
PLOD2 Is a Prognostic Marker in Glioblastoma That Modulates the Immune Microenvironment and Tumor Progression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
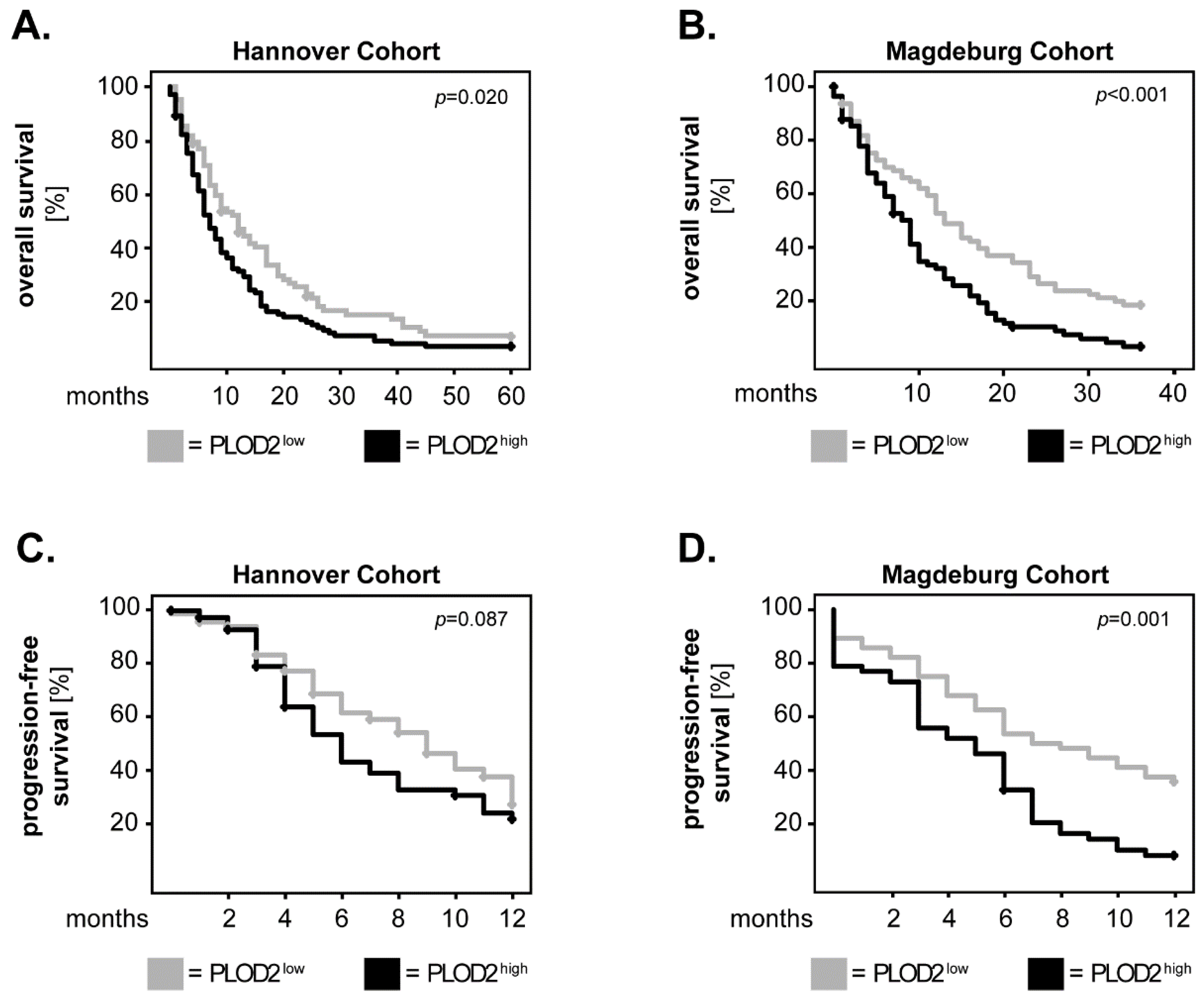
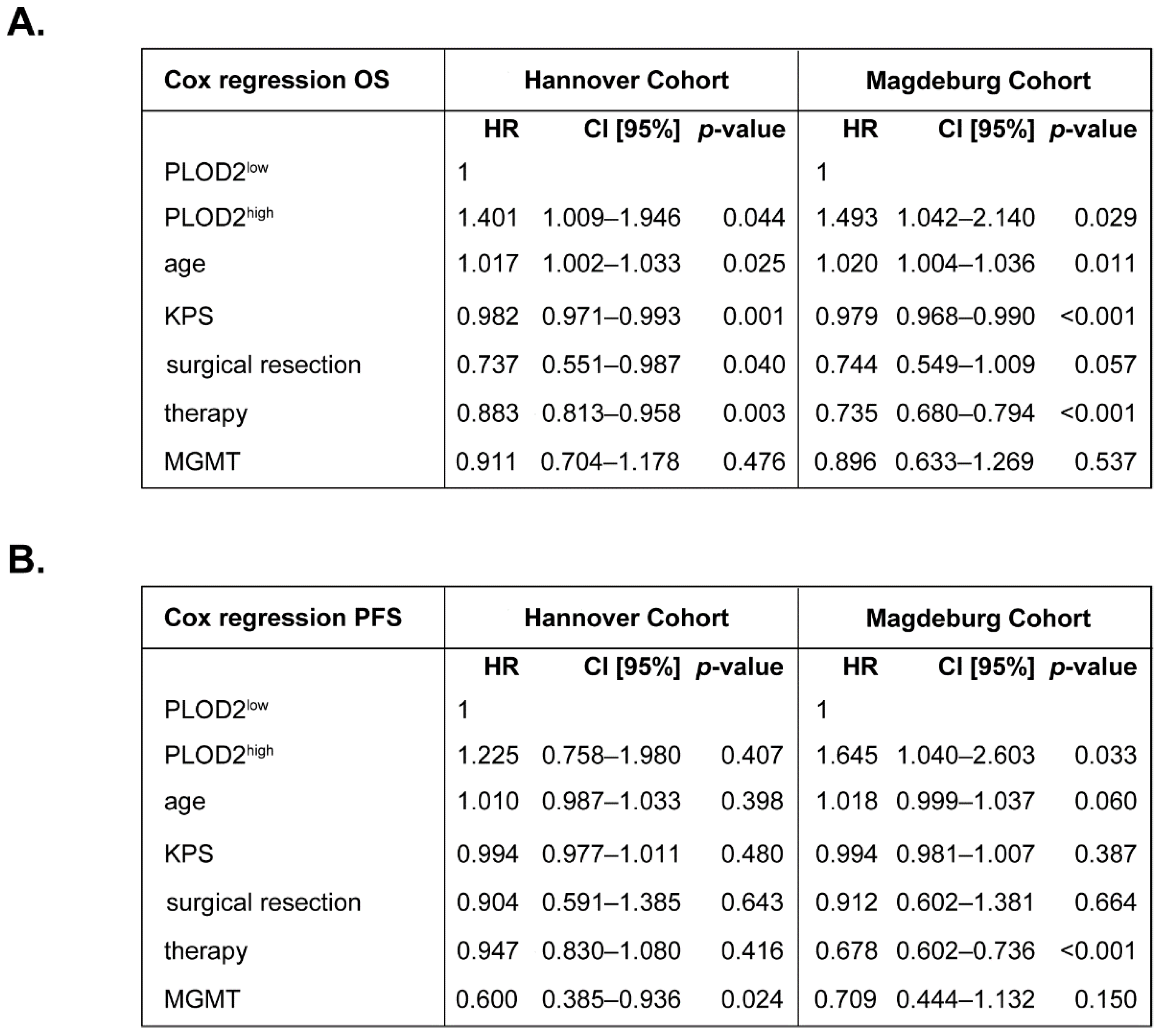
2.1. PLOD2 Associates with and Predicts Poor Overall Survival of GBM Patients
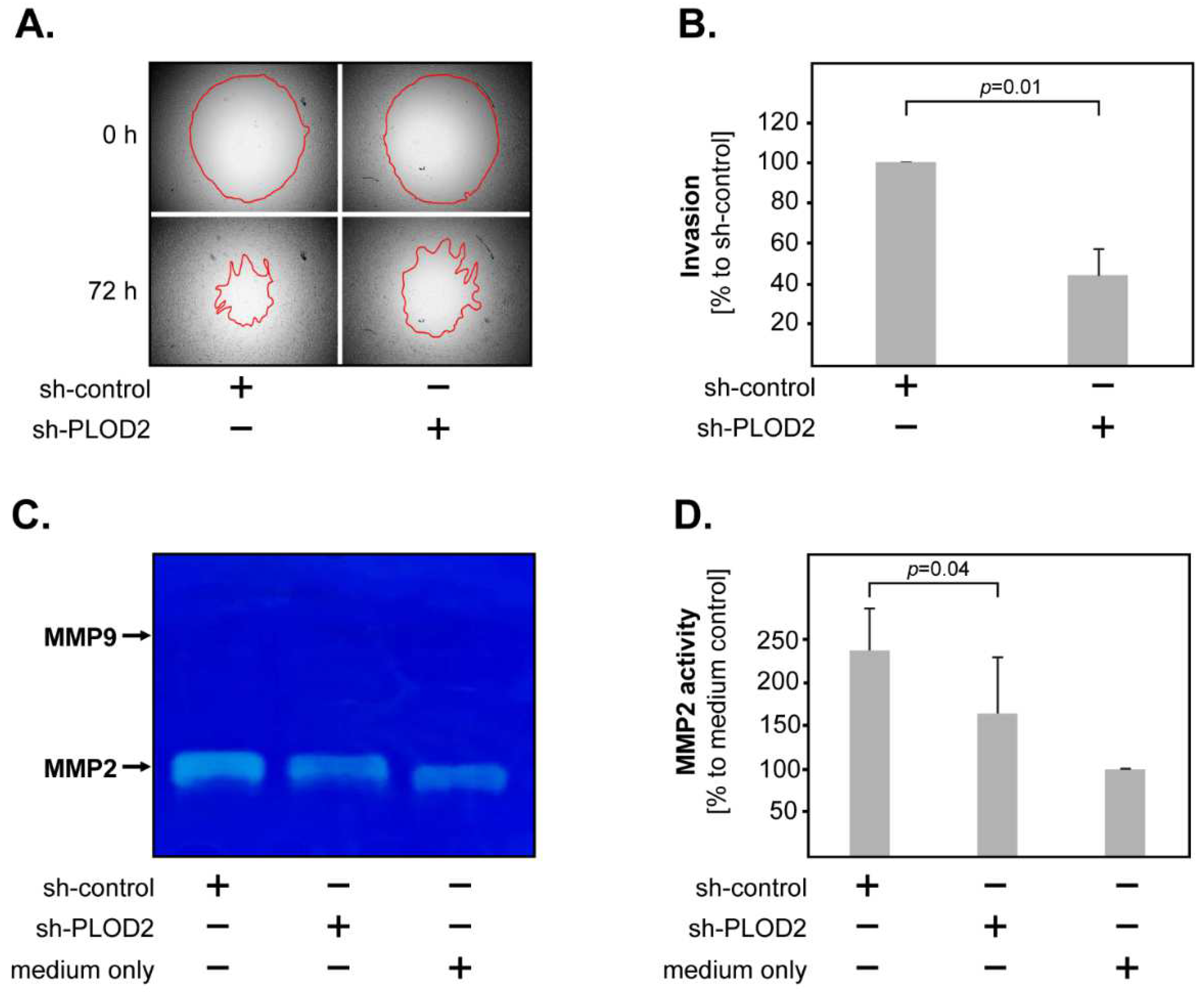
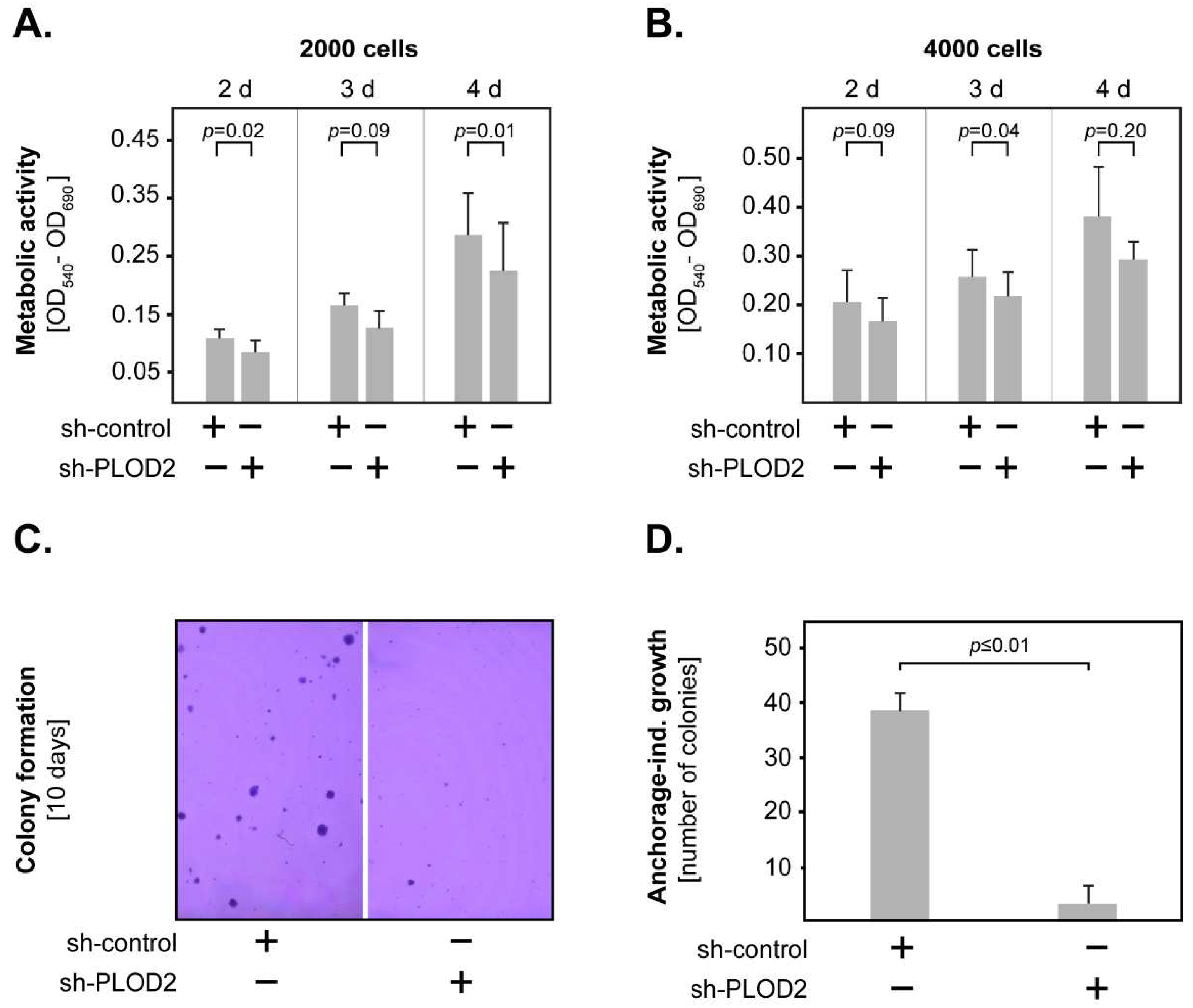
2.2. PLOD2 Promotes the Invasion, Proliferation and Anchorage-Independent Growth of GBM Cells
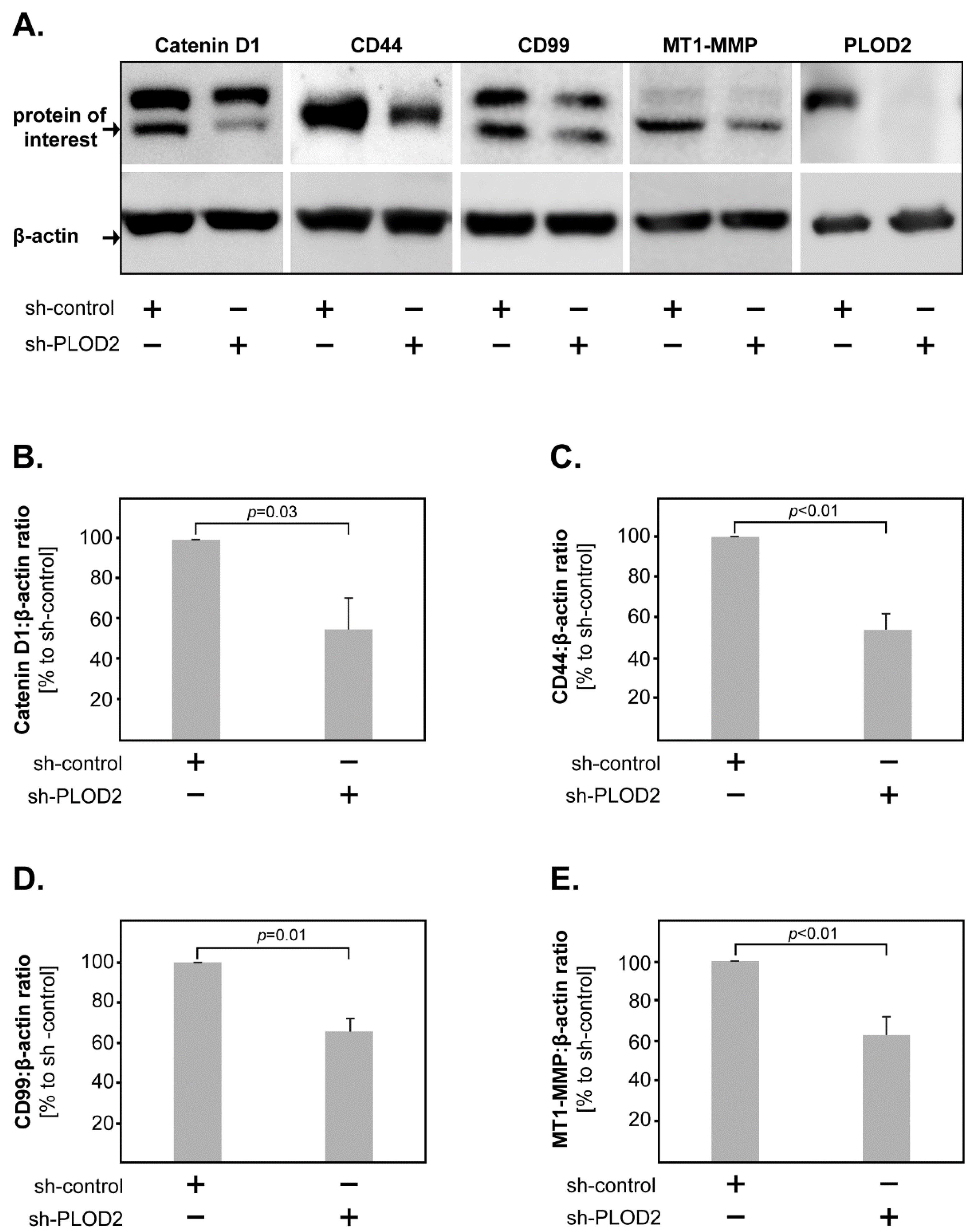
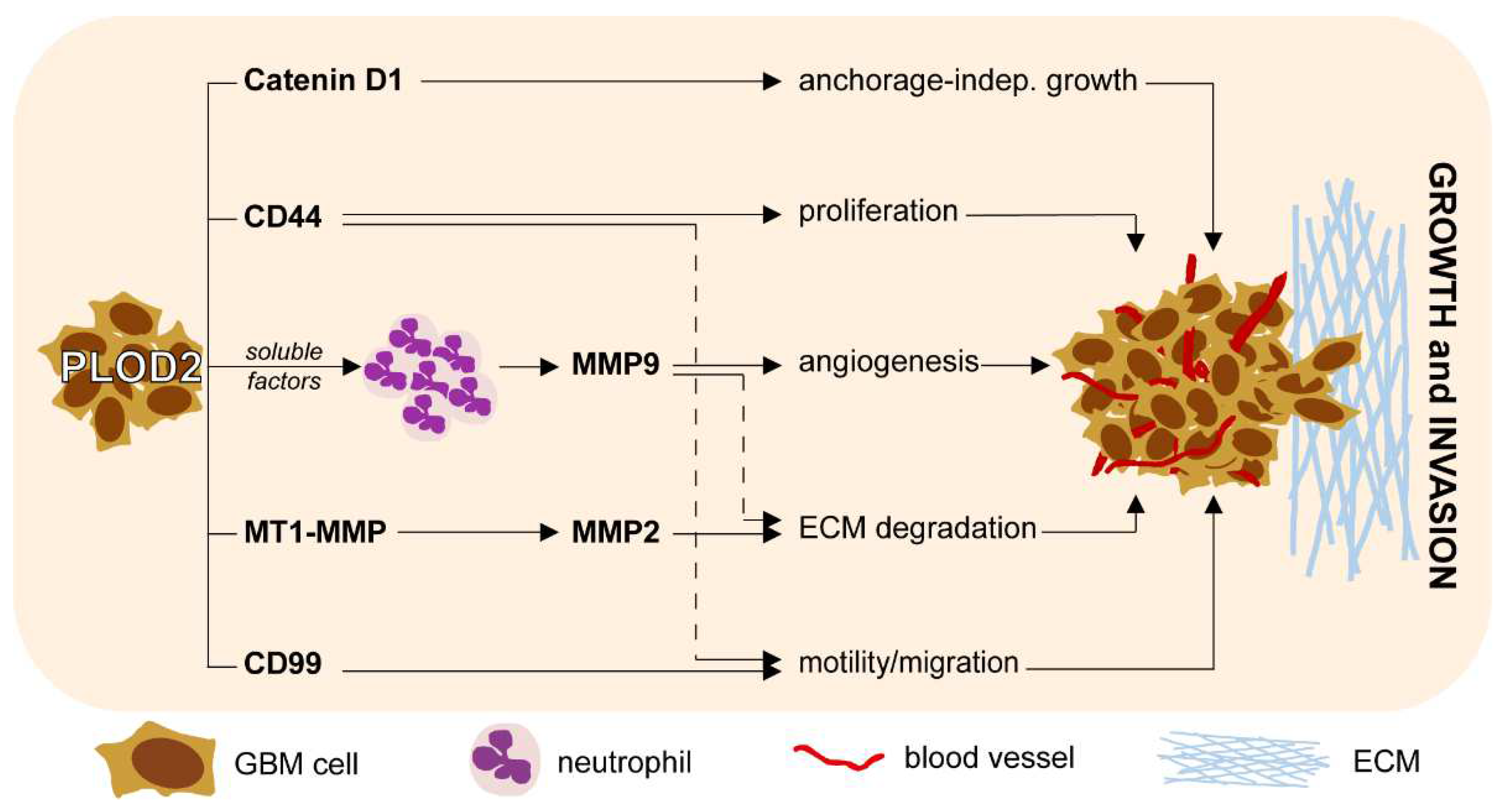
2.3. PLOD2 Modulates the Expression of Catenin D1, CD44, CD99 and MT1-MMP in GBM Cells
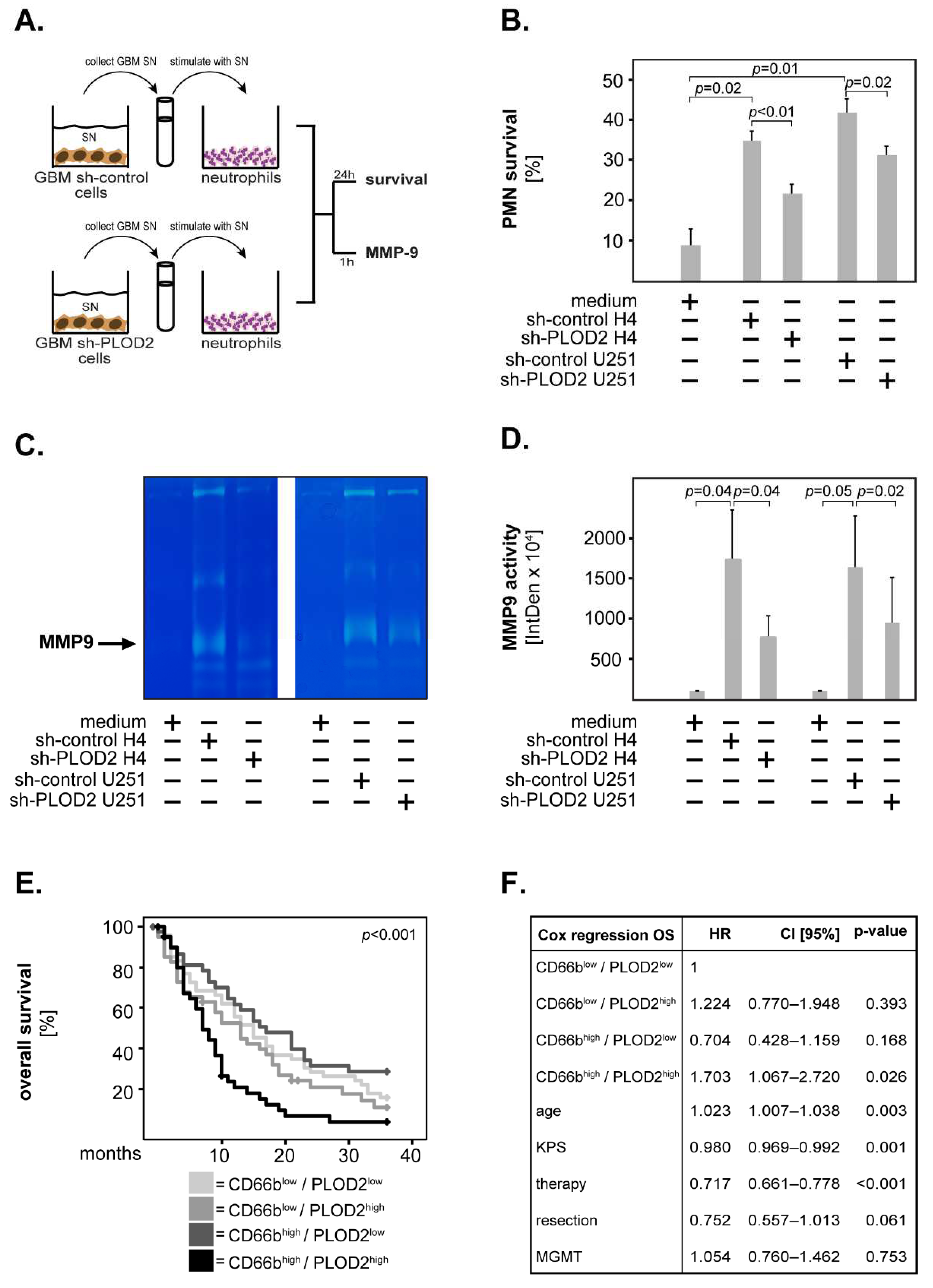
2.4. GBM-Associated PLOD2 Induces Neutrophil Granulocytes to Aquire a Pro-Tumor Phenotype
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Tissue Microarrays (TMA): Immunhistochemistry and Scoring
4.3. Cell Lines and Stable Transfection
4.4. SDS-PAGE and Western Blot
4.5. Gene Expression Analysis
4.6. MTT Assay
4.7. Soft Agar Clonogenic Assay
4.8. Invasion Assay
4.9. Gelatin Zymography
4.10. Isolation of Neutrophils from Peripheral Blood
4.11. Apoptosis Assays
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21 (Suppl. 5), v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-Year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [PubMed]
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Di Lullo, G.A.; Sweeney, S.M.; Korkko, J.; Ala-Kokko, L.; San Antonio, J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002, 277, 4223–4231. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Reid, S.E.; Kay, E.J.; Neilson, L.J.; Henze, A.T.; Serneels, J.; McGhee, E.J.; Dhayade, S.; Nixon, C.; Mackey, J.B.; Santi, A.; et al. Tumor matrix stiffness promotes metastatic cancer cell interaction with the endothelium. EMBO J. 2017, 36, 2373–2389. [Google Scholar] [CrossRef]
- van der Slot, A.J.; Zuurmond, A.M.; Bardoel, A.F.; Wijmenga, C.; Pruijs, H.E.; Sillence, D.O.; Brinckmann, J.; Abraham, D.J.; Black, C.M.; Verzijl, N.; et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 2003, 278, 40967–40972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Guo, H.; Terajima, M.; Banerjee, P.; Liu, X.; Yu, J.; Momin, A.A.; Katayama, H.; Hanash, S.M.; Burns, A.R.; et al. Lysyl Hydroxylase 2 Is Secreted by Tumor Cells and Can Modify Collagen in the Extracellular Space. J. Biol. Chem. 2016, 291, 25799–25808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, K.; Seki, N.; Matsushita, R.; Yonemori, M.; Yoshino, H.; Nakagawa, M.; Enokida, H. Tumour-suppressive miRNA-26a-5p and miR-26b-5p inhibit cell aggressiveness by regulating PLOD2 in bladder cancer. Br. J. Cancer 2016, 115, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Zhao, Y.; Wang, L.; Zhao, Y.; Wei, L.; Chen, D.; Chen, Z. Identification of PLOD Family Genes as Novel Prognostic Biomarkers for Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 1695. [Google Scholar] [CrossRef]
- Noda, T.; Yamamoto, H.; Takemasa, I.; Yamada, D.; Uemura, M.; Wada, H.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Tanemura, M.; et al. PLOD2 induced under hypoxia is a novel prognostic factor for hepatocellular carcinoma after curative resection. Liver Int. 2012, 32, 110–118. [Google Scholar] [CrossRef]
- Hu, H.L.; Wang, C.F.; Wei, X.H.; Lv, J.X.; Cao, X.H.; Shi, Y.Y.; Han, L.F.; Zhang, Y.N. Correlation between procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 and breast cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 1015–1021. [Google Scholar]
- Du, H.; Pang, M.; Hou, X.; Yuan, S.; Sun, L. PLOD2 in cancer research. Biomed. Pharmacother. 2017, 90, 670–676. [Google Scholar] [CrossRef]
- Dong, S.; Nutt, C.L.; Betensky, R.A.; Stemmer-Rachamimov, A.O.; Denko, N.C.; Ligon, K.L.; Rowitch, D.H.; Louis, D.N. Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J. Neuropathol. Exp. Neurol. 2005, 64, 948–955. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, L.; Wei, Y.; Zhang, X.; Xu, R.; Han, M.; Huang, B.; Chen, A.; Li, W.; Zhang, Q.; et al. Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 promotes hypoxia-induced glioma migration and invasion. Oncotarget 2017, 8, 23401–23413. [Google Scholar] [CrossRef]
- Song, Y.; Zheng, S.; Wang, J.; Long, H.; Fang, L.; Wang, G.; Li, Z.; Que, T.; Liu, Y.; Li, Y.; et al. Hypoxia-induced PLOD2 promotes proliferation, migration and invasion via PI3K/Akt signaling in glioma. Oncotarget 2017, 8, 41947–41962. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Kang, X.H.; Cui, Y.H.; Wang, Y.; Zhao, K.L.; Wang, Y.N.; Kou, W.Z.; Miao, Z.H.; Cao, X.J. Upregulation of PLOD2 promotes invasion and metastasis of osteosarcoma cells. Zhonghua Zhong Liu Za Zhi 2019, 41, 435–440. [Google Scholar] [PubMed]
- Zha, C.; Meng, X.; Li, L.; Mi, S.; Qian, D.; Li, Z.; Wu, P.; Hu, S.; Zhao, S.; Cai, J.; et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol. Med. 2020, 17, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, J.K.; Jeon, H.Y.; Ham, S.W.; Kim, H. CD133 Regulates IL-1beta Signaling and Neutrophil Recruitment in Glioblastoma. Mol. Cells 2017, 40, 515–522. [Google Scholar] [PubMed]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Siker, M.L.; Wang, M.; Porter, K.; Nelson, D.F.; Curran, W.J.; Michalski, J.M.; Souhami, L.; Chakravarti, A.; Yung, W.K.; Delrowe, J.; et al. Age as an independent prognostic factor in patients with glioblastoma: A Radiation Therapy Oncology Group and American College of Surgeons National Cancer Data Base comparison. J. Neurooncol. 2011, 104, 351–356. [Google Scholar] [CrossRef]
- Chambless, L.B.; Kistka, H.M.; Parker, S.L.; Hassam-Malani, L.; McGirt, M.J.; Thompson, R.C. The relative value of postoperative versus preoperative Karnofsky Performance Scale scores as a predictor of survival after surgical resection of glioblastoma multiforme. J. Neurooncol. 2015, 121, 359–364. [Google Scholar] [CrossRef]
- Han, Q.; Liang, H.; Cheng, P.; Yang, H.; Zhao, P. Gross Total vs. Subtotal Resection on Survival Outcomes in Elderly Patients With High-Grade Glioma: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Filippini, G.; Falcone, C.; Boiardi, A.; Broggi, G.; Bruzzone, M.G.; Caldiroli, D.; Farina, R.; Farinotti, M.; Fariselli, L.; Finocchiaro, G.; et al. Brain Cancer Register of the Fondazione, I.I.N.C.B. Prognostic factors for survival in 676 consecutive patients with newly diagnosed primary glioblastoma. Neuro Oncol. 2008, 10, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Felsberg, J.; Hartmann, C.; Berger, H.; Steinbach, J.P.; Schramm, J.; Westphal, M.; Schackert, G.; Simon, M.; Tonn, J.C.; et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: A prospective translational study of the German Glioma Network. J. Clin. Oncol. 2009, 27, 5743–5750. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, C.; Anacker, J.; Ernestus, R.I.; Vince, G.H. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 2012, 3, 67–79. [Google Scholar] [CrossRef]
- Li, G.; Wang, X.; Liu, G. PLOD2 Is a Potent Prognostic Marker and Associates with Immune Infiltration in Cervical Cancer. Biomed. Res. Int. 2021, 2021, 5512340. [Google Scholar] [PubMed]
- Eisinger-Mathason, T.S.; Zhang, M.; Qiu, Q.; Skuli, N.; Nakazawa, M.S.; Karakasheva, T.; Mucaj, V.; Shay, J.E.; Stangenberg, L.; Sadri, N.; et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013, 3, 1190–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargiotis, O.; Chetty, C.; Gondi, C.S.; Tsung, A.J.; Dinh, D.H.; Gujrati, M.; Lakka, S.S.; Kyritsis, A.P.; Rao, J.S. Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene 2008, 27, 4830–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsyth, P.A.; Wong, H.; Laing, T.D.; Rewcastle, N.B.; Morris, D.G.; Muzik, H.; Leco, K.J.; Johnston, R.N.; Brasher, P.M.; Suth-erland, G.; et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br. J. Cancer 1999, 79, 1828–1835. [Google Scholar] [CrossRef]
- Sinceviciute, R.; Vaitkiene, P.; Urbanaviciute, R.; Steponaitis, G.; Tamasauskas, A.; Skiriute, D. MMP2 is associated with glioma malignancy and patient outcome. Int. J. Clin. Exp. Pathol. 2018, 11, 3010–3018. [Google Scholar]
- Georgescu, M.M.; Islam, M.Z.; Li, Y.; Traylor, J.; Nanda, A. Novel targetable FGFR2 and FGFR3 alterations in glioblastoma associate with aggressive phenotype and distinct gene expression programs. Acta Neuropathol. Commun. 2021, 9, 69. [Google Scholar] [CrossRef]
- Belien, A.T.; Paganetti, P.A.; Schwab, M.E. Membrane-type 1 matrix metalloprotease (MT1-MMP) enables invasive migration of glioma cells in central nervous system white matter. J. Cell Biol. 1999, 144, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Hotary, K.; Allen, E.; Punturieri, A.; Yana, I.; Weiss, S.J. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 2000, 149, 1309–1323. [Google Scholar] [CrossRef]
- Itoh, Y.; Takamura, A.; Ito, N.; Maru, Y.; Sato, H.; Suenaga, N.; Aoki, T.; Seiki, M. Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J. 2001, 20, 4782–4793. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef] [PubMed]
- Rozanov, D.V.; Savinov, A.Y.; Williams, R.; Liu, K.; Golubkov, V.S.; Krajewski, S.; Strongin, A.Y. Molecular signature of MT1-MMP: Transactivation of the downstream universal gene network in cancer. Cancer Res. 2008, 68, 4086–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, K.L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T.T.; Voth, B.; Barnette, N.; Yang, I. The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Si, D.; Yin, F.; Peng, J.; Zhang, G. High Expression of CD44 Predicts a Poor Prognosis in Glioblastomas. Cancer Manag. Res. 2020, 12, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Li, Y.; Li, Y.; Liu, W.; Lu, Z.; Zhan, J.; Xu, M.; Chen, L.; Luo, X.; Cai, G.; et al. PLOD2 contributes to drug resistance in laryngeal cancer by promoting cancer stem cell-like characteristics. BMC Cancer 2019, 19, 840. [Google Scholar] [CrossRef] [Green Version]
- Seol, H.J.; Chang, J.H.; Yamamoto, J.; Romagnuolo, R.; Suh, Y.; Weeks, A.; Agnihotri, S.; Smith, C.A.; Rutka, J.T. Overexpression of CD99 Increases the Migration and Invasiveness of Human Malignant Glioma Cells. Genes Cancer 2012, 3, 535–549. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, L.C.; Soares, R.D.S.; Laurentino, T.S.; Lerario, A.M.; Marie, S.K.N.; Oba-Shinjo, S.M. CD99 Expression in Glioblastoma Molecular Subtypes and Role in Migration and Invasion. Int. J. Mol. Sci. 2019, 20, 1137. [Google Scholar] [CrossRef] [Green Version]
- Dohn, M.R.; Brown, M.V.; Reynolds, A.B. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 2009, 184, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Qi, Q.; Huang, W.; Zhang, H.; Zhang, B.; Sun, X.; Ma, J.; Zhu, C.; Wang, C. Bioinformatic analysis of PLOD family member expression and prognostic value in non-small cell lung cancer. Transl. Cancer Res. 2021, 10, 2707–2724. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Lang, S.; Brandau, S. Modulation of neutrophil granulocytes in the tumor microenvironment: Mechanisms and consequences for tumor progression. Semin. Cancer Biol. 2013, 23, 141–148. [Google Scholar] [CrossRef]
- Du, H.; Chen, Y.; Hou, X.; Huang, Y.; Wei, X.; Yu, X.; Feng, S.; Wu, Y.; Zhan, M.; Shi, X.; et al. PLOD2 regulated by transcription factor FOXA1 promotes metastasis in NSCLC. Cell Death. Dis. 2017, 8, e3143. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.T.; Xue, Y.X.; Wang, Y.F.; Wang, J.H.; Chen, X.; ShangGuan, Q.R.; Lian, Y.; Zhong, L.; Meng, Y.N. Minoxidil sulfate induced the increase in blood-brain tumor barrier permeability through ROS/RhoA/PI3K/PKB signaling pathway. Neuropharmacology 2013, 75, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Ningaraj, N.S.; Sankpal, U.T.; Khaitan, D.; Meister, E.A.; Vats, T. Activation of KATP channels increases anticancer drug delivery to brain tumors and survival. Eur. J. Pharmacol. 2009, 602, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Brouwer, E.; Stelzer, T.; Nocerino, S.; Rading, S.; Wilkens, L.; Sandalcioglu, I.E.; Karsak, M. Dynein Light Chain Protein Tctex1: A Novel Prognostic Marker and Molecular Mediator in Glioblastoma. Cancers 2021, 13, 2624. [Google Scholar] [CrossRef] [PubMed]
- Schiffgens, S.; Wilkens, L.; Brandes, A.A.; Meier, T.; Franceschi, E.; Ermani, M.; Hartmann, C.; Sandalcioglu, I.E.; Dumitru, C.A. Sex-specific clinicopathological significance of novel (Frizzled-7) and established (MGMT, IDH1) biomarkers in glioblastoma. Oncotarget 2016, 7, 55169–55180. [Google Scholar] [CrossRef] [Green Version]
- Dumitru, C.A.; Gholaman, H.; Trellakis, S.; Bruderek, K.; Dominas, N.; Gu, X.; Bankfalvi, A.; Whiteside, T.L.; Lang, S.; Brandau, S. Tumor-derived macrophage migration inhibitory factor modulates the biology of head and neck cancer cells via neutrophil activation. Int. J. Cancer 2011, 129, 859–869. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreße, N.; Schröder, H.; Stein, K.-P.; Wilkens, L.; Mawrin, C.; Sandalcioglu, I.E.; Dumitru, C.A. PLOD2 Is a Prognostic Marker in Glioblastoma That Modulates the Immune Microenvironment and Tumor Progression. Int. J. Mol. Sci. 2022, 23, 6037. https://doi.org/10.3390/ijms23116037
Kreße N, Schröder H, Stein K-P, Wilkens L, Mawrin C, Sandalcioglu IE, Dumitru CA. PLOD2 Is a Prognostic Marker in Glioblastoma That Modulates the Immune Microenvironment and Tumor Progression. International Journal of Molecular Sciences. 2022; 23(11):6037. https://doi.org/10.3390/ijms23116037
Chicago/Turabian StyleKreße, Nina, Hannah Schröder, Klaus-Peter Stein, Ludwig Wilkens, Christian Mawrin, Ibrahim Erol Sandalcioglu, and Claudia Alexandra Dumitru. 2022. "PLOD2 Is a Prognostic Marker in Glioblastoma That Modulates the Immune Microenvironment and Tumor Progression" International Journal of Molecular Sciences 23, no. 11: 6037. https://doi.org/10.3390/ijms23116037
APA StyleKreße, N., Schröder, H., Stein, K.-P., Wilkens, L., Mawrin, C., Sandalcioglu, I. E., & Dumitru, C. A. (2022). PLOD2 Is a Prognostic Marker in Glioblastoma That Modulates the Immune Microenvironment and Tumor Progression. International Journal of Molecular Sciences, 23(11), 6037. https://doi.org/10.3390/ijms23116037