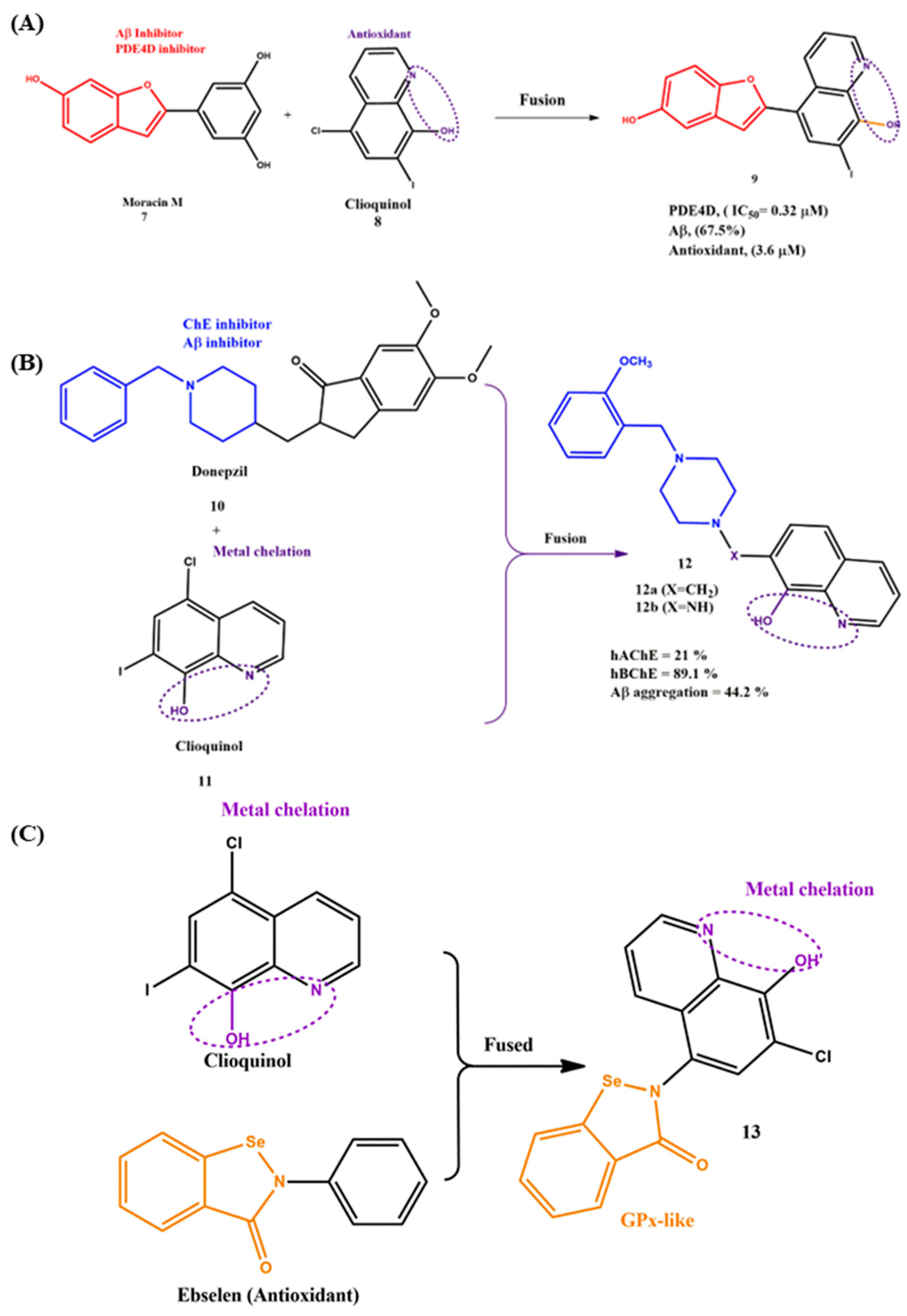
2.3. Fused MTDLs for AD Targets
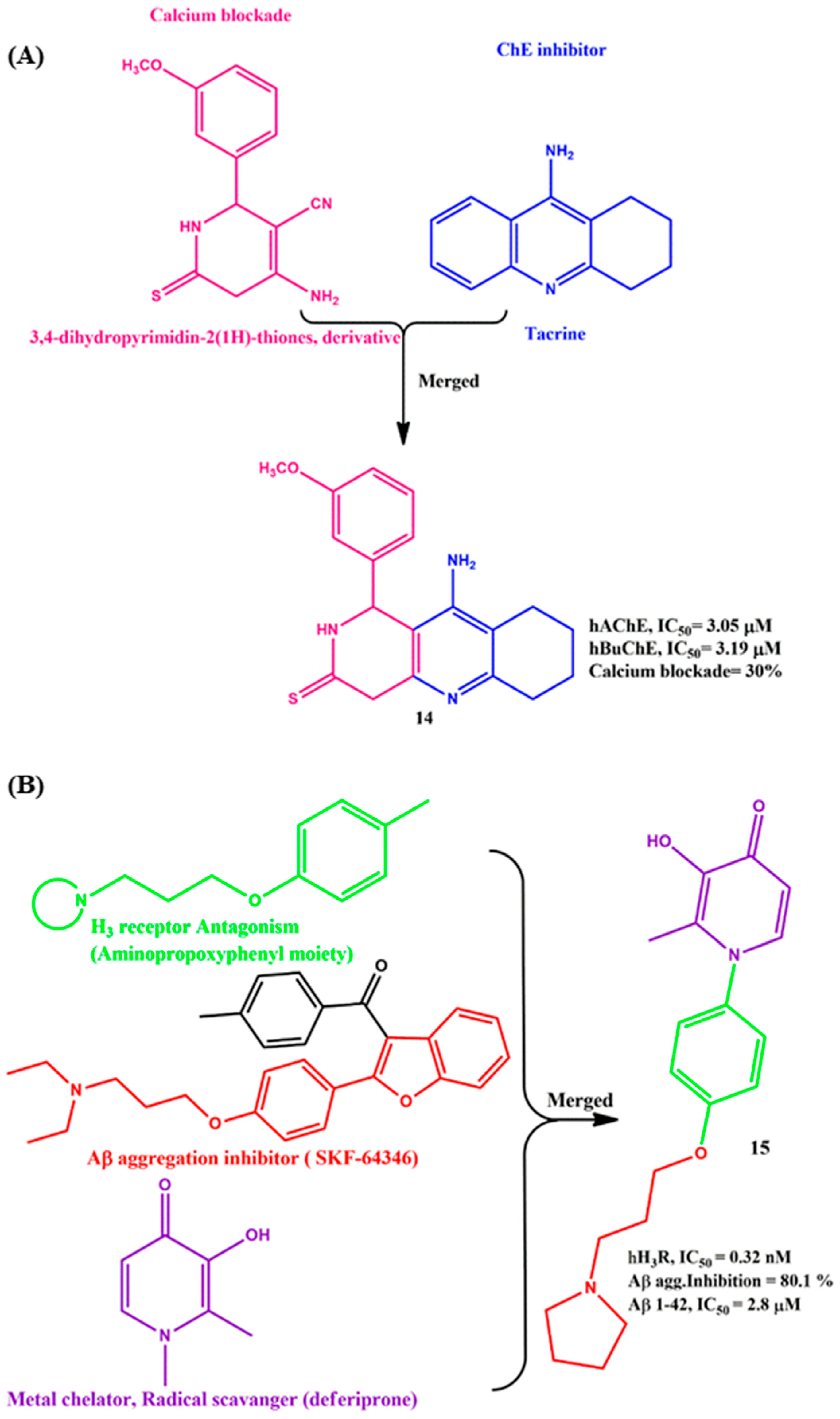
2.4. Merged MTDLs for AD Targets
3. Examples of MTD Pharmacophores
4. Effects of the Linkers and Substitutions on Pyrazolopyrimidinone Derived MTDLs
5. Effects of the Linkers and Substitutions on Donepezil-Derived MTDLs
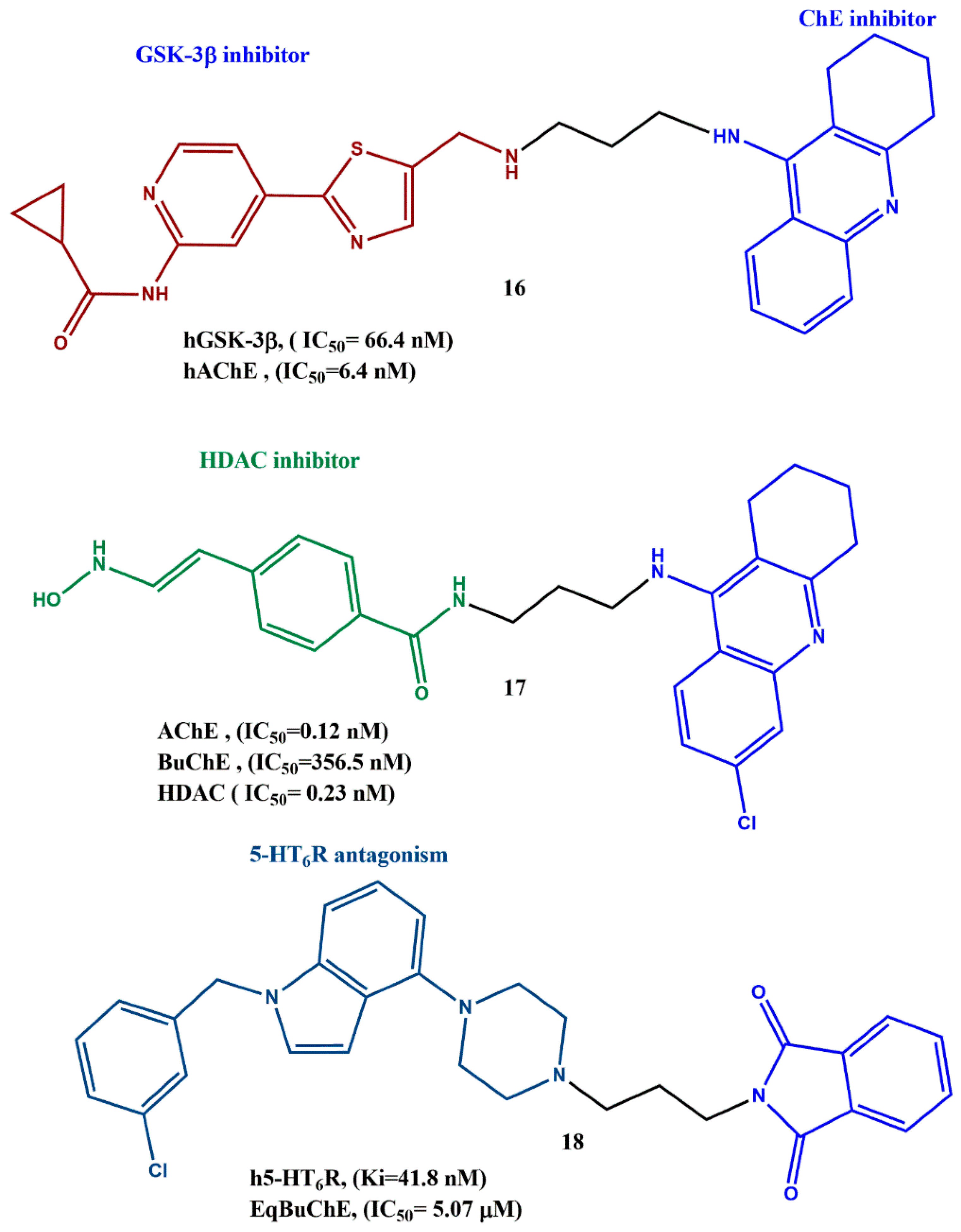
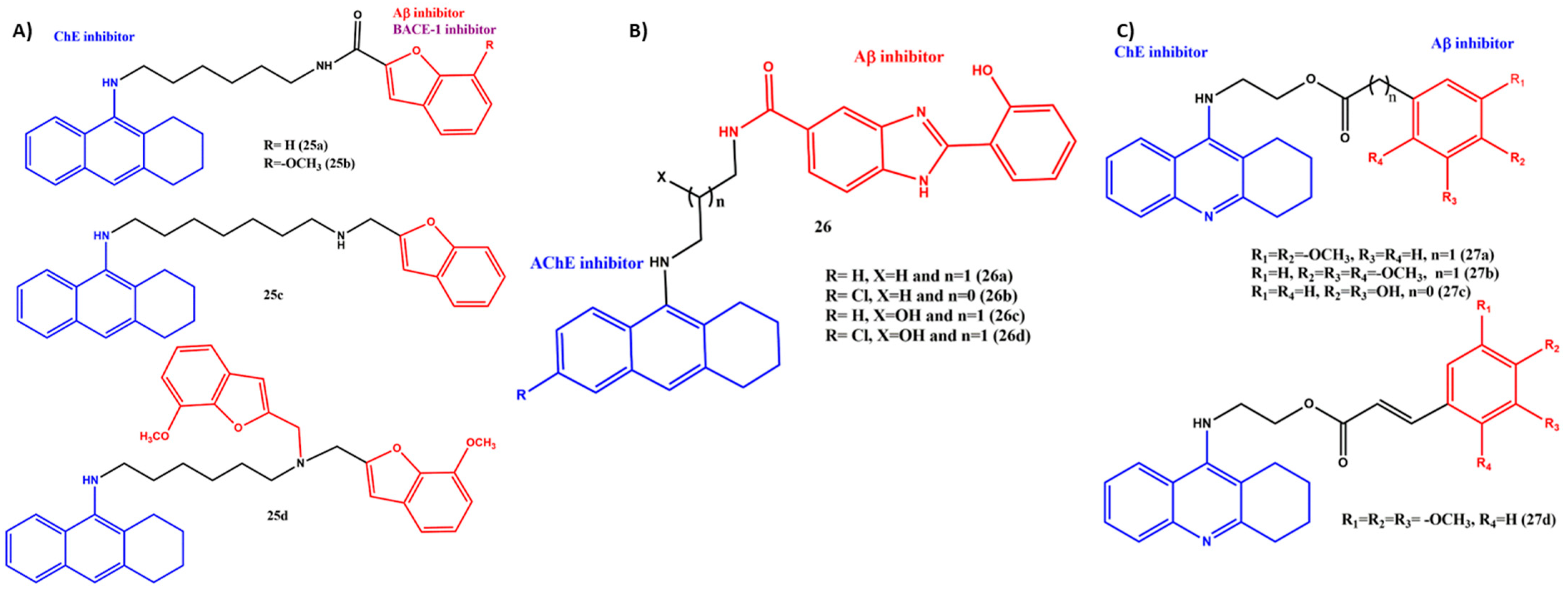
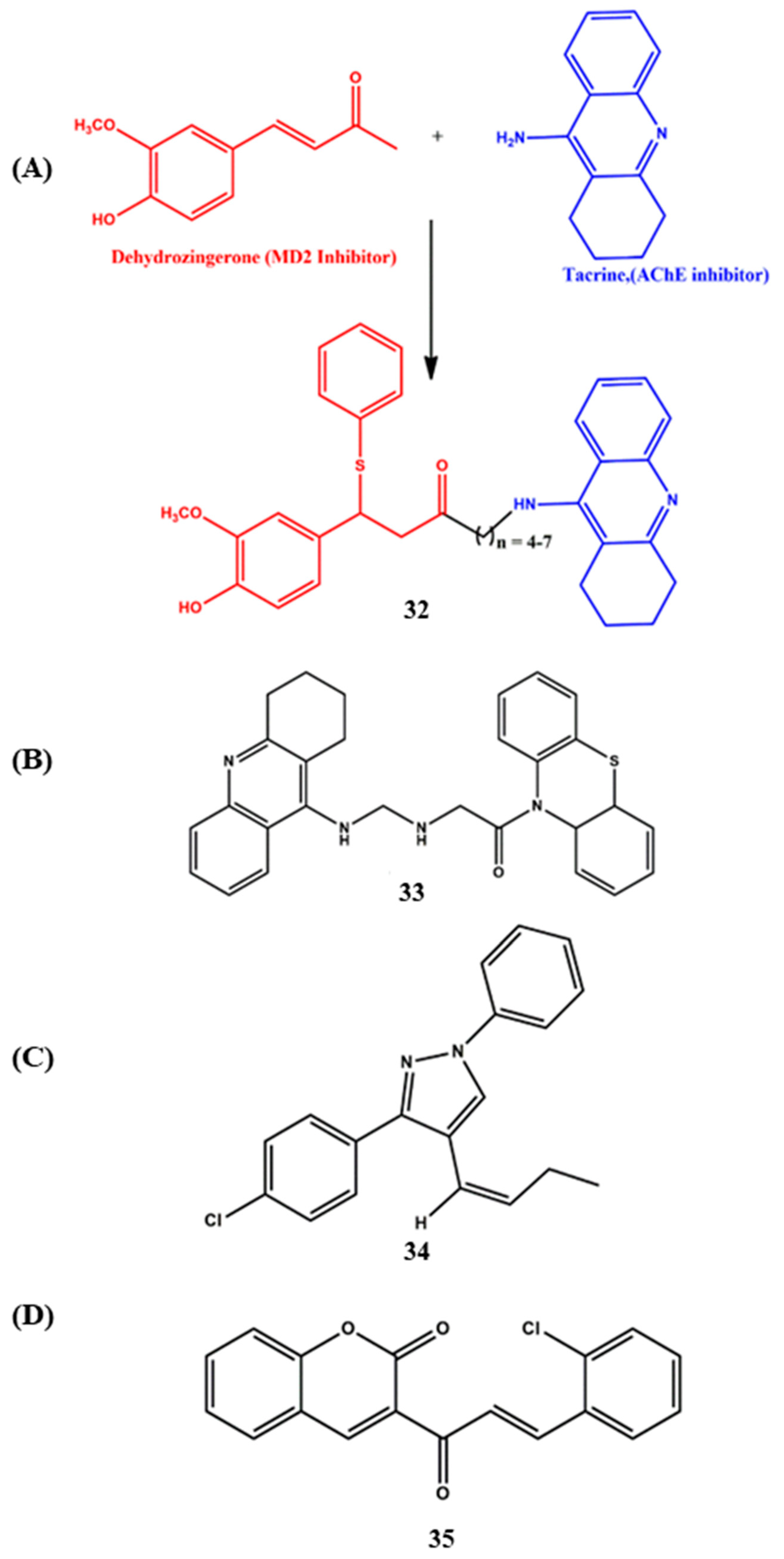
6. Effects of the Linkers and Substitutions on Tacrine-Derived MTDLs
7. Effects of the Linkers and Substitutions on Rivastigmine-Derived MTDLs
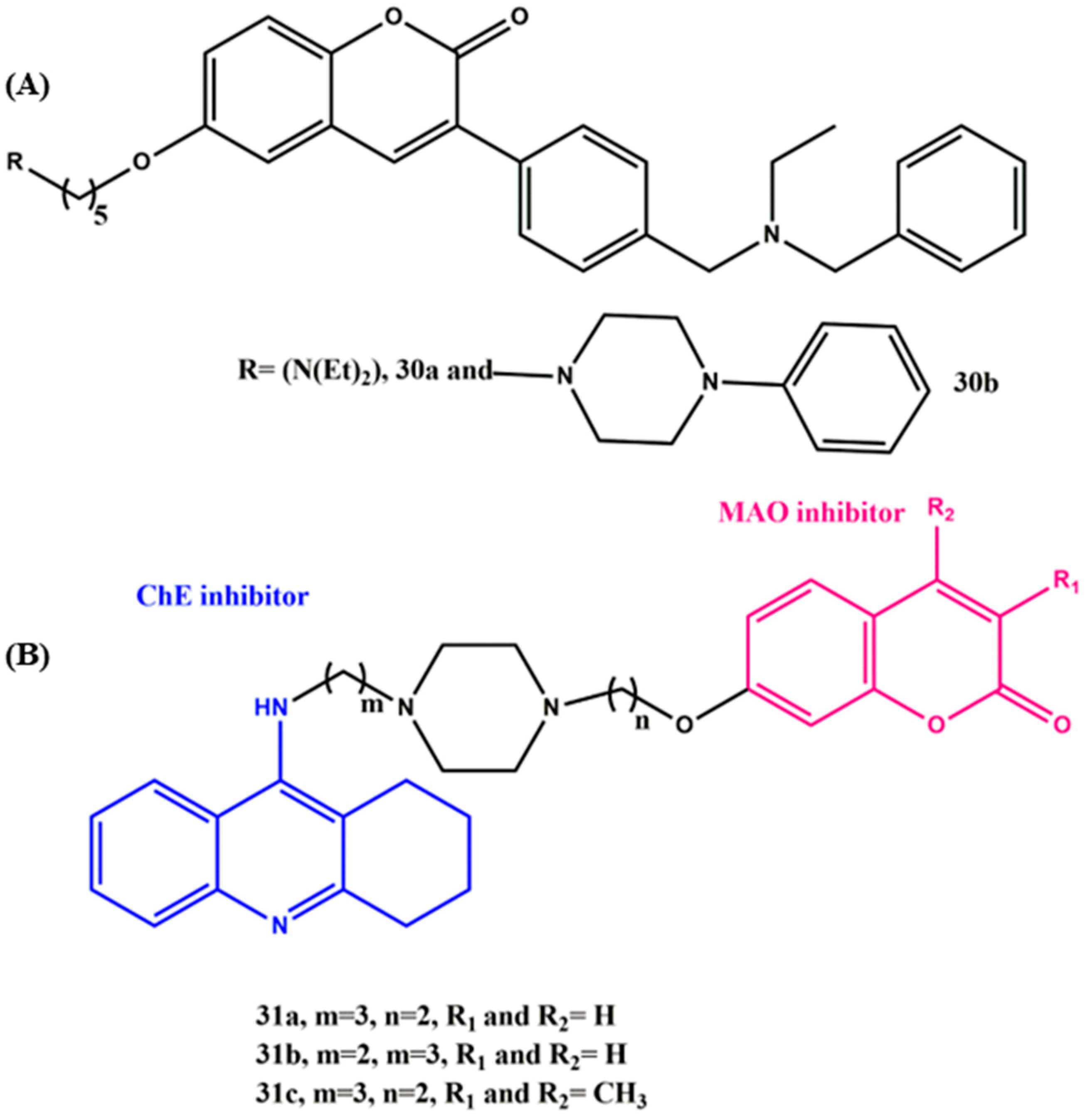
8. Effects of the Linkers and Substitutions on Coumarin-Derived MTDLs
9. Computational Strategies in MTDLs for AD Drug Design
10. Conclusions and Future Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| Ac-CoA | Acetyl-Coenzyme A |
| ACh | Acetylcholine |
| AD | Alzheimer’s Disease |
| APP | Amyloid Precursor Protein |
| Aβ | Amyloid-β-peptide |
| BACE-1 | Beta-secretase 1 |
| BBB | Blood Brain Barrier |
| BuChE | Butyrylcholinesterase |
| CNS | Central Nervous System |
| ChEs | Cholinesterases |
| CAS | Catalytic Active Site |
| ChAT | choline acetyltransferase |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
| EGFR | epidermal growth factor receptor |
| FDA | Food and Drug Administration |
| GSK-3β | Glycogen Synthase Kinase 3 beta |
| GPX-like | glutathione peroxidase-like |
| hAChE | human acetylcholinesterase |
| hBuChE | human butyrylcholinesterase |
| HER2 | human epidermal growth factor receptor 2 |
| MTDLs | Multitarget Directed Ligands |
| MAO-B | Monoamine oxidase B |
| MD | Molecular Dynamics |
| MVD | MolegroVirtual Docker |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NMDAR | N-methyl-D-aspartate receptor |
| PDE4D | Phosphodiesterase 4D |
| PAS | Peripheral Anionic Site |
| PDE9A | Phosphodiesterase 9A |
| PROTAC | Proteolysis Targeting Chimera |
| ROS | Reactive Oxygen Species |
| SGLT1 | sodium glucose co-transporter-1 |
| SGLT2 | sodium glucose co-transporter-2 |
| UV | Ultraviolet |
| WHO | World Health Organization |
| XO | Xanthine Oxidase |
References
- Medina-Franco, J.L.; Giulianotti, M.A.; Welmaker, G.S.; Houghten, R.A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov. Today 2013, 18, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- van der Schyf, C.J. The use of multi-target drugs in the treatment of neurodegenerative diseases. Expert Rev. Clin. Pharmacol. 2011, 4, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Sun, Y.; Sheng, W.; Liao, D. Designing multi-targeted agents: An emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 2017, 136, 195–211. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Gaugler, J.; James, B.; Johnson, T.; Scholz, K.; Weuve, J. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Dementia—World Health Organization 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 5 April 2022).
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative Stress Hypothesis in Alzheimer’s Disease. Free. Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Bolognesi, M.L. Polypharmacology in a Single Drug: Multitarget Drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef]
- Mi, J.; He, Y.; Yang, J.; Zhou, Y.; Zhu, G.; Wu, A.; Liu, W.; Sang, Z. Development of naringenin-O-carbamate derivatives as multi-target-directed liagnds for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2022, 60, 128574. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kumar, V.; Anand, P.; Kumar, V.; Dwivedi, A.R.; Kumar, V. Advancements in the development of multi-target directed ligands for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. 2022, 61, 116742. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Ji, L.; Wang, C.; Luo, H.; Li, S.; Peng, W.; Yin, F.; Lu, D.; Liu, X.; Kong, L.; et al. Synthesis and evaluation of multi-target-directed ligands with BACE-1 inhibitory and Nrf2 agonist activities as potential agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 219, 113441. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Ladostigil: A novel multimodal neuroprotective drug with cholinesterase and brain-selective monoamine oxidase inhibitory activities for Alzheimer’s disease treatment. Curr. Drug Targets 2012, 13, 483–494. [Google Scholar] [CrossRef]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef]
- Li, X.; Li, J.; Huang, Y.; Gong, Q.; Fu, Y.; Xu, Y.; Huang, J.; You, H.; Zhang, D.; Zhang, D.; et al. The novel therapeutic strategy of vilazodone-donepezil chimeras as potent triple-target ligands for the potential treatment of Alzheimer’s disease with comorbid depression. Eur. J. Med. Chem. 2022, 229, 114045. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Lushchekina, S.V.; Kovaleva, N.V.; Astakhova, T.Y.; Boltneva, N.P.; Rudakova, E.V.; Serebryakova, O.G.; Proshin, A.N.; Serkov, I.V.; Trofimova, T.P.; et al. Amiridine-piperazine hybrids as cholinesterase inhibitors and potential multitarget agents for Alzheimer’s disease treatment. Bioorganic Chem. 2021, 112, 104974. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Ivanco, L.S.; Lopresti, B.; Davis, J.G.; Constantine, G.; Mathis, C.A.; Moore, R.Y.; DeKosky, S.T. Cognitive correlates of alterations in acetylcholinesterase in Alzheimer’s disease. Neurosci. Lett. 2005, 380, 127–132. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Bibl, M.; Mollenhauer, B.; Lewczuk, P.; Esselmann, H.; Wolf, S.; Trenkwalder, C.; Otto, M.; Stiens, G.; Rüther, E.; Kornhuber, J.; et al. Validation of amyloid-β peptides in CSF diagnosis of neurodegenerative dementias. Mol. Psychiatry 2007, 12, 671–680. [Google Scholar] [CrossRef]
- Bibl, M.; Mollenhauer, B.; Esselmann, H.; Lewczuk, P.; Klafki, H.W.; Sparbier, K.; Smirnov, A.; Cepek, L.; Trenkwalder, C.; Rüther, E.; et al. CSF amyloid-β-peptides in Alzheimer’s disease, dementia with Lewy bodies and Parkinson’s disease dementia. Brain 2006, 129, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. Mens Sana Monogr. 2010, 8, 146. [Google Scholar] [CrossRef] [Green Version]
- Drug Approval Package: Aduhelm (Aducanumab-Avwa). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000TOC.cfm (accessed on 5 April 2022).
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Hung, C.H.L.; Cheng, S.S.Y.; Cheung, Y.T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.S.; Lee, S.M.Y.; Chang, R.C.C. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef]
- Collin, F.; Cheignon, C.; Hureau, C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomark. Med. 2018, 12, 201–203. [Google Scholar] [CrossRef]
- Wojtunik-Kulesza, K.A.; Oniszczuk, A.; Oniszczuk, T.; Waksmundzka-Hajnos, M. The influence of common free radicals and antioxidants on development of Alzheimer’s Disease. Biomed. Pharmacother. 2016, 78, 39–49. [Google Scholar] [CrossRef]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-Active Metals, Oxidative Stress, and Alzheimer’s Disease Pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.; Weinman, J. Adherence to cholinesterase inhibitors in Alzheimer’s Disease: A review. Dement. Geriatr. Cogn. Disord. 2013, 35, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Derrick, J.S.; Lim, M.H. Tools of the Trade: Investigations into Design Strategies of Small Molecules to Target Components in Alzheimer’s Disease. ChemBioChem 2015, 16, 887–898. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Mìnarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Carreiras, M.; Mendes, E.; Perry, M.; Francisco, A.; Marco-Contelles, J. The Multifactorial Nature of Alzheimer’s Disease for Developing Potential Therapeutics. Curr. Top. Med. Chem. 2014, 13, 1745–1770. [Google Scholar] [CrossRef]
- Schugar, H.; Green, D.E.; Bowen, M.L.; Scott, L.E.; Storr, T.; Böhmerle, K.; Thomas, F.; Allen, D.D.; Lockman, P.R.; Merkel, M.; et al. Lockman, Combating Alzheimer’s Disease with Multifunctional Molecules Designed for Metal Passivation. Angew. Chem. Int. Ed. 2007, 46, 1716–1718. [Google Scholar] [CrossRef]
- Han, J.; Lee, H.J.; Kim, K.Y.; Lee, S.J.C.; Suh, J.M.; Cho, J.; Chae, J.; Lim, M.H. Tuning Structures and Properties for Developing Novel Chemical Tools toward Distinct Pathogenic Elements in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 800–808. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Detoma, A.S.; Derrick, J.S.; Lim, M.H. The ongoing search for small molecules to study metal-Associated amyloid-β species in alzheimers disease. Acc. Chem. Res. 2014, 47, 2475–2482. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, G.T.; Edwards, K.R.; Zhao, Q. Rationale for Combination Therapy with Galantamine and Memantine in Alzheimer’s Disease. J. Clin. Pharmacol. 2006, 46, 17S–26S. [Google Scholar] [CrossRef] [PubMed]
- Simoni, E.; Daniele, S.; Bottegoni, G.; Pizzirani, D.; Trincavelli, M.L.; Goldoni, L.; Tarozzo, G.; Reggiani, A.; Martini, C.; Piomelli, D.; et al. Combining galantamine and memantine in multitargeted, new chemical entities potentially useful in Alzheimer’s disease. J. Med. Chem. 2012, 55, 9708–9721. [Google Scholar] [CrossRef] [Green Version]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine-Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low in Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Wang, B.; Li, W.; Huang, L.; Li, X. Design, synthesis, and evaluation of orally available clioquinol-moracin M hybrids as multitarget-directed ligands for cognitive improvement in a rat model of neurodegeneration in Alzheimer’s disease. J. Med. Chem. 2015, 58, 8616–8637. [Google Scholar] [CrossRef]
- Prati, F.; Bergamini, C.; Fato, R.; Soukup, O.; Korabecny, J.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L. Novel 8-Hydroxyquinoline Derivatives as Multitarget Compounds for the Treatment of Alzheimer′s Disease. ChemMedChem 2016, 11, 1284–1295. [Google Scholar] [CrossRef]
- Wang, Z.; Li, W.; Wang, Y.; Li, X.; Huang, L.; Li, X. Design, synthesis and evaluation of clioquinol-ebselen hybrids as multi-target-directed ligands against Alzheimer’s disease. RSC Adv. 2016, 6, 7139–7158. [Google Scholar] [CrossRef]
- Chioua, M.; Buzzi, E.; Moraleda, I.; Iriepa, I.; Maj, M.; Wnorowski, A.; Giovannini, C.; Tramarin, A.; Portali, F.; Ismaili, L.; et al. Tacripyrimidines, the first tacrine-dihydropyrimidine hybrids, as multi-target-directed ligands for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 155, 839–846. [Google Scholar] [CrossRef]
- Sheng, R.; Tang, L.; Jiang, L.; Hong, L.; Shi, Y.; Zhou, N.; Hu, Y. Novel 1-Phenyl-3-hydroxy-4-pyridinone Derivatives as Multifunctional Agents for the Therapy of Alzheimer’s Disease. ACS Chem. Neurosci. 2016, 7, 69–81. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Chen, T.K.; Zhou, J.T.; He, S.Y.; Yang, H.Y.; Chen, Y.; Qu, W.; Feng, F.; Sun, H.P. Dual GSK-3β/AChE Inhibitors as a New Strategy for Multitargeting Anti-Alzheimer’s Disease Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 171–176. [Google Scholar] [CrossRef]
- Xu, A.; He, F.; Zhang, X.; Li, X.; Ran, Y.; Wei, C.; Chou, C.J.; Zhang, R.; Wu, J. Tacrine-hydroxamate derivatives as multitarget-directed ligands for the treatment of Alzheimer’s disease: Design, synthesis, and biological evaluation. Bioorganic Chem. 2020, 98, 103721. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska, M.; Bucki, A.; Panek, D.; Siwek, A.; Fajkis, N.; Bednarski, M.; Zygmunt, M.; Godyń, J.; del Rio Valdivieso, A.; Kotańska, M.; et al. Anti-Alzheimer’s multitarget-directed ligands with serotonin 5-HT6 antagonist, butyrylcholinesterase inhibitory, and antioxidant activity. Arch. Pharm. 2019, 352, 1900041. [Google Scholar] [CrossRef] [PubMed]
- Suzen, S.; Cihaner, S.S.; Coban, T. Synthesis and Comparison of Antioxidant Properties of Indole-Based Melatonin Analogue Indole Amino Acid Derivatives. Chem. Biol. Drug Des. 2012, 79, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Kelada, M.; Walsh, J.M.D.; Devine, R.W.; McArdle, P.; Stephens, J.C. Synthesis of pyrazolopyrimidinones using a “one-pot” approach under microwave irradiation. Beilstein J. Org. Chem. 2018, 14, 1222–1228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Jiang, M.Y.; Le, M.L.; Zhang, B.; Zhou, Q.; Wu, Y.; Zhang, C.; Luo, H.B. Design, synthesis and evaluation of pyrazolopyrimidinone derivatives as novel PDE9A inhibitors for treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2020, 30, 127254. [Google Scholar] [CrossRef]
- Yu, Y.F.; Huang, Y.D.; Zhang, C.; Wu, X.N.; Zhou, Q.; Wu, D.; Wu, Y.; Luo, H.B. Discovery of Novel Pyrazolopyrimidinone Derivatives as Phosphodiesterase 9A Inhibitors Capable of Inhibiting Butyrylcholinesterase for Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2017, 8, 2522–2534. [Google Scholar] [CrossRef]
- Hu, J.; Huang, Y.D.; Pan, T.; Zhang, T.; Su, T.; Li, X.; Luo, H.B.; Huang, L. Design, Synthesis, and Biological Evaluation of Dual-Target Inhibitors of Acetylcholinesterase (AChE) and Phosphodiesterase 9A (PDE9A) for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 537–551. [Google Scholar] [CrossRef]
- Cai, P.; Fang, S.Q.; Yang, H.L.; Yang, X.L.; Liu, Q.H.; Kong, L.Y.; Wang, X.B. Donepezil-butylated hydroxytoluene (BHT) hybrids as Anti-Alzheimer’s disease agents with cholinergic, antioxidant, and neuroprotective properties. Eur. J. Med. Chem. 2018, 157, 161–176. [Google Scholar] [CrossRef]
- Korabecny, J.; Spilovska, K.; Mezeiova, E.; Benek, O.; Juza, R.; Kaping, D.; Soukup, O. A Systematic Review on Donepezil-based Derivatives as Potential Cholinesterase Inhibitors for Alzheimer’s Disease. Curr. Med. Chem. 2018, 26, 5625–5648. [Google Scholar] [CrossRef]
- Queda, F.; Calò, S.; Gwizdala, K.; Magalhães, J.D.; Cardoso, S.M.; Chaves, S.; Piemontese, L.; Santos, M.A. Novel Donepezil–Arylsulfonamide Hybrids as Multitarget-Directed Ligands for Potential Treatment of Alzheimer’s Disease. Molecules 2021, 26, 1658. [Google Scholar] [CrossRef]
- Mezeiova, E.; Chalupova, K.; Nepovimova, E.; Gorecki, L.; Prchal, L.; Malinak, D.; Kuca, K.; Soukup, O.; Korabecny, J. Donepezil Derivatives Targeting Amyloid-β Cascade in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 772–800. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Sheng, J.; Sun, Y.; Lu, C.; Yan, J.; Liu, A.; Luo, H.B.; Huang, L.; Li, X. Synthesis and evaluation of multi-target-directed ligands against Alzheimer’s disease based on the fusion of donepezil and ebselen. J. Med. Chem. 2013, 56, 9089–9099. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Tomás, D.; Hiremathad, A.; Capriati, V.; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J. Enzym. Inhib. Med. Chem. 2018, 33, 1212–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Liu, X.; Xie, J.; Ma, F. Novel Deoxyvasicinone-Donepezil Hybrids as Potential Multitarget Drug Candidates for Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Dias, K.S.T.; de Paula, C.T.; dos Santos, T.; Souza, I.N.O.; Boni, M.S.; Guimarães, M.J.R.; da Silva, F.M.R.; Castro, N.G.; Neves, G.A.; Veloso, C.C.; et al. Design, synthesis and evaluation of novel feruloyl-donepezil hybrids as potential multitarget drugs for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 130, 440–457. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.M.; Li, X.M.; Li, F.; Wu, J.J.; Kong, L.Y.; Wang, X.B. Synthesis and evaluation of multi-target-directed ligands for the treatment of Alzheimer’s disease based on the fusion of donepezil and melatonin. Bioorganic Med. Chem. 2016, 24, 4324–4338. [Google Scholar] [CrossRef]
- Cai, P.; Fang, S.Q.; Yang, X.L.; Wu, J.J.; Liu, Q.H.; Hong, H.; Wang, X.B.; Kong, L.Y. Rational Design and Multibiological Profiling of Novel Donepezil-Trolox Hybrids against Alzheimer’s Disease, with Cholinergic, Antioxidant, Neuroprotective, and Cognition Enhancing Properties. ACS Chem. Neurosci. 2017, 8, 2496–2511. [Google Scholar] [CrossRef]
- Eckroat, T.J.; Manross, D.L.; Cowan, S.C. Merged Tacrine-Based, Multitarget-Directed Acetylcholinesterase Inhibitors 2015–Present: Synthesis and Biological Activity. Int. J. Mol. Sci. 2020, 21, 5965. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, H.; Zhu, J.; Gu, K.; Li, Q.; He, S.; Lu, X.; Tan, R.; Pei, Y.; Wu, L.; et al. Design, synthesis, in vitro and in vivo evaluation of tacrine-cinnamic acid hybrids as multi-target acetyl- and butyrylcholinesterase inhibitors against Alzheimer’s disease. RSC Adv. 2017, 7, 33851–33867. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Bolognesi, Sustainable Drug Discovery of Multi-Target-Directed Ligands for Alzheimer’s Disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef]
- Javed, M.A.; Ashraf, N.; Jan, M.S.; Mahnashi, M.H.; Alqahtani, Y.S.; Alyami, B.A.; Alqarni, A.O.; Asiri, Y.I.; Ikram, M.; Sadiq, A.; et al. Structural Modification, in Vitro, in Vivo, Ex Vivo, and in Silico Exploration of Pyrimidine and Pyrrolidine Cores for Targeting Enzymes Associated with Neuroinflammation and Cholinergic Deficit in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 4123–4143. [Google Scholar] [CrossRef] [PubMed]
- Sameem, B.; Saeedi, M.; Mahdavi, M.; Shafiee, A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 128, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Lamba, D.; Zhang, L.; Lou, Y.; Xu, C.; Kang, D.; Chen, L.; Xu, Y.; Zhang, L.; de Simone, A.; et al. Novel Tacrine-Benzofuran Hybrids as Potent Multitarget-Directed Ligands for the Treatment of Alzheimers Disease: Design, Synthesis, Biological Evaluation, and X-ray Crystallography. J. Med. Chem. 2016, 59, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel Tacrine-Hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 148, 255–267. [Google Scholar] [CrossRef]
- Zhang, C.; Du, Q.Y.; Chen, L.; Wu, W.H.; Liao, S.Y.; Yu, L.H.; Liang, X.T. Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as multi-targeted compounds against Alzheimer’s disease. Eur. J. Med. Chem. 2016, 116, 200–209. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Shi, J.; Cheng, X.; Zhu, G.; Wei, R.; Ma, Q.; Yu, L.; Zhao, Y.; Tan, Z.; et al. Apigenin-rivastigmine hybrids as multi-target-directed liagnds for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2020, 187, 111958. [Google Scholar] [CrossRef]
- Sang, Z.; Li, Y.; Qiang, X.; Xiao, G.; Liu, Q.; Tan, Z.; Deng, Y. Multifunctional scutellarin–rivastigmine hybrids with cholinergic, antioxidant, biometal chelating and neuroprotective properties for the treatment of Alzheimer’s disease. Bioorganic Med. Chem. 2015, 23, 668–680. [Google Scholar] [CrossRef]
- de Souza, L.G.; Rennã, M.N.; Figueroa-Villar, J.D. Coumarins as cholinesterase inhibitors: A review. Chem. Biol. Interact. 2016, 254, 11–23. [Google Scholar] [CrossRef]
- Pisani, L.; Farina, R.; Nicolotti, O.; Gadaleta, D.; Soto-Otero, R.; Catto, M.; di Braccio, M.; Mendez-Alvarez, E.; Carotti, A. In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur. J. Med. Chem. 2015, 89, 98–105. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Lan, J.S.; Ding, J.; Sun, Y.; Fang, Y.; Jiang, N.; Yang, Z.; Sun, L.; Jin, Y.; et al. Coumarin-dithiocarbamate hybrids as novel multitarget AChE and MAO-B inhibitors against Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorganic Chem. 2018, 81, 512–528. [Google Scholar] [CrossRef]
- Montanari, S.; Bartolini, M.; Neviani, P.; Belluti, F.; Gobbi, S.; Pruccoli, L.; Tarozzi, A.; Falchi, F.; Andrisano, V.; Miszta, P.; et al. Multitarget Strategy to Address Alzheimer’s Disease: Design, Synthesis, Biological Evaluation, and Computational Studies of Coumarin-Based Derivatives. ChemMedChem 2016, 11, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.S.; Wang, X.; Jiang, N.; Yu, W.; Wang, K.D.G.; Lan, J.S.; Li, Z.R.; Kong, L.Y. Multi-target tacrine-coumarin hybrids: Cholinesterase and monoamine oxidase B inhibition properties against Alzheimer’s disease. Eur. J. Med. Chem. 2015, 95, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Eslami, M.; Nezafat, N.; Negahdaripour, M.; Ghasemi, Y. Computational approach to suggest a new multi-target-directed ligand as a potential medication for Alzheimer’s disease. J. Biomol. Struct. Dyn. 2019, 37, 4825–4839. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.-L.; Chen, Y.; Zhu, S.-J.; Gan, C.-S.; Pan, J.; Zhou, A. Design and synthesis of tacrine-phenothiazine hybrids as multitarget drugs for Alzheimer’s disease. Med. Chem. Res. 2014, 23, 3546–3557. [Google Scholar] [CrossRef]
- Kumar, A.; Jain, S.; Parle, M.; Jain, N.; Kumar, P. 3-Aryl-1-Phenyl-1H-Pyrazole Derivatives as New Multitarget Directed Ligands for the Treatment of Alzheimer’s Disease, with Acetylcholinesterase and Monoamine Oxidase Inhibitory Properties. EXCLI J. 2013, 12, 1030. Available online: https://pmc/articles/PMC4904743/ (accessed on 6 April 2022).
- Rehuman, N.A.; Oh, J.M.; Nath, L.R.; Khames, A.; Abdelgawad, M.A.; Gambacorta, N.; Nicolotti, O.; Jat, R.K.; Kim, H.; Mathew, B. Halogenated Coumarin−Chalcones as Multifunctional Monoamine Oxidase-B and Butyrylcholinesterase Inhibitors. ACS Omega 2021, 6, 28182–28193. [Google Scholar] [CrossRef]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the Dual Kinase Inhibitor Lapatinib (GW572016) against HER-2-Overexpressing and Trastuzumab-Treated Breast Cancer Cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2016, 17, 907–918. [Google Scholar] [CrossRef]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; Davidson, M.B.; Einhorn, D.; Garvey, W.T.; et al. American Association of Clinical Endocrinologists’ comprehensive diabetes management algorithm 2013 consensus statement—Executive summary. Endocr. Pract. 2013, 19, 536–557. [Google Scholar] [CrossRef] [Green Version]
- Cefalo, C.M.A.; Cinti, F.; Moffa, S.; Impronta, F.; Sorice, G.P.; Mezza, T.; Pontecorvi, A.; Giaccari, A. Sotagliflozin, the first dual SGLT inhibitor: Current outlook and perspectives. Cardiovasc. Diabetol. 2019, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Hussein, Z.; Wentworth, J.M.; Nankervis, A.J.; Proietto, J.; Colman, P.G. Effectiveness and side effects of thiazolidinediones for type 2 diabetes: Real-life experience from a tertiary hospital. Med. J. Aust. 2004, 181, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Trial of ARV-110 in Patients with Metastatic Castration Resistant Prostate Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03888612 (accessed on 6 April 2022).
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.Ü.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, 5154. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pravin, N.; Jozwiak, K. Effects of Linkers and Substitutions on Multitarget Directed Ligands for Alzheimer’s Diseases: Emerging Paradigms and Strategies. Int. J. Mol. Sci. 2022, 23, 6085. https://doi.org/10.3390/ijms23116085
Pravin N, Jozwiak K. Effects of Linkers and Substitutions on Multitarget Directed Ligands for Alzheimer’s Diseases: Emerging Paradigms and Strategies. International Journal of Molecular Sciences. 2022; 23(11):6085. https://doi.org/10.3390/ijms23116085
Chicago/Turabian StylePravin, Narayanaperumal, and Krzysztof Jozwiak. 2022. "Effects of Linkers and Substitutions on Multitarget Directed Ligands for Alzheimer’s Diseases: Emerging Paradigms and Strategies" International Journal of Molecular Sciences 23, no. 11: 6085. https://doi.org/10.3390/ijms23116085
APA StylePravin, N., & Jozwiak, K. (2022). Effects of Linkers and Substitutions on Multitarget Directed Ligands for Alzheimer’s Diseases: Emerging Paradigms and Strategies. International Journal of Molecular Sciences, 23(11), 6085. https://doi.org/10.3390/ijms23116085