
Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies

 , , , , ,
, , , , ,  and
and
Abstract
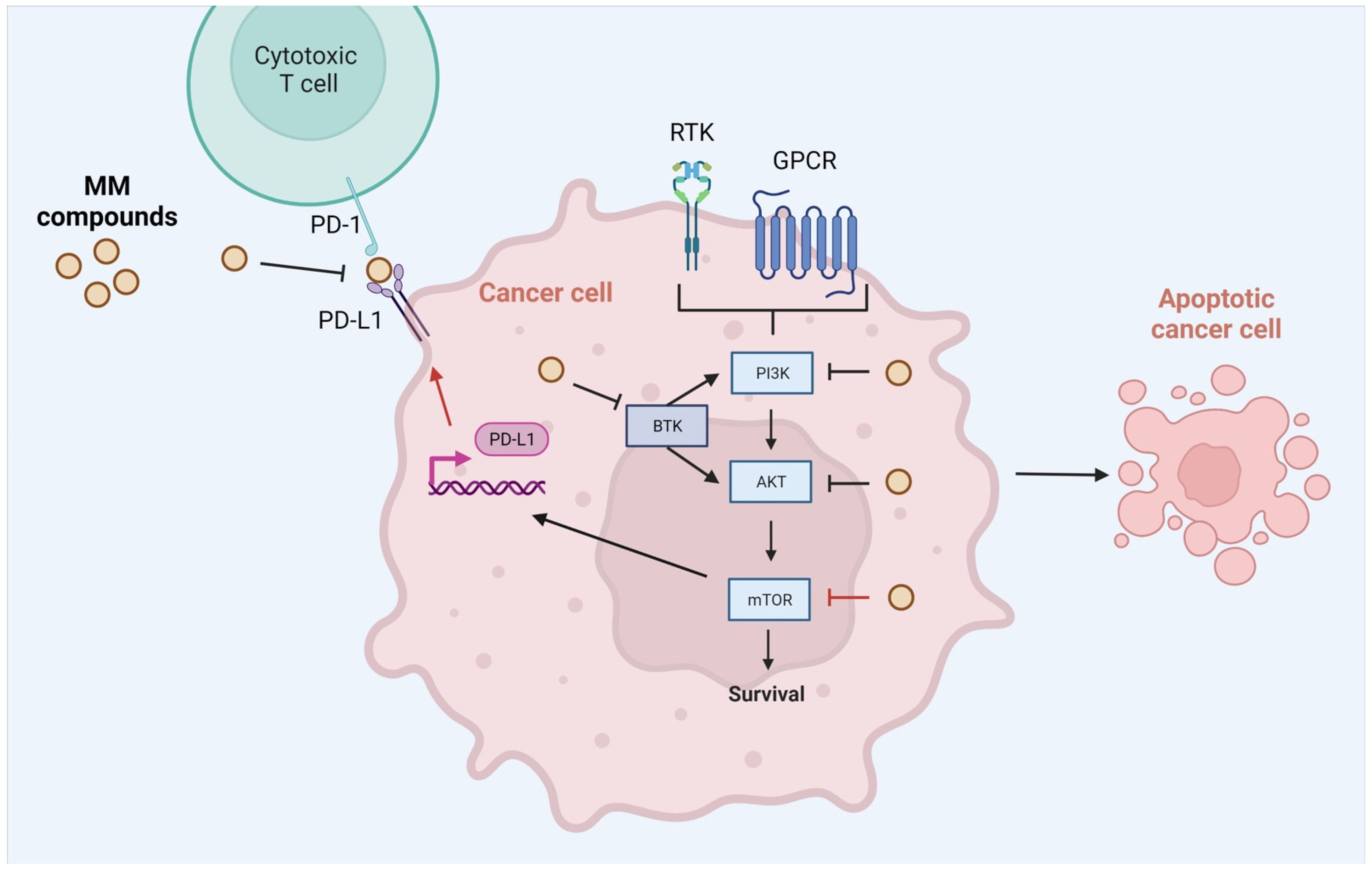
:1. Introduction
2. Results
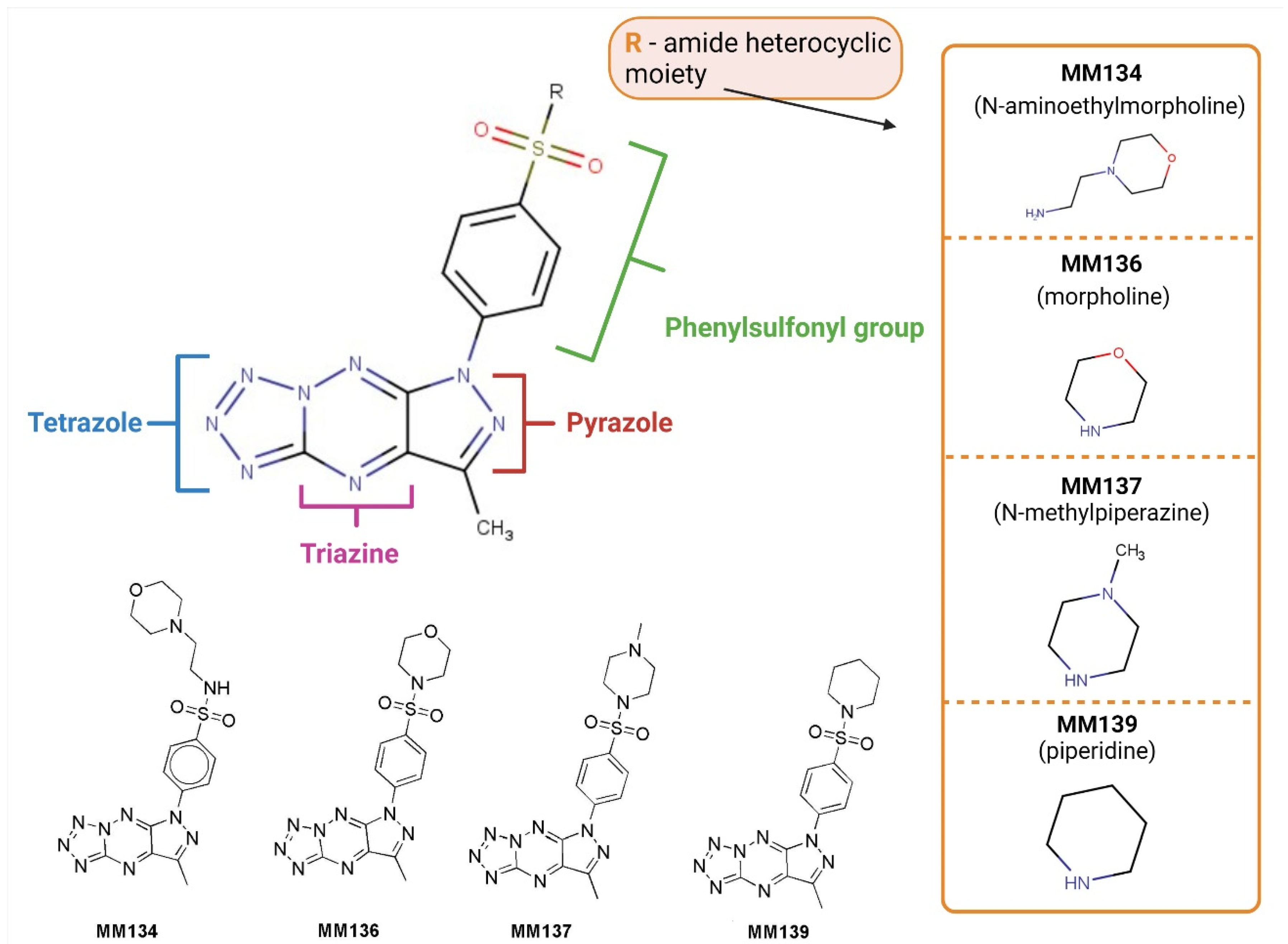
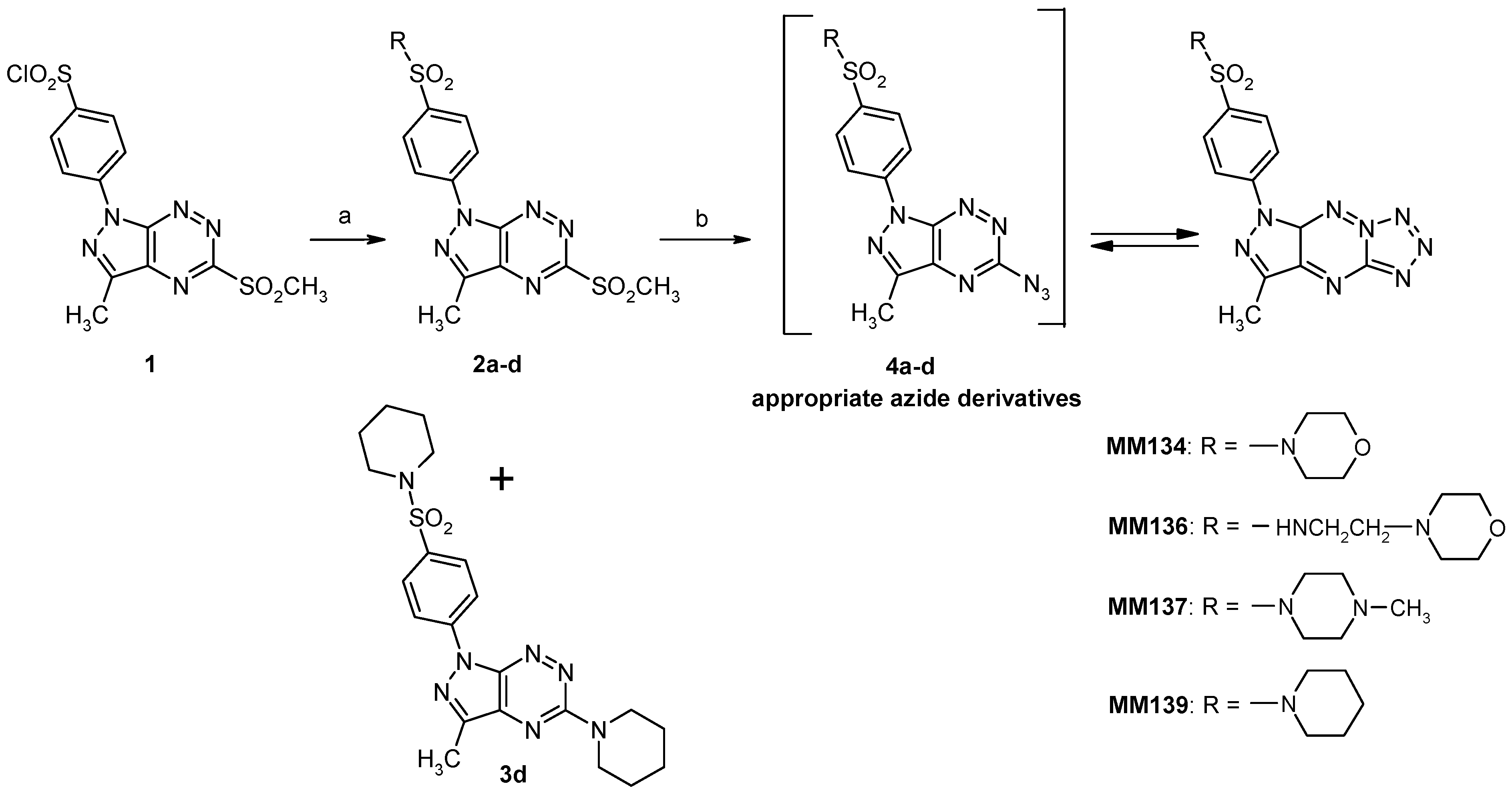
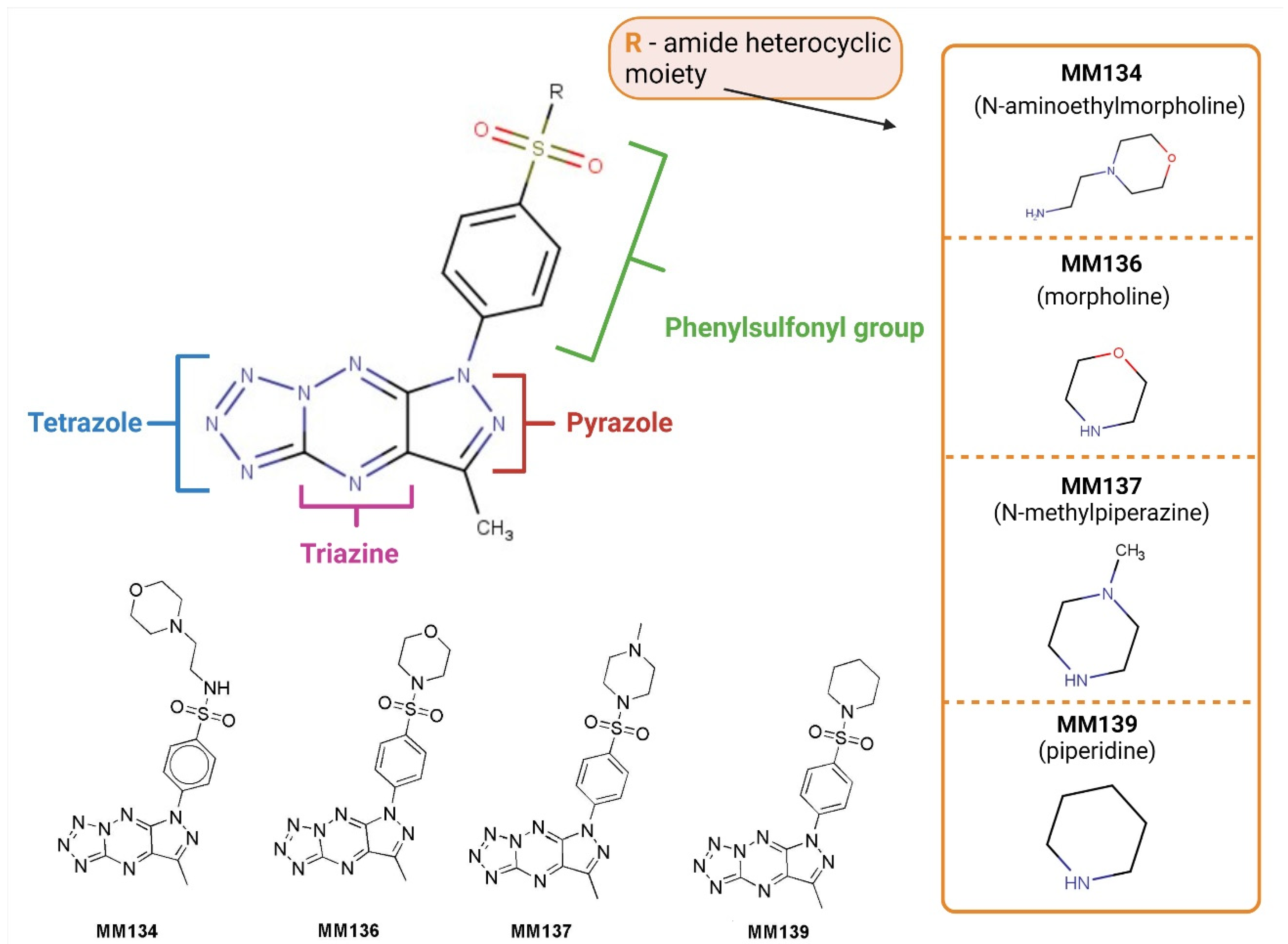
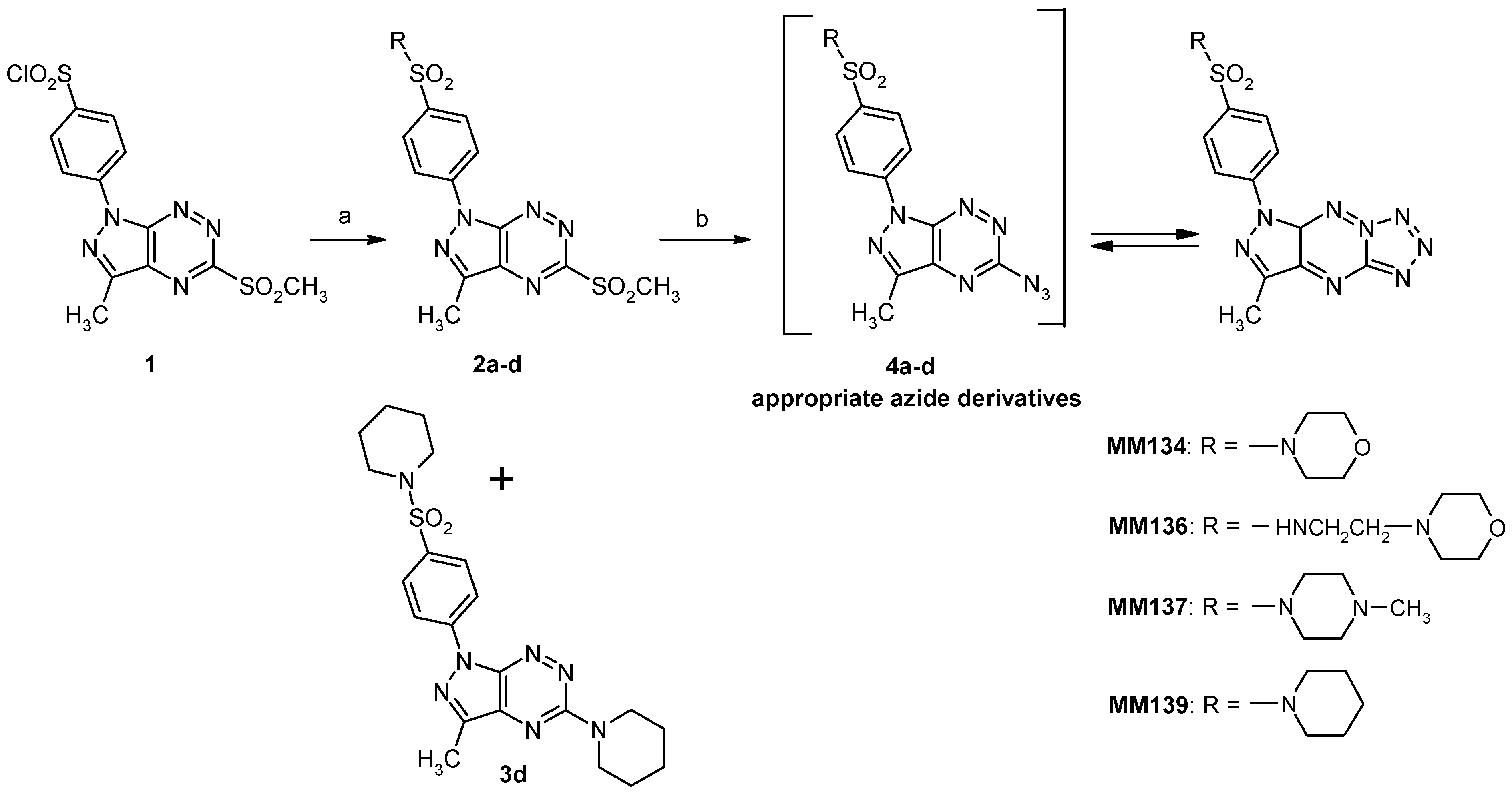
2.1. Chemistry
2.2. Biological Studies
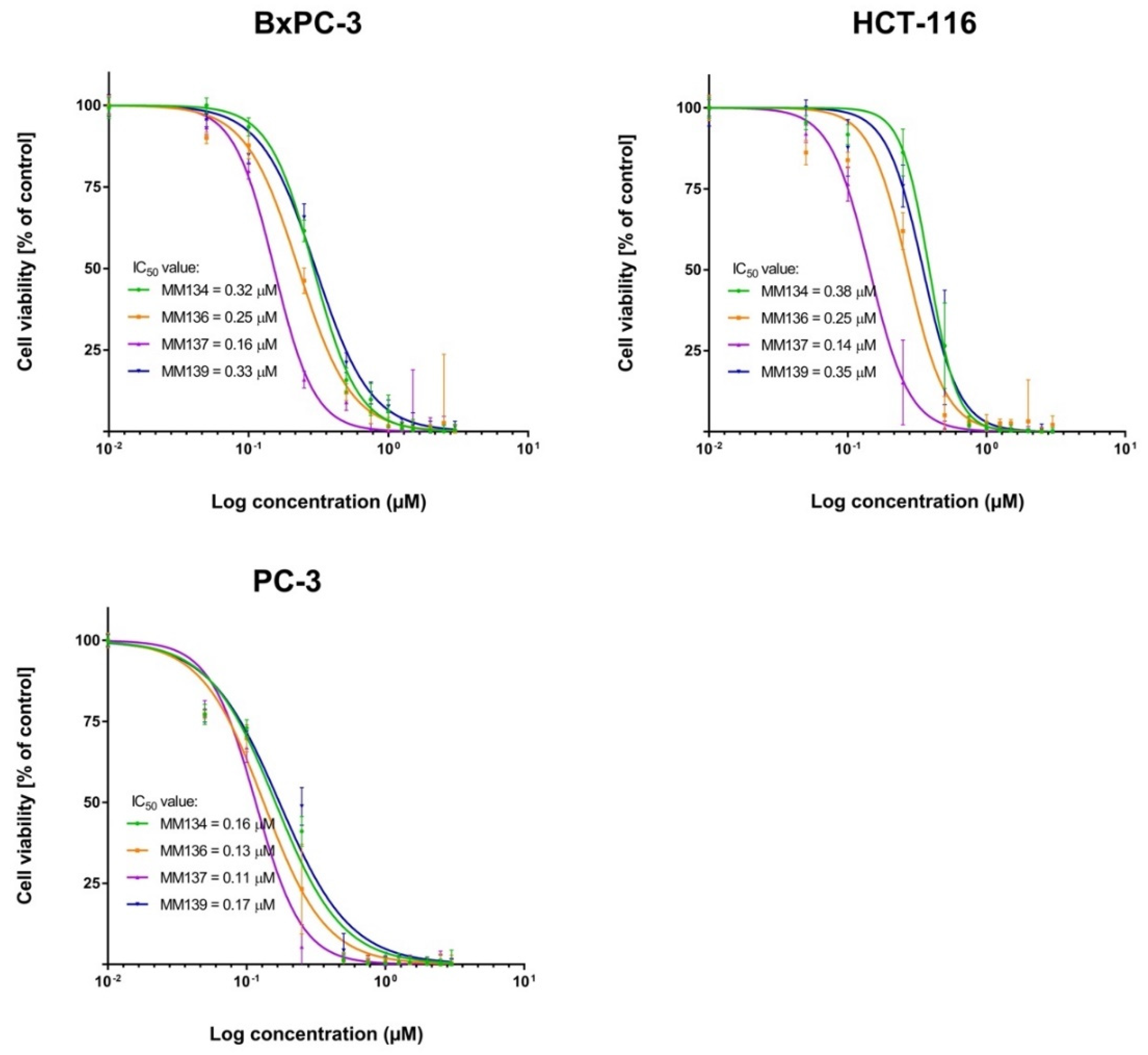
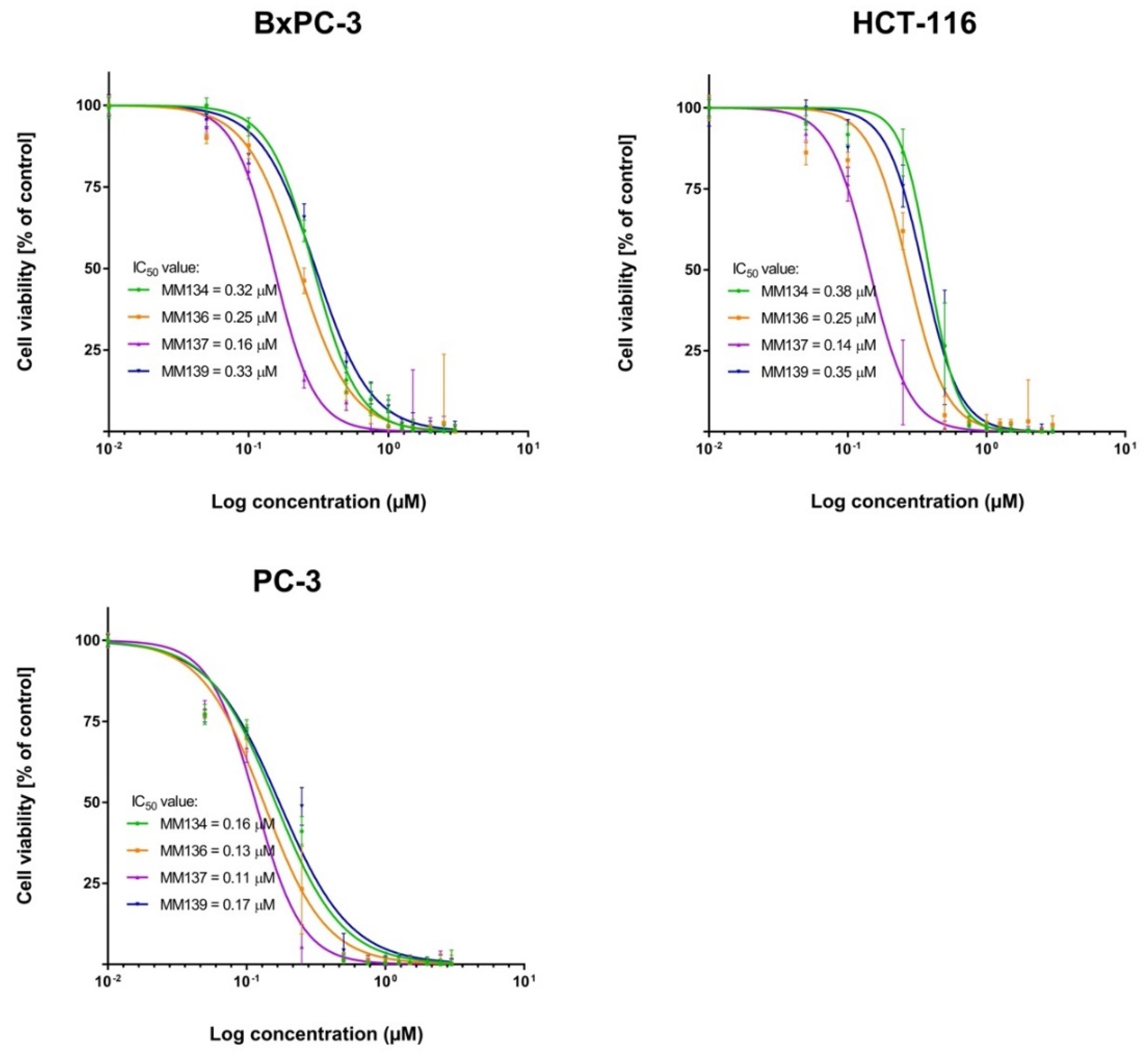
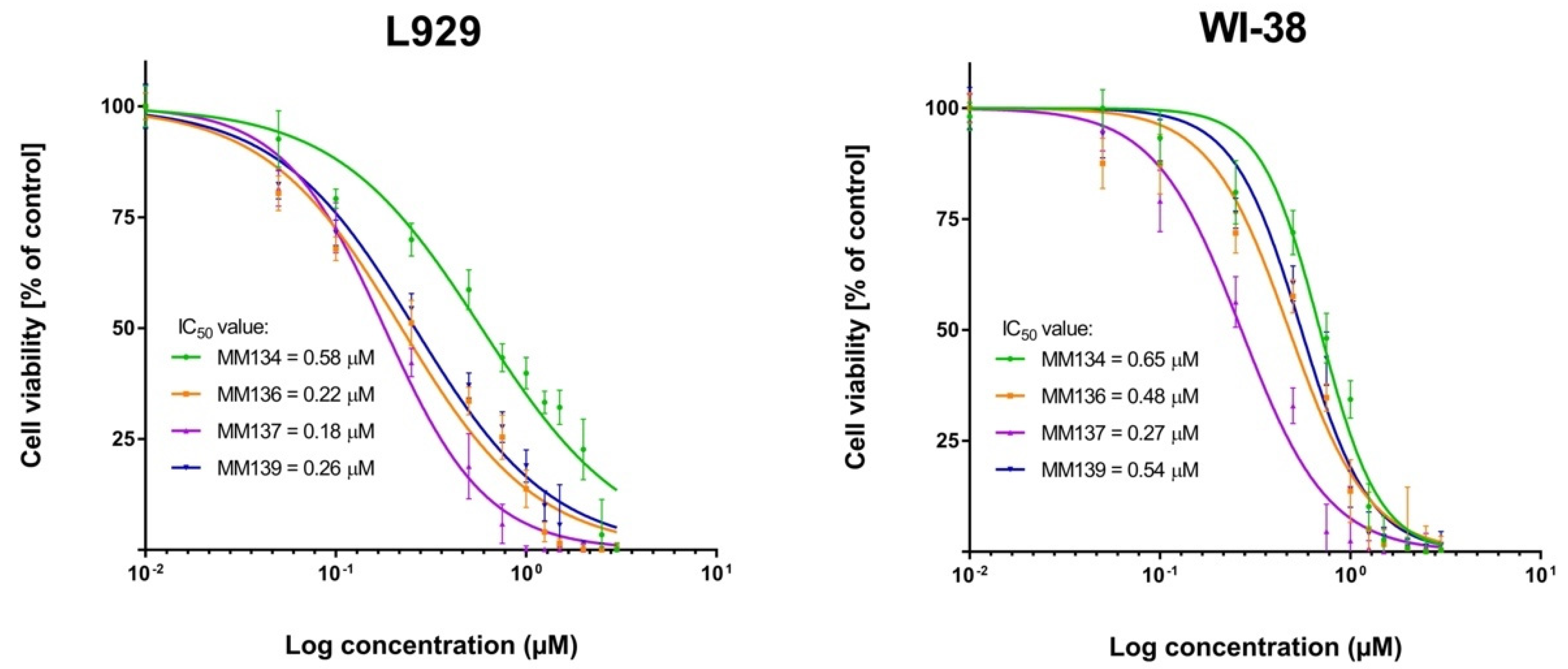
2.2.1. MTT Assay
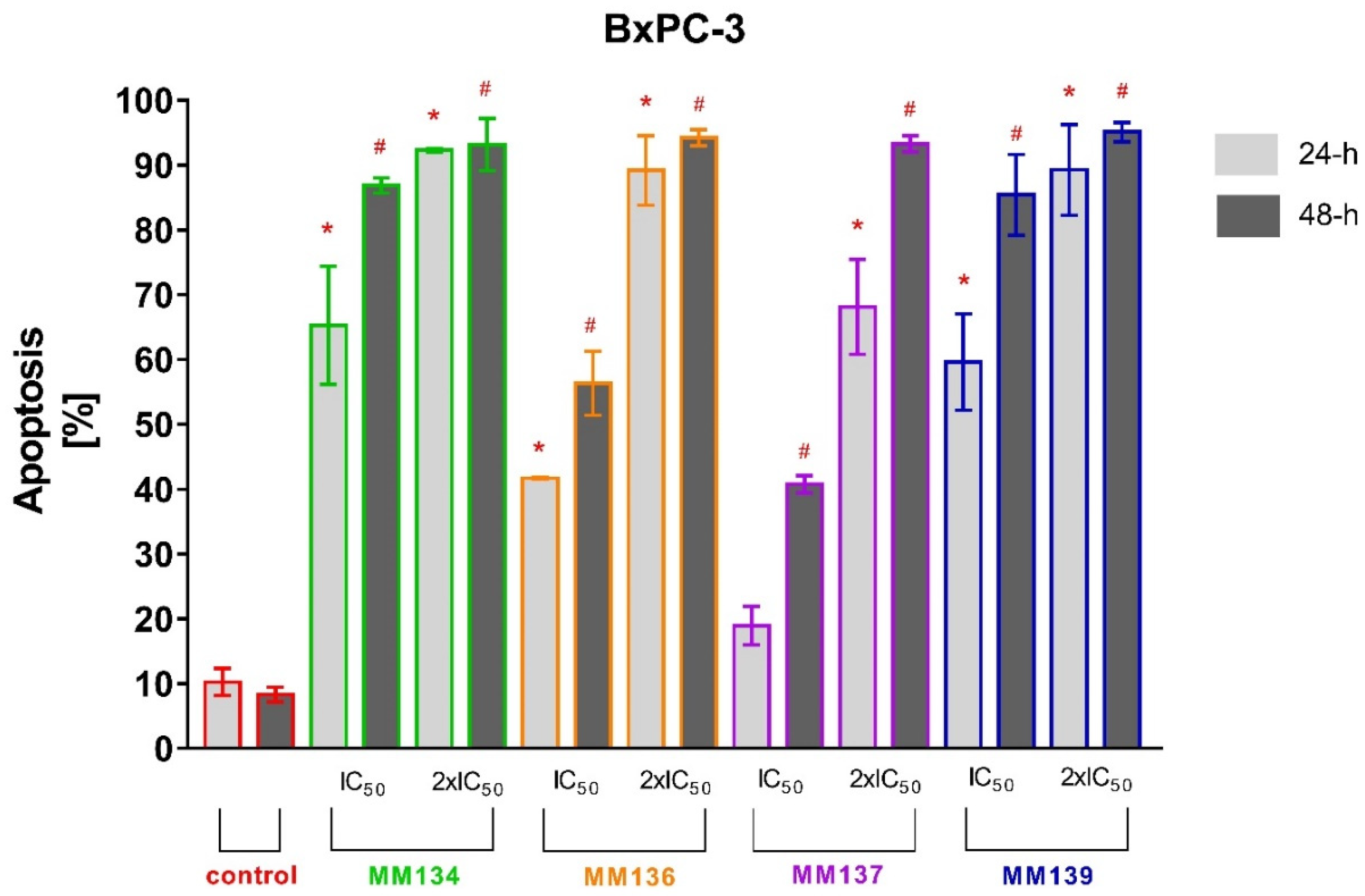
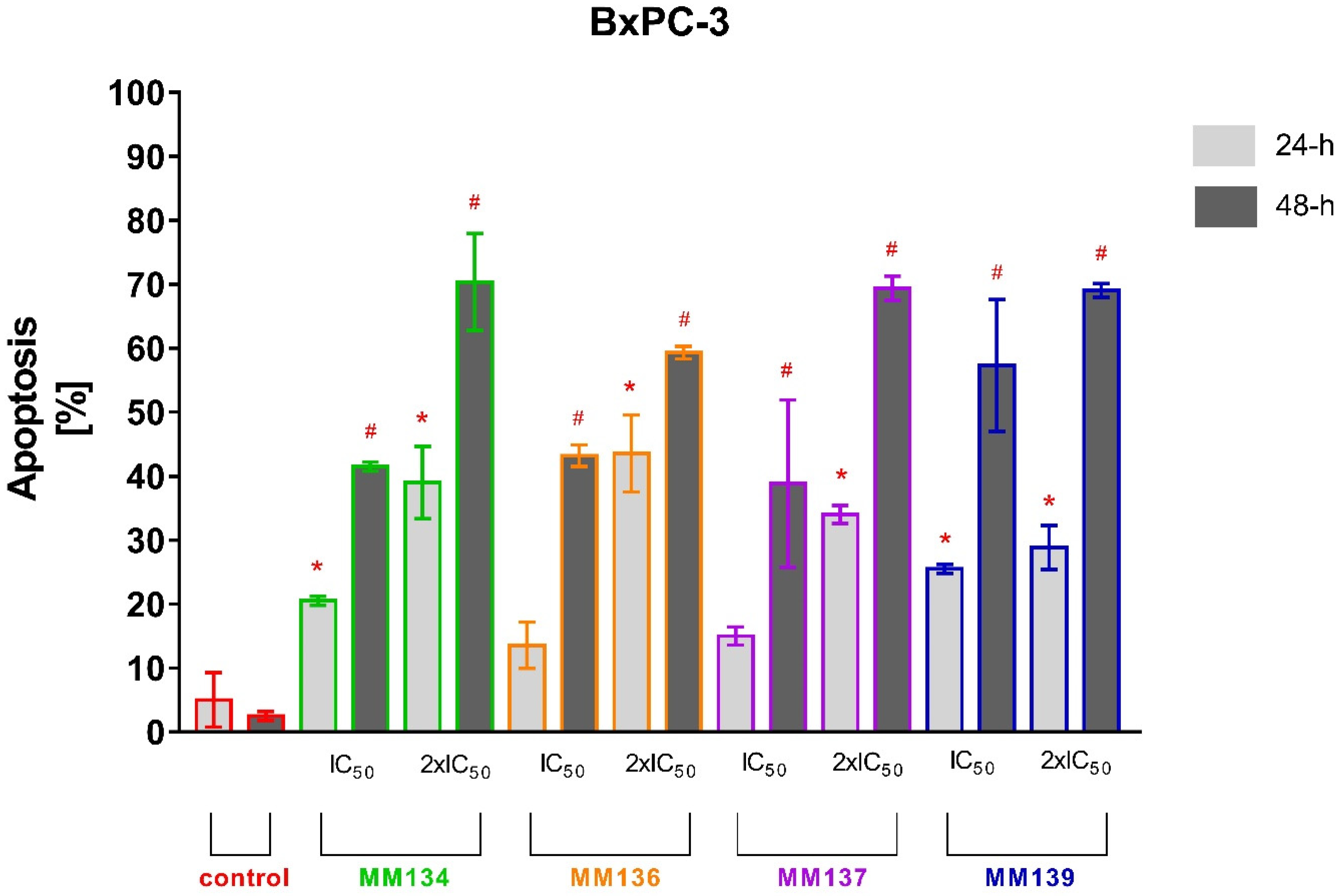
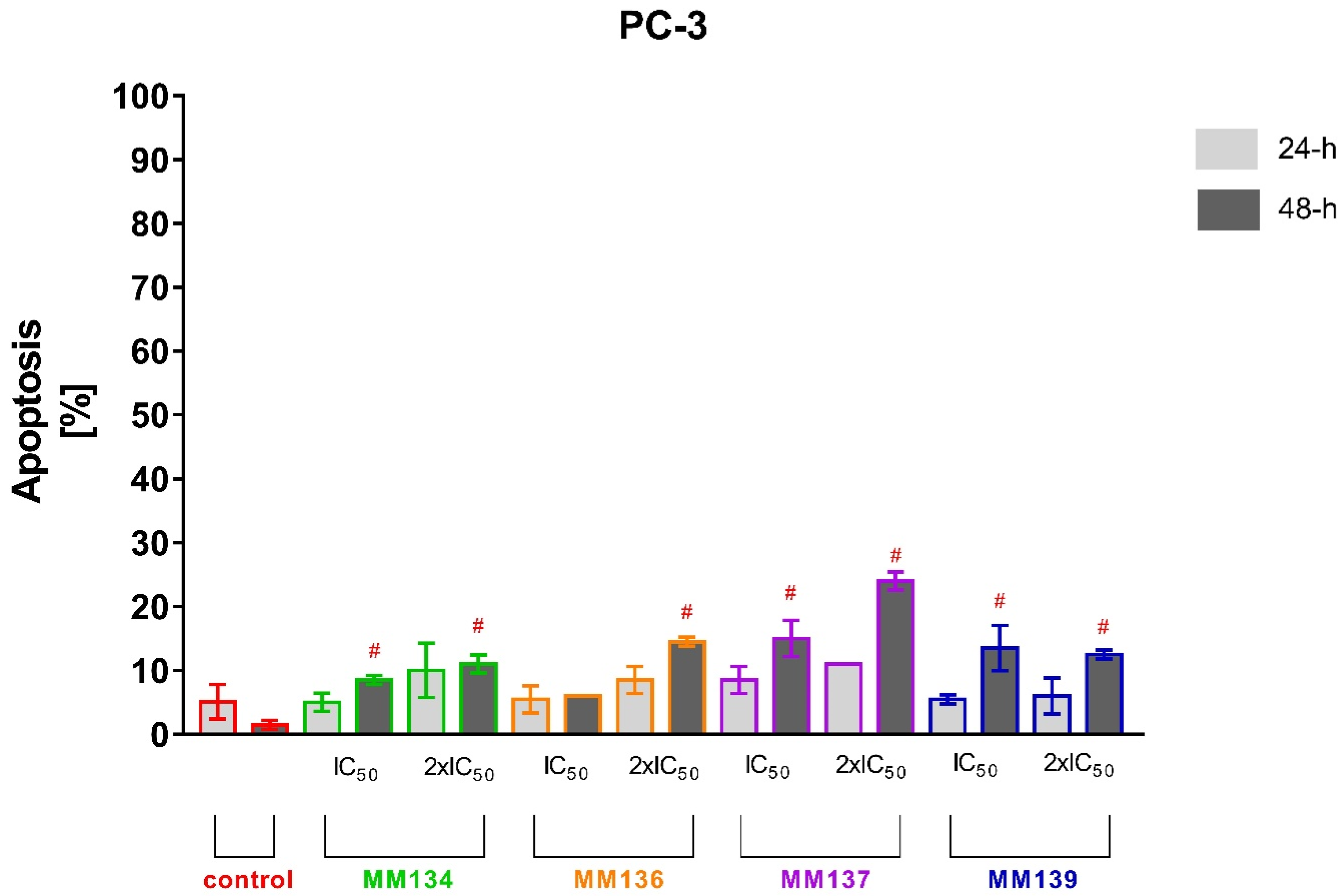
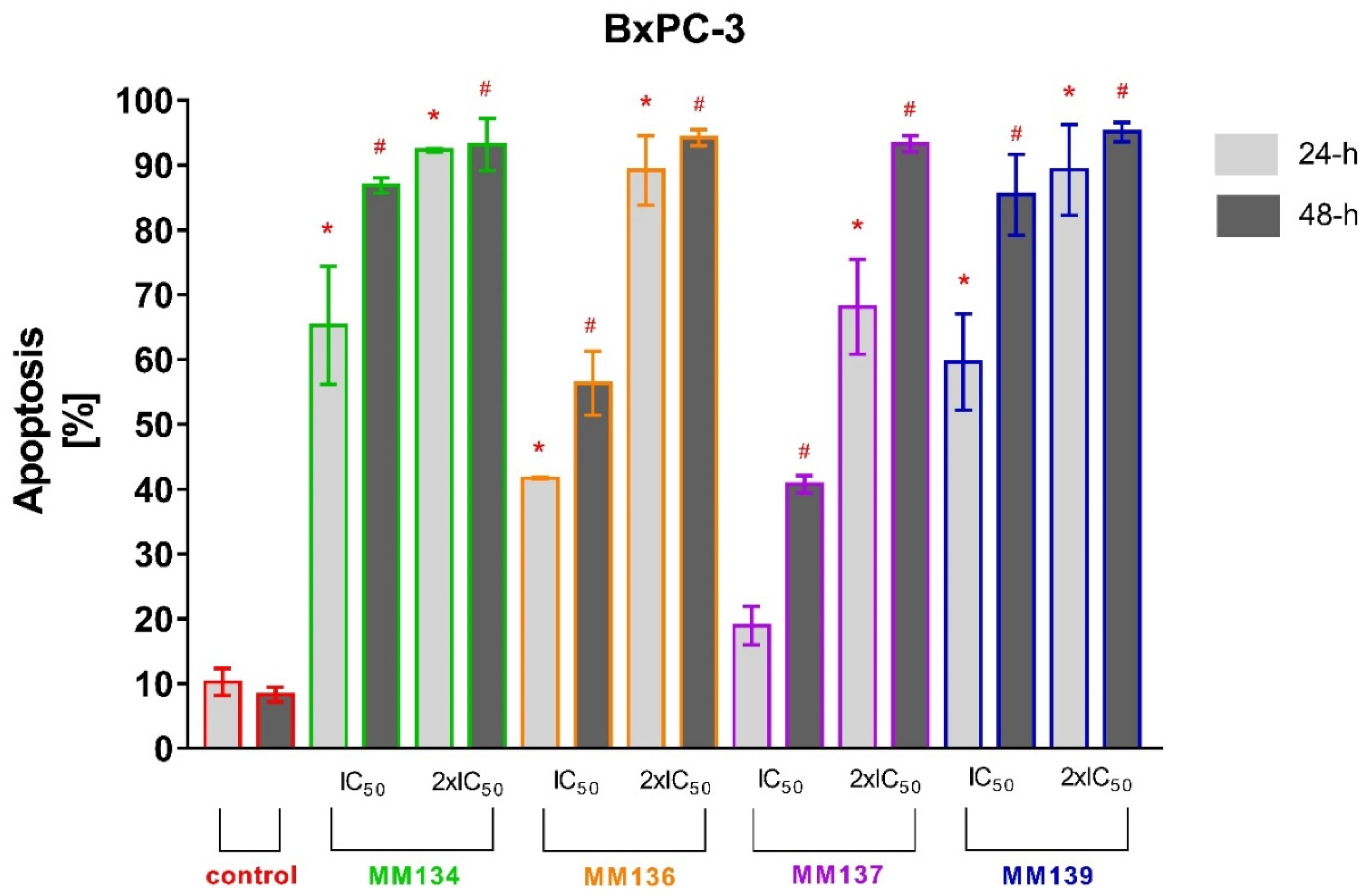
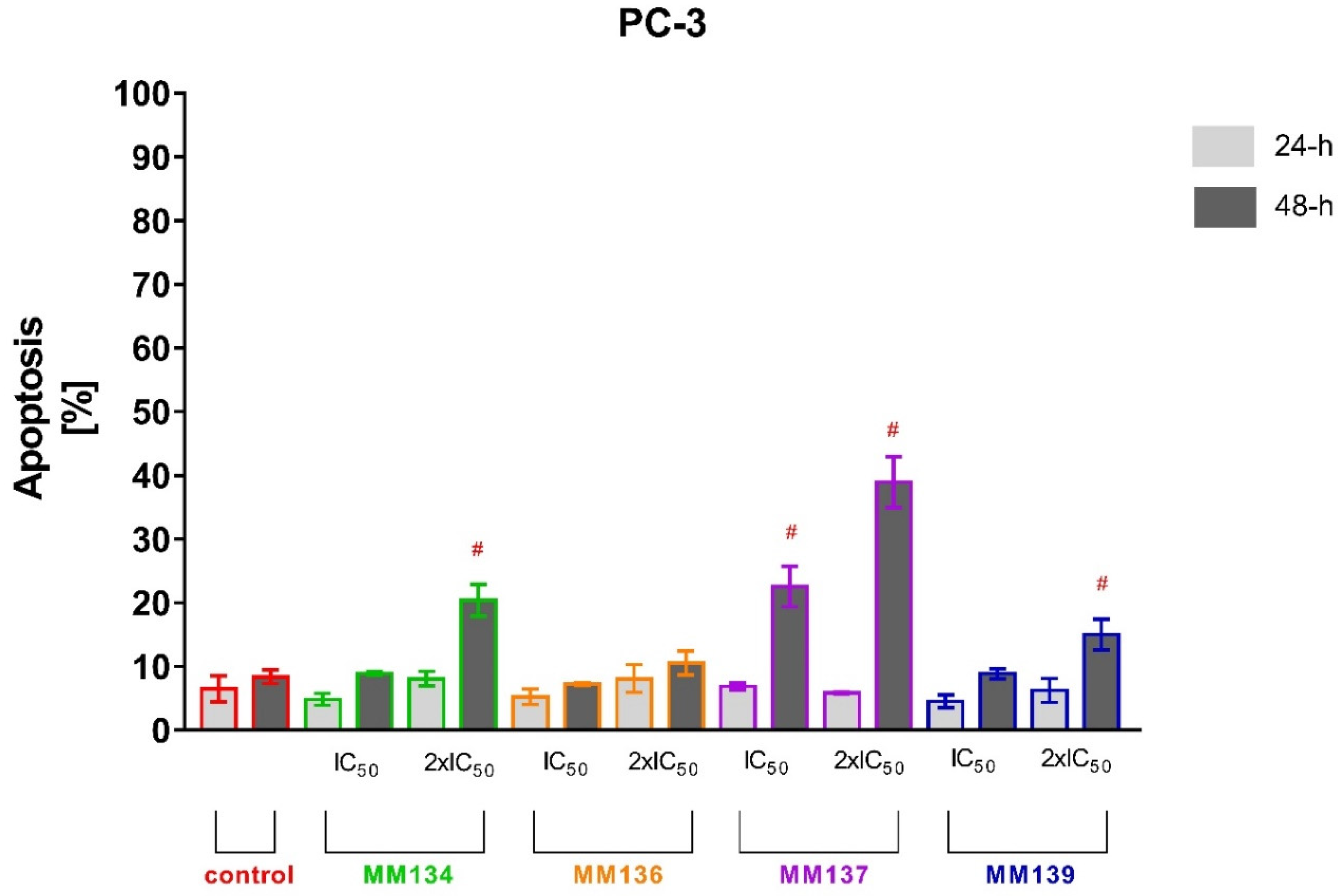
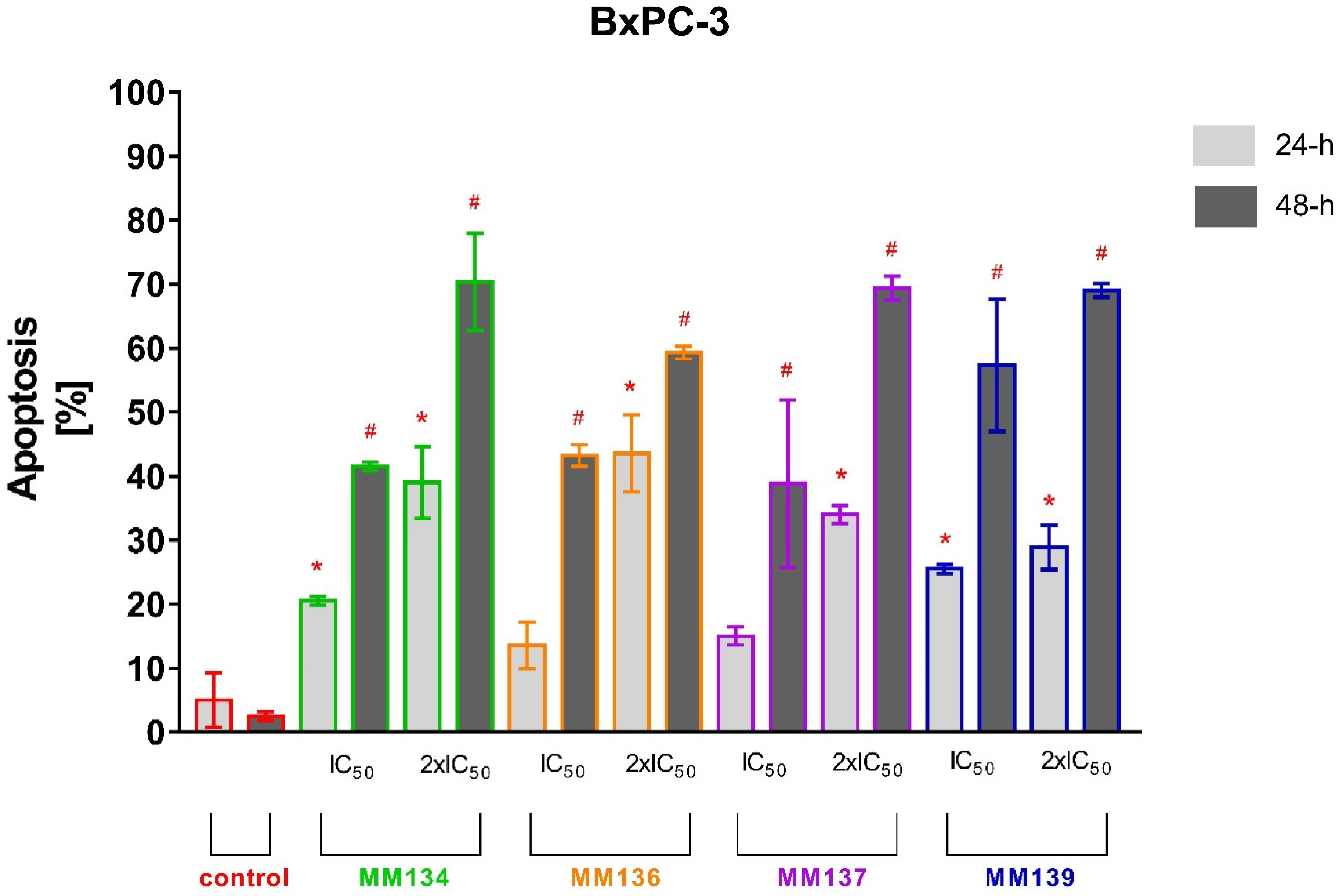
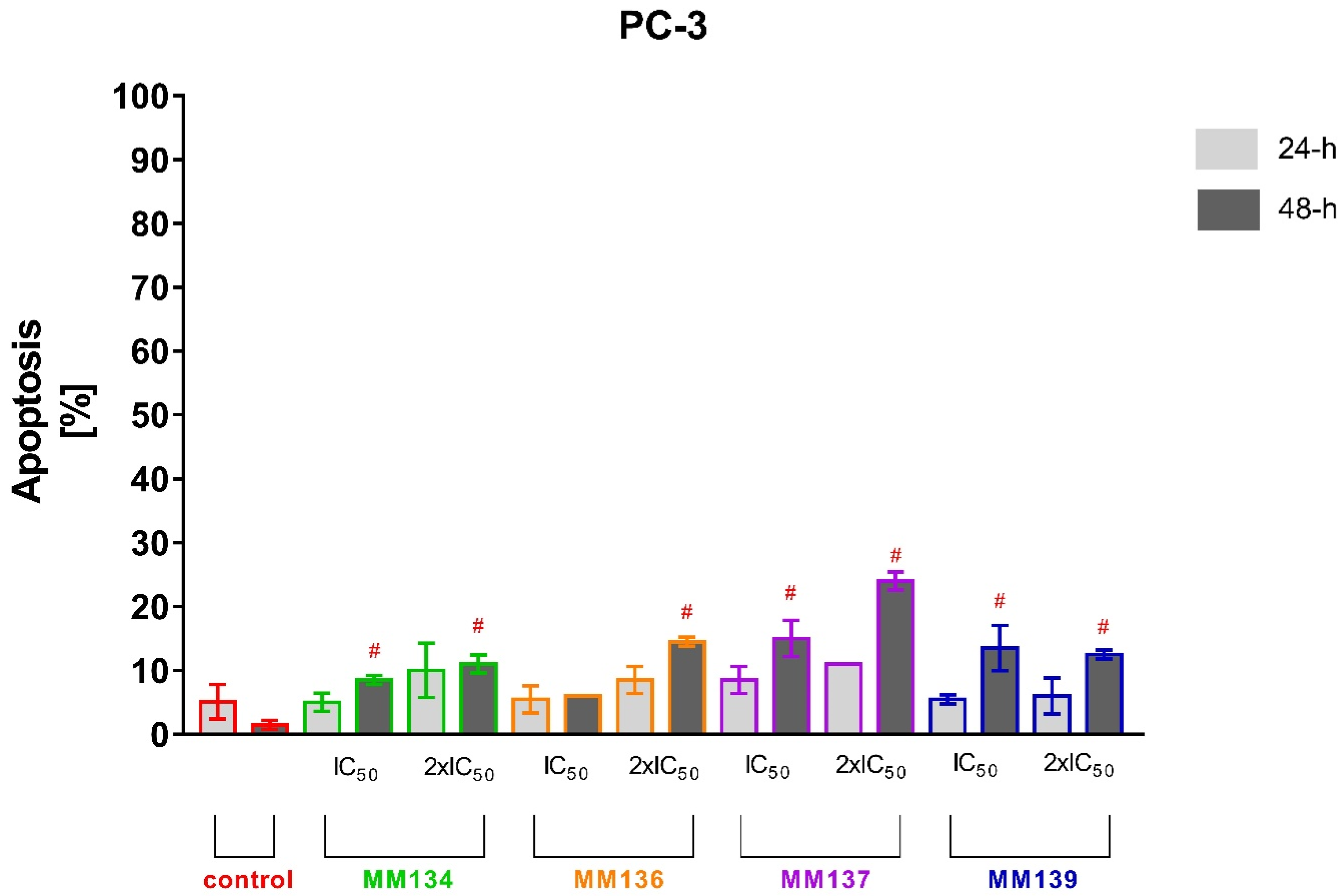
2.2.2. Annexin V and Propidium Iodide Flow Cytometry Analysis
2.2.3. Dual Acridine Orange/Ethidium Bromide (AO/EB) Fluorescent Staining
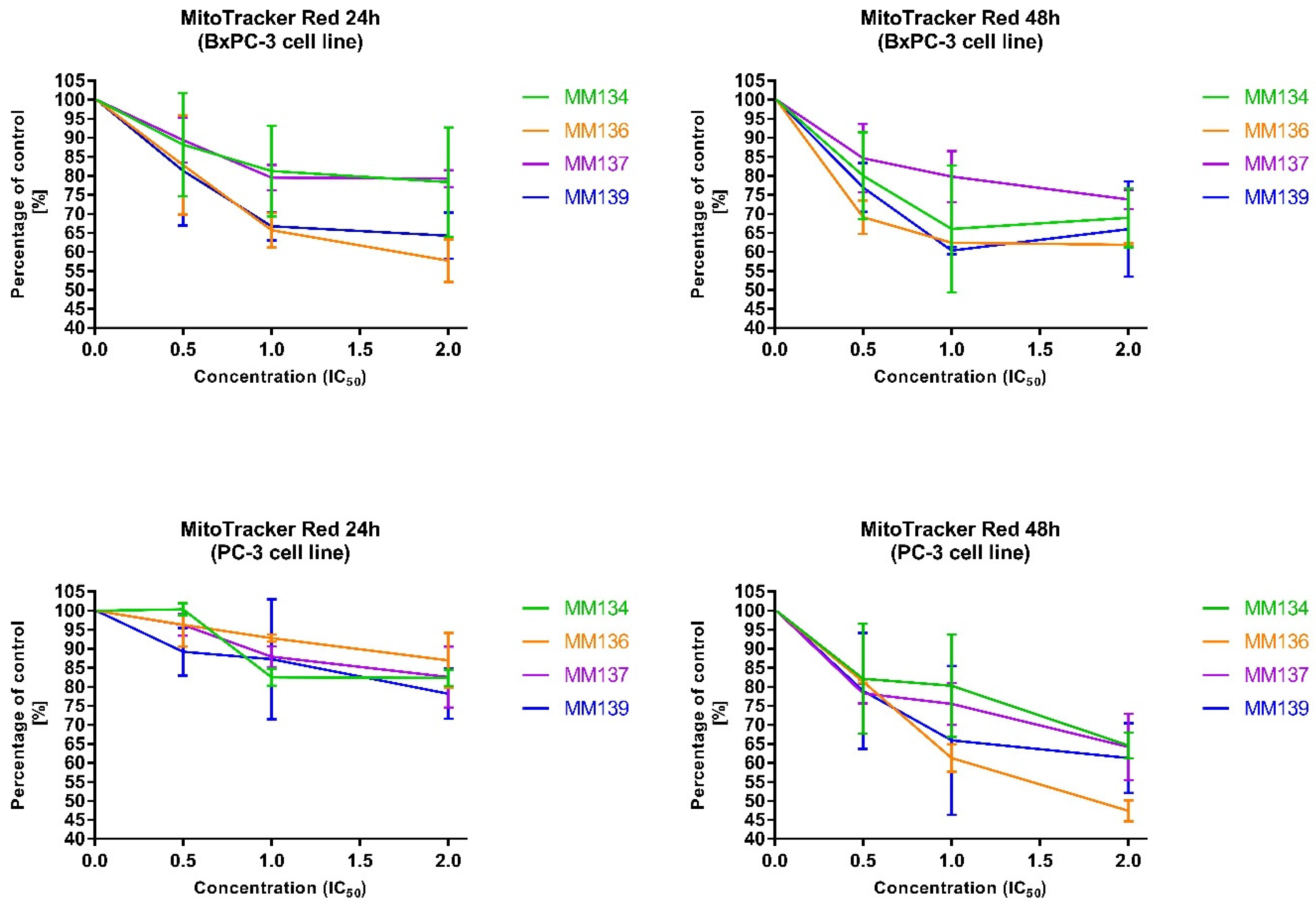
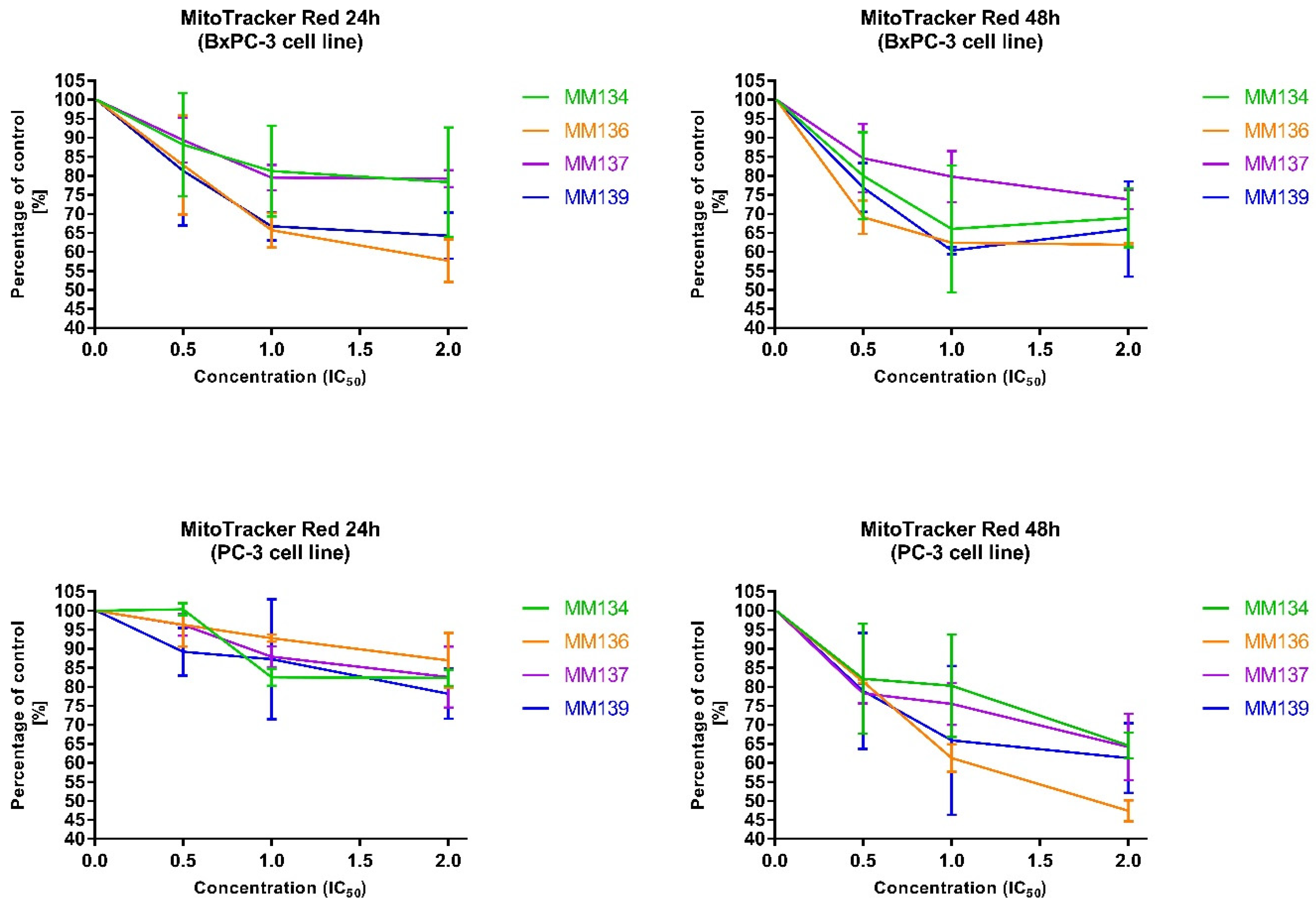
2.2.4. Changes in Transmembrane Mitochondrial Potential-MitoTracker Red
2.3. Data Analysis
2.3.1. MTT Assay
2.3.2. Apoptosis Detection
2.4. Computational Studies
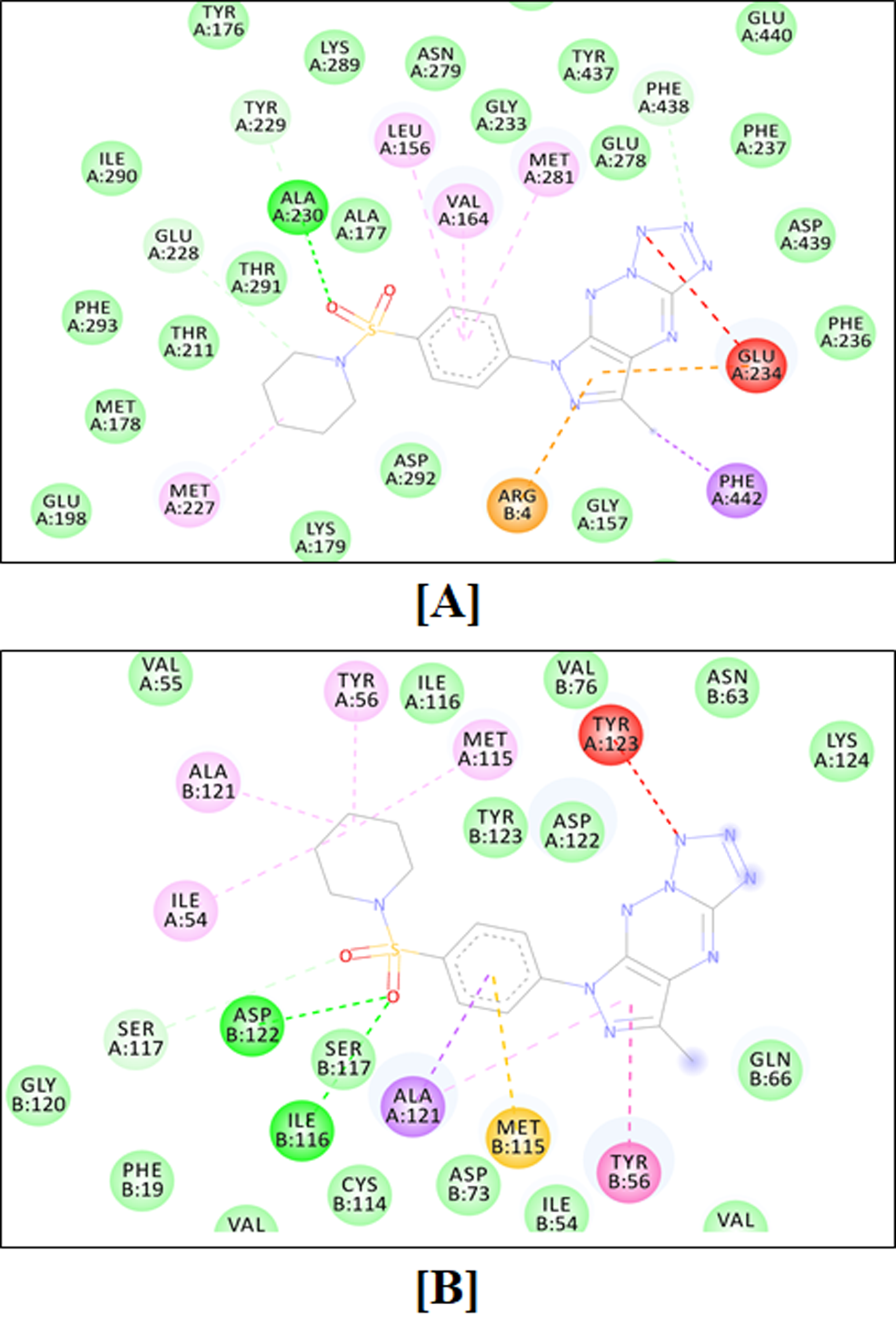
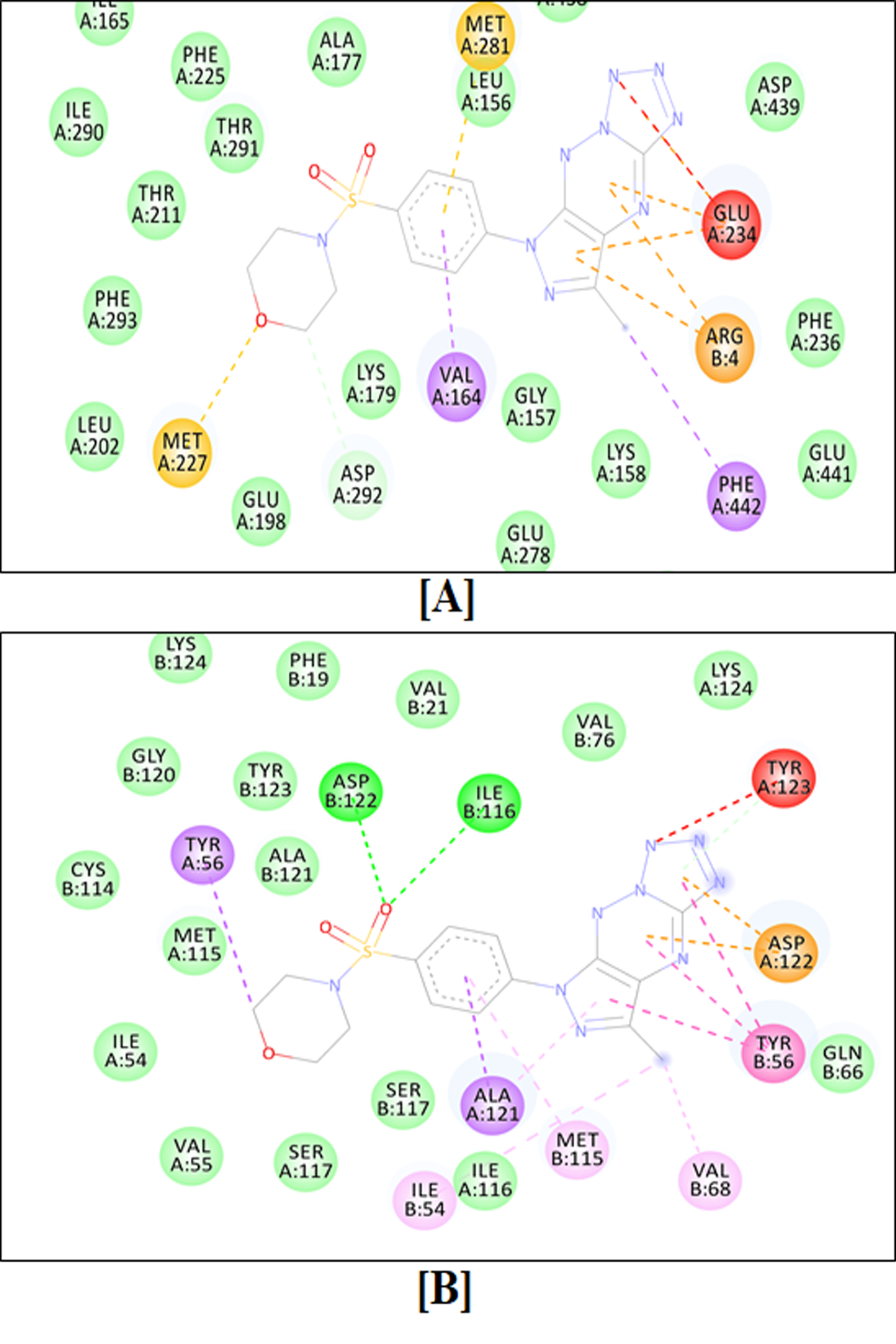
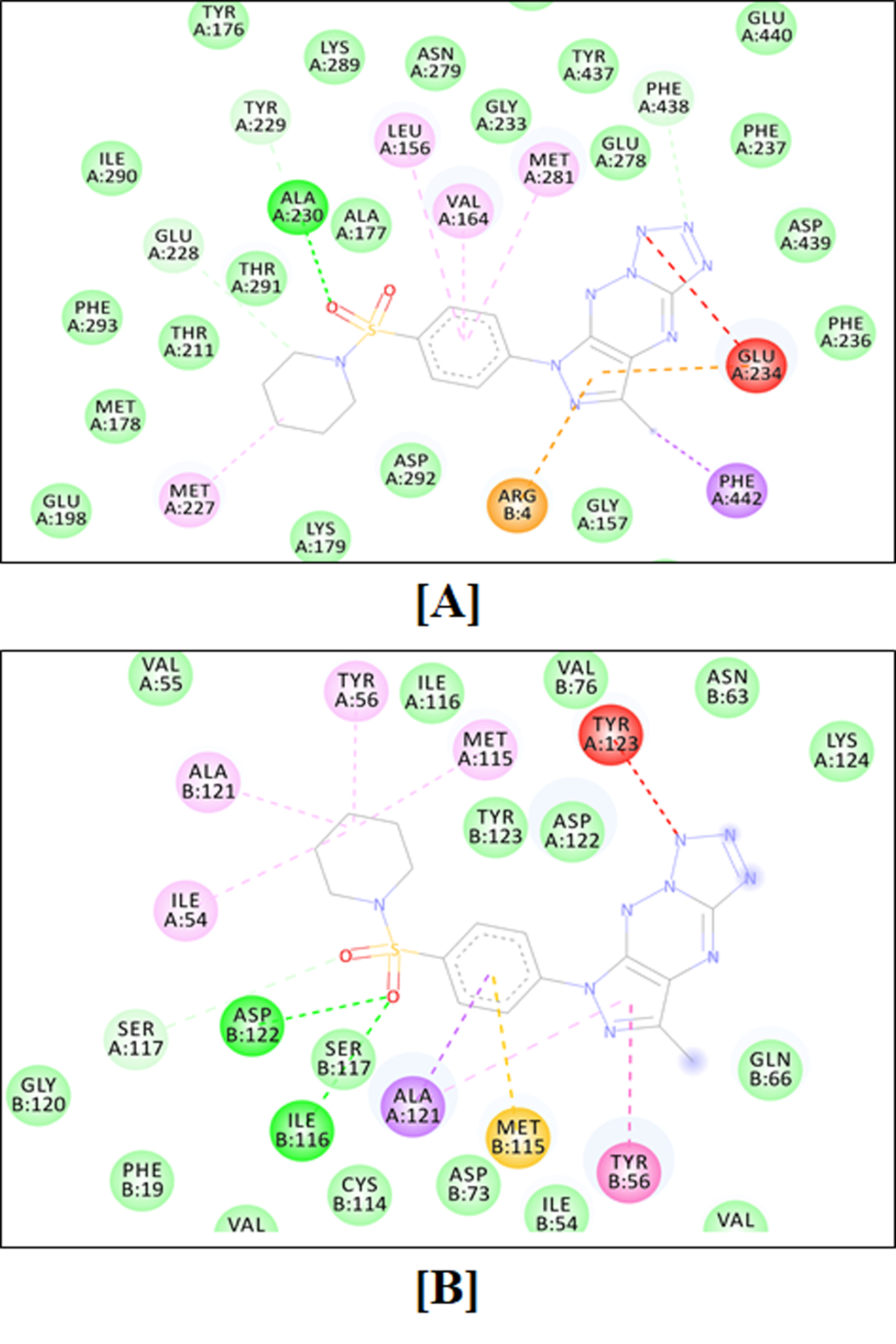
2.4.1. Molecular Docking Simulation
2.4.2. Molecular Dynamics Simulations
2.4.3. Drug Likeness and ADMET
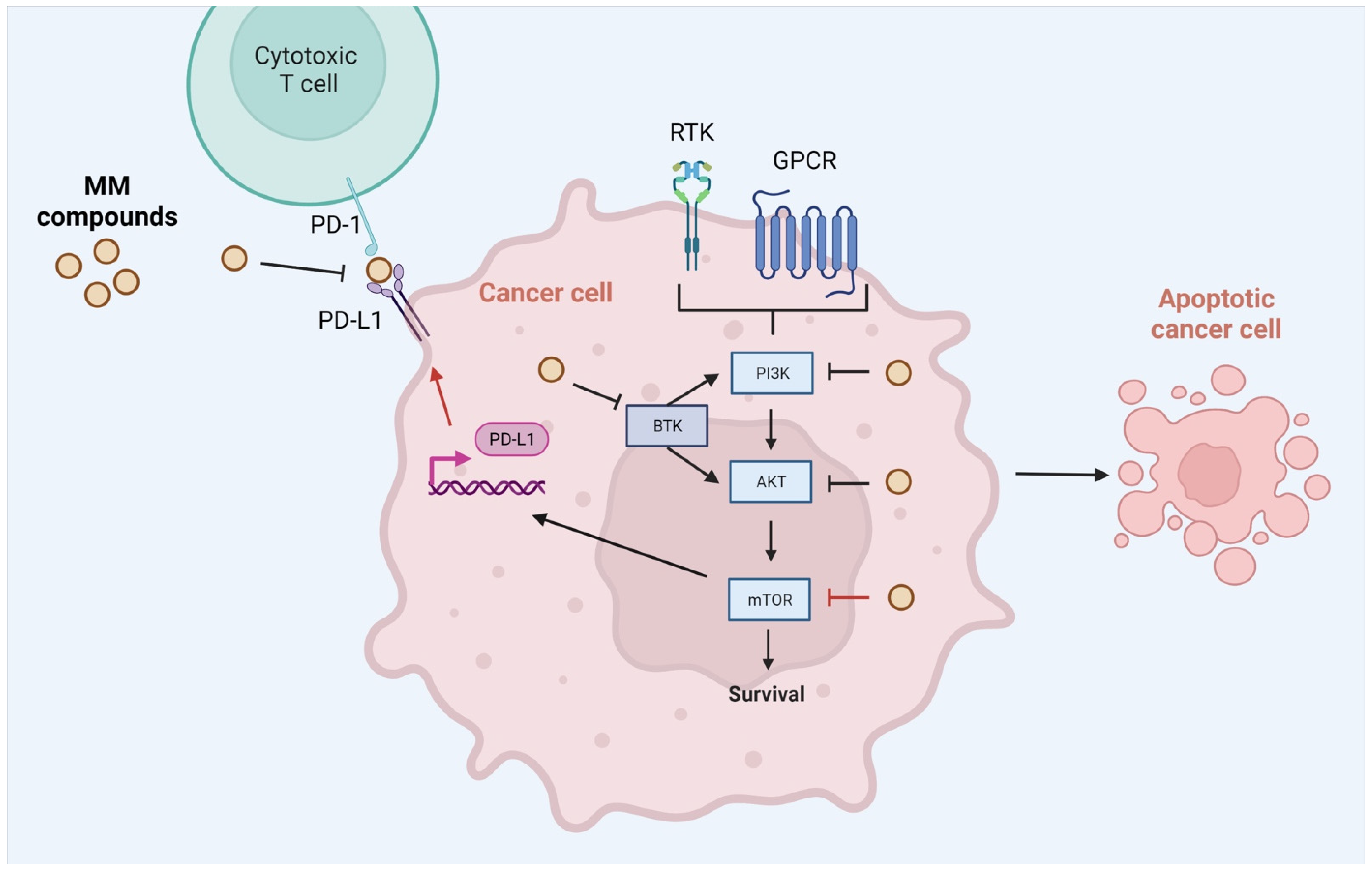
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.1.1. Synthesis of Sulfonamides (2a–d)
4.1.2. Synthesis of Tricyclic Suflonamides (MMs)
4.2. Biological Studies
4.2.1. Chemicals
4.2.2. Cell Culture
4.2.3. MTT Assay
4.3. Apoptosis Detection
4.3.1. Flow Cytometry Analysis with Annexin V-FITC Staining
4.3.2. Dual Acridine Orange/Ethidium Bromide (AO/EB) Fluorescent Staining
4.3.3. Changes in Transmembrane Mitochondrial Potential-MitoTracker Red (ΔΨm)
4.4. Computational Studies
4.4.1. Molecular Docking Simulations
4.4.2. Molecular Dynamics Simulations
4.4.3. Drug Likeness and ADMET Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABL | ABL tyrosine-protein kinase |
| AIF | apoptosis-inducing factor |
| AKT | serine/threonine-protein kinase |
| AO/EB | Acridine orange/ethidium bromide |
| BBB | blood–brain barrier |
| BTK | Bruton’s tyrosine kinase |
| CA | carbonic anhydrase |
| CDK | cyclin-dependent kinases |
| Csp3 | carbons in the sp3 hybridization |
| CYP | cytochrome P450 |
| DMSO | dimethyl sulfoxide |
| FBS | fetal bovine serum |
| HIA | human gastrointestinal absorption |
| ICAM-1 | intercellular adhesion molecule 1 |
| MEM | amino acids solution |
| MMP | mitochondria membrane potential |
| mTOR | mechanistic target of the rapamycin |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,3-diphenyltetrazolium bromide |
| MW | molecular weight |
| PBS | buffered saline |
| PD-1 | programmed death receptor-1 |
| PDB | protein data bank |
| PD-L1 | programmed death ligand-1 |
| PGP | glycoprotein P |
| PI3K | phosphoinositide-3-kinase |
| PTEN | phosphatase and tensin homolog |
| RMSD | root mean square deviation |
| RMSF | root mean square fluctuation |
| TP53 | cellular tumor antigen p53 |
| TPSA | topological polar surface area |
References
- Lee, C.-I.; Lee, C.-L.; Hwang, J.-F.; Lee, Y.-H.; Wang, J.-J. Monascus-fermented red mold rice exhibits cytotoxic effect and induces apoptosis on human breast cancer cells. Appl. Microbiol. Biotechnol. 2012, 97, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Hatse, S.; De Clercq, E.; Balzarini, J. Role of antimetabolites of purine and pyrimidine nucleotide metabolism in tumor cell differentiation. Biochem. Pharmacol. 1999, 58, 539–555. [Google Scholar] [CrossRef]
- Parker, W.B. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzorno, G.; Diasio, R.B.; Cheng, Y.-C. Pyrimidine and Purine Antimetabolites. In Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, Canada, 2003; Chapter 50. Available online: https://www.ncbi.nlm.nih.gov/books/NBK13028/ (accessed on 1 May 2022).
- Brunelle, J.K.; Zhang, B. Apoptosis assays for quantifying the bioactivity of anticancer drug products. Drug Resist. Updates 2010, 13, 172–179. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Crowley, L.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-rot087288. [Google Scholar] [CrossRef]
- Bouchain, G.; Delorme, D. Novel hydroxamate and anilide derivatives as potent histone deacetylase inhibitors: Synthesis and antiproliferative evaluation. Curr. Med. Chem. 2003, 10, 2359–2372. [Google Scholar] [CrossRef]
- Cheng, X.-C.; Wang, Q.; Fang, H.; Xu, W.-F. Role of sulfonamide group in matrix metalloproteinase inhibitors. Curr. Med. Chem. 2008, 15, 368–373. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [Green Version]
- Mojzych, M.; Dolashki, A.; Voelter, W. Synthesis of pyrazolo[4,3-e][1,2,4]triazine sulfonamides, novel Sildenafil analogs with tyrosinase inhibitory activity. Bioorg. Med. Chem. 2014, 22, 6616–6624. [Google Scholar] [CrossRef] [PubMed]
- Mojzych, M.; Tarasiuk, P.; Kotwica-Mojzych, K.; Rafiq, M.; Seo, S.-Y.; Nicewicz, M.; Fornal, E. Synthesis of chiral pyrazolo[4,3-e][1,2,4]triazine sulfonamides with tyrosinase and urease inhibitory activity. J. Enzym. Inhib. Med. Chem. 2016, 32, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojzych, M.; Šubertová, V.; Bielawska, A.; Bielawski, K.; Bazgier, V.; Berka, K.; Gucký, T.; Fornal, E.; Kryštof, V. Synthesis and kinase inhibitory activity of new sulfonamide derivatives of pyrazolo[4,3-e][1,2,4]triazines. Eur. J. Med. Chem. 2014, 78, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Cowan-Jacob, S.W.; Fendrich, G.; Floersheimer, A.; Furet, P.; Liebetanz, J.; Rummel, G.; Rheinberger, P.; Centeleghe, M.; Fabbro, D.; Manley, P.W. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 63, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Bernat, Z.; Szymanowska, A.; Kciuk, M.; Kotwica-Mojzych, K.; Mojzych, M. Review of the Synthesis and Anticancer Properties of Pyrazolo[4,3-e][1,2,4]triazine Derivatives. Molecules 2020, 25, 3948. [Google Scholar] [CrossRef]
- Byth, K.F.; Cooper, N.; Culshaw, J.D.; Heaton, D.W.; Oakes, S.E.; Minshull, C.A.; Norman, R.A.; Pauptit, R.A.; Tucker, J.A.; Breed, J.; et al. Imidazo[1,2-b]pyridazines: A potent and selective class of cyclin-dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 2249–2252. [Google Scholar] [CrossRef]
- Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Almassy, R.; Lu, J.; Averill, A.; Yager, K.M.; Chu, S. Structure-based design, synthesis, and study of pyrazolo[1,5-a][1,3,5]triazine derivatives as potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195. [Google Scholar] [CrossRef]
- Filhol, O.; Cochet, C. Protein Kinase CK2 in Health and Disease. Exp. 2009, 66, 1830–1839. [Google Scholar] [CrossRef]
- Sciú, M.L.; Sebastián-Pérez, V.; Martinez-Gonzalez, L.; Benitez, R.; Perez, D.I.; Pérez, C.; Campillo, N.E.; Martinez, A.; Moyano, E.L. Computer-aided molecular design of pyrazolotriazines targeting glycogen synthase kinase 3. J. Enzym. Inhib. Med. Chem. 2018, 34, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and Antiviral Sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Marciniak, B.; Drozda, R.; Kontek, R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1278–1298. [Google Scholar] [CrossRef] [PubMed]
- Mojzych, M.; Bielawska, A.; Bielawski, K.; Ceruso, M.; Supuran, C.T. Pyrazolo[4,3-e][1,2,4]triazine sulfonamides as carbonic anhydrase inhibitors with antitumor activity. Bioorg. Med. Chem. 2014, 22, 2643–2647. [Google Scholar] [CrossRef] [PubMed]
- Mojzych, M.; Karczmarzyk, Z.; Wysocki, W.; Ceruso, M.; Supuran, C.T.; Kryštof, V.; Urbańczyk-Lipkowska, Z.; Kalicki, P. New approaches to the synthesis of sildenafil analogues and their enzyme inhibitory activity. Bioorg. Med. Chem. 2015, 23, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Hermanowicz, J.M.; Szymanowska, A.; Sieklucka, B.; Czarnomysy, R.; Pawlak, K.; Bielawska, A.; Bielawski, K.; Kalafut, J.; Przybyszewska, A.; Surazynski, A.; et al. Exploration of novel heterofused 1,2,4-triazine derivative in colorectal cancer. J. Enzym. Inhib. Med. Chem. 2021, 36, 535–548. [Google Scholar] [CrossRef]
- Hermanowicz, J.; Pawlak, K.; Sieklucka, B.; Czarnomysy, R.; Kwiatkowska, I.; Kazberuk, A.; Surazynski, A.; Mojzych, M.; Pawlak, D. MM-129 as a Novel Inhibitor Targeting PI3K/AKT/mTOR and PD-L1 in Colorectal Cancer. Cancers 2021, 13, 3203. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell. Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Guan, J.; Lim, K.S.; Mekhail, T.; Chang, C.-C. Programmed Death Ligand-1 (PD-L1) Expression in the Programmed Death Receptor-1 (PD-1)/PD-L1 Blockade: A Key Player Against Various Cancers. Arch. Pathol. Lab. Med. 2017, 141, 851–861. [Google Scholar] [CrossRef]
- Hassan, B.; Akcakanat, A.; Holder, A.M.; Meric-Bernstam, F. Targeting the PI3-Kinase/Akt/mTOR Signaling Pathway. Surg. Oncol. Clin. N. Am. 2013, 22, 641–664. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. BioSyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Hermanowicz, J.M.; Kalaska, B.; Pawlak, K.; Sieklucka, B.; Miklosz, J.; Mojzych, M.; Pawlak, D. Preclinical Toxicity and Safety of MM-129—First-in-Class BTK/PD-L1 Inhibitor as a Potential Candidate against Colon Cancer. Pharmaceutics 2021, 13, 1222. [Google Scholar] [CrossRef] [PubMed]
- Gornowicz, A.; Szymanowska, A.; Mojzych, M.; Czarnomysy, R.; Bielawski, K.; Bielawska, A. The Anticancer Action of a Novel 1,2,4-Triazine Sulfonamide Derivative in Colon Cancer Cells. Molecules 2021, 26, 2045. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blázquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazotte, B. Labeling Mitochondria with MitoTracker Dyes. Cold Spring Harb. Protoc. 2011, 2011, 990–992. [Google Scholar] [CrossRef]
- Ricci, J.E.; Muñoz-Pinedo, C.; Fitzgerald, P.; Bailly-Maitre, B.; Perkins, G.A.; Yadava, N.; Scheffler, I.E.; Ellisman, M.H.; Green, D.R. Disruption of Mitochondrial Function during Apoptosis Is Mediated by Caspase Cleavage of the p75 Subunit of Complex I of the Electron Transport Chain. Cell 2004, 117, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Krysko, D.V.; Roels, F.; Leybaert, L.; D’Herde, K. Mitochondrial Transmembrane Potential Changes Support the Concept of Mitochondrial Heterogeneity During Apoptosis. J. Histochem. Cytochem. 2001, 49, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Gornowicz, A.; Szymanowska, A.; Mojzych, M.; Bielawski, K.; Bielawska, A. The Effect of Novel 7-methyl-5-phenyl-pyrazolo[4,3-e]tetrazolo[4,5-b][1,2,4]triazine Sulfonamide Derivatives on Apoptosis and Autophagy in DLD-1 and HT-29 Colon Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5221. [Google Scholar] [CrossRef]
- Mojzych, M.; Karczmarzyk, Z.; Rykowski, A. Synthesis and structure of 7-methyl-5-phenyl-1H-pyrazolo[4,3-e]tetrazolo[4,5-b][1,2,4]triazine(1). J. Chem. Crystallogr. 2005, 35, 151–155. [Google Scholar] [CrossRef]
- Karczmarzyk, Z.; Mojzych, M.; Rykowski, A. Synthesis and structure of a novel mesomeric betaine 6,7-dimethyl-2H-pyrazolo[4,3-e]tetrazolo[4,5-b][1,2,4]triazine. J. Mol. Struct. 2007, 829, 22–28. [Google Scholar] [CrossRef]
- Mojzych, M.; Karczmarzyk, Z.; Wysocki, W.; Urbańczyk-Lipkowska, Z.; Żaczek, N. Valence tautomerism of new pyrazolo[4,3-e]tetrazole[4,5-b][1,2,4]triazines. J. Mol. Struct. 2014, 1067, 147–153. [Google Scholar] [CrossRef]
- Deev, S.L.; Shestakova, T.S.; Charushin, V.N.; Chupakhin, O.N. Synthesis and azido-tetrazole tautomerism of 3-azido-1,2,4-triazines. Chem. Heterocycl. Compd. 2017, 53, 963–975. [Google Scholar] [CrossRef]
- Kasibhatla, S.; Amarante-Mendes, G.P.; Finucane, D.; Brunner, T.; Bossy-Wetzel, E.; Green, D.R. Acridine Orange/Ethidium Bromide (AO/EB) Staining to Detect Apoptosis. Cold Spring Harb. Protoc. 2006. [CrossRef]
- Pemovska, T.; Johnson, E.; Kontro, M.; Repasky, G.A.; Chen, J.; Wells, P.; Cronin, C.N.; McTigue, M.; Kallioniemi, O.; Porkka, K.; et al. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nat. Cell Biol. 2015, 519, 102–105. [Google Scholar] [CrossRef]
- Salah, E.; Ugochukwu, E.; Barr, A.J.; von Delft, F.; Knapp, S.; Elkins, J.M. Crystal Structures of ABL-Related Gene (ABL2) in Complex with Imatinib, Tozasertib (VX-680), and a Type I Inhibitor of the Triazole Carbothioamide Class. J. Med. Chem. 2011, 54, 2359–2367. [Google Scholar] [CrossRef]
- Freeman-Cook, K.D.; Autry, C.; Borzillo, G.; Gordon, D.; Barbacci-Tobin, E.; Bernardo, V.; Briere, D.; Clark, T.; Corbett, M.; Jakubczak, J.; et al. Design of Selective, ATP-Competitive Inhibitors of Akt. J. Med. Chem. 2010, 53, 4615–4622. [Google Scholar] [CrossRef]
- Heerding, D.A.; Rhodes, N.; Leber, J.D.; Clark, T.J.; Keenan, R.M.; Lafrance, L.V.; Li, M.; Safonov, I.G.; Takata, D.T.; Venslavsky, J.W.; et al. Identification of 4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}-1H-imidazo [4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a Novel Inhibitor of AKT Kinase. J. Med. Chem. 2008, 51, 5663–5679. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Reckel, S.; Gehin, C.; Tardivon, D.; Georgeon, S.; Kükenshöner, T.; Löhr, F.; Koide, A.; Buchner, L.; Panjkovich, A.; Reynaud, A.; et al. Structural and functional dissection of the DH and PH domains of oncogenic Bcr-Abl tyrosine kinase. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.J.; Liu, Y.-T.; Arduini, R.M.; Hession, C.A.; Miatkowski, K.; Wildes, C.P.; Cullen, P.F.; Hong, V.; Hopkins, B.T.; Mertsching, E.; et al. Structures of human Bruton’s tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 2010, 19, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakšauskas, A.; Čapkauskaitė, E.; Jezepčikas, L.; Linkuvienė, V.; Paketurytė, V.; Smirnov, A.; Leitans, J.; Kazaks, A.; Dvinskis, E.; Manakova, E.; et al. Halogenated and di-substituted benzenesulfonamides as selective inhibitors of carbonic anhydrase isoforms. Eur. J. Med. Chem. 2020, 185, 111825. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Tirado, O.M.; Lambot, S.M.; Ferandin, Y.; Lozach, O.; Morris, J.; Mateo-Lozano, S.; Drueckes, P.; Schächtele, C.; Kubbutat, M.H.; et al. Meriolins, a New Class of Cell Death–Inducing Kinase Inhibitors with Enhanced Selectivity for Cyclin-Dependent Kinases. Cancer Res. 2007, 67, 8325–8334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, P.J.; Cleasby, A.; Tickle, I.J.; O’Reilly, M.; Coyle, J.E.; Holding, F.P.; McMenamin, R.L.; Yon, J.; Chopra, R.; Lengauer, C.; et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. USA 2009, 106, 4166–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.-A.; et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lolli, G.; Lowe, E.D.; Brown, N.R.; Johnson, L.N. The Crystal Structure of Human CDK7 and Its Protein Recognition Properties. Structure 2004, 12, 2067–2079. [Google Scholar] [CrossRef]
- Lennartz, F.; Adams, Y.; Bengtsson, A.; Olsen, R.W.; Turner, L.; Ndam, N.T.; Ecklu-Mensah, G.; Moussiliou, A.; Ofori, M.F.; Gamain, B.; et al. Structure-Guided Identification of a Family of Dual Receptor-Binding PfEMP1 that Is Associated with Cerebral Malaria. Cell Host Microbe 2017, 21, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Scaiola, A.; Mangia, F.; Imseng, S.; Boehringer, D.; Berneiser, K.; Shimobayashi, M.; Stuttfeld, E.; Hall, M.N.; Ban, N.; Maier, T. The 3.2-Å resolution structure of human mTORC2. Sci. Adv. 2020, 6, eabc1251. [Google Scholar] [CrossRef]
- Butera, R.; Ważyńska, M.; Magiera-Mularz, K.; Plewka, J.; Musielak, B.; Surmiak, E.; Sala, D.; Kitel, R.; de Bruyn, M.; Nijman, H.W.; et al. Design, Synthesis, and Biological Evaluation of Imidazopyridines as PD-1/PD-L1 Antagonists. ACS Med. Chem. Lett. 2021, 12, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitencourt-Ferreira, G.; Pintro, V.O.; de Azevedo, W.F. Docking with AutoDock4. Methods Mol. Biol. 2019, 2053, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Mujwar, S. Repurposing metocurine as main protease inhibitor to develop novel antiviral therapy for COVID-19. Struct. Chem. 2020, 31, 2487–2499. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S.; Kumar, V. Computational Drug Repurposing Approach to Identify Potential Fatty Acid-Binding Protein-4 Inhibitors to Develop Novel Antiobesity Therapy. ASSAY Drug Dev. Technol. 2020, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Mujwar, S.; Krishna, G.; Gupta, J.K. Computational Design and Biological Depiction of Novel Naproxen Derivative. ASSAY Drug Dev. Technol. 2020, 18, 308–317. [Google Scholar] [CrossRef]
- Shah, K.; Mujwar, S.; Gupta, J.K.; Shrivastava, S.K.; Mishra, P. Molecular Docking and In Silico Cogitation Validate Mefenamic Acid Prodrugs as Human Cyclooxygenase-2 Inhibitor. ASSAY Drug Dev. Technol. 2019, 17, 285–291. [Google Scholar] [CrossRef]
- Mujwar, S.; Deshmukh, R.; Harwansh, R.K.; Gupta, J.K.; Gour, A. Drug Repurposing Approach for Developing Novel Therapy Against Mupirocin-Resistant Staphylococcus aureus. ASSAY Drug Dev. Technol. 2019, 17, 298–309. [Google Scholar] [CrossRef]
- Mishra, I.; Mishra, R.; Mujwar, S.; Chandra, P.; Sachan, N. A retrospect on antimicrobial potential of thiazole scaffold. J. Heterocycl. Chem. 2020, 57, 2304–2329. [Google Scholar] [CrossRef]
- Mujwar, S.; Shah, K.; Gupta, J.K.; Gour, A. Docking based screening of curcumin derivatives: A novel approach in the inhibition of tubercular DHFR. Int. J. Comput. Biol. Drug Des. 2021, 14, 297. [Google Scholar] [CrossRef]
- Sharma, K.K.; Singh, B.; Mujwar, S.; Bisen, P.; Kaushal, S.K.; Brijendra, S.; Prakash, B.S. Molecular Docking Based Analysis to Elucidate the DNA Topoisomerase IIβ as the Potential Target for the Ganoderic Acid; A Natural Therapeutic Agent in Cancer Therapy. Curr. Comput. Aided-Drug Des. 2020, 16, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Minaz, N.; Razdan, R.; Hammock, B.D.; Mujwar, S.; Goswami, S.K. Impact of diabetes on male sexual function in streptozotocin-induced diabetic rats: Protective role of soluble epoxide hydrolase inhibitor. Biomed. Pharmacother. 2019, 115, 108897. [Google Scholar] [CrossRef] [PubMed]
- SwissADME. Available online: http://www.swissadme.ch (accessed on 4 May 2022).
- Dresser, G.K.; Spence, J.D.; Bailey, D. Pharmacokinetic-Pharmacodynamic Consequences and Clinical Relevance of Cytochrome P450 3A4 Inhibition. Clin. Pharmacokinet. 2000, 38, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Fontana, E.; Dansette, P.M.; Poli, S.M. Cytochrome P450 Enzymes Mechanism Based Inhibitors: Common Sub-Structures and Reactivity. Curr. Drug Metab. 2005, 6, 413–451. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F. Drugs Behave as Substrates, Inhibitors and Inducers of Human Cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef]
- Miller, D.S.; Bauer, B.; Hartz, A.M.S. Modulation of P-Glycoprotein at the Blood-Brain Barrier: Opportunities to Improve Central Nervous System Pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Robinson, K.; Tiriveedhi, V. Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front. Oncol. 2020, 10, 265. [Google Scholar] [CrossRef]
- Kim, E.-J.; Kang, G.-J.; Kang, J.-I.; Boo, H.-J.; Hyun, J.W.; Koh, Y.S.; Chang, W.-Y.; Kim, Y.R.; Kwon, J.-M.; Maeng, Y.H.; et al. Over-activation of AKT signaling leading to 5-Fluorouracil resistance in SNU-C5/5-FU cells. Oncotarget 2018, 9, 19911–19928. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., III; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT–mTOR Pathway in Non–Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Van Der Kraak, L.; Goel, G.; Ramanan, K.; Kaltenmeier, C.; Zhang, L.; Normolle, D.P.; Freeman, G.J.; Tang, D.; Nason, K.S.; Davison, J.M.; et al. 5-Fluorouracil upregulates cell surface B7-H1 (PD-L1) expression in gastrointestinal cancers. J. Immunother. Cancer 2016, 4, 65. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Sun, S.-Y. Searching for the real function of mTOR signaling in the regulation of PD-L1 expression. Transl. Oncol. 2020, 13, 100847. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.; Hughes, A.; Taylor, M.A.; Kuczynski, E.A.; Mele, D.A.; Delpuech, O.; Jarvis, L.; Staniszewska, A.; Cosulich, S.; Carnevalli, L.S.; et al. Combination of dual mTORC1/2 inhibition and immune-checkpoint blockade potentiates anti-tumour immunity. OncoImmunology 2018, 7, e1458810. [Google Scholar] [CrossRef] [Green Version]
- Mojzych, M.; Rykowski, A. Synthesis of Functionalized 1H-Pyrazolo[4,3-e][1,2,4]triazines and Their Fused Derivatives via Ipso-Substitution of Methylsulfonyl Group with O-, N-, S- and C-Nucleophiles. Heterocycles 2004, 63, 1829–1838. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Da Violante, G.; Zerrouk, N.; Richard, I.; Provot, G.; Chaumeil, J.C.; Arnaud, P. Evaluation of the Cytotoxicity Effect of Dimethyl Sulfoxide (DMSO) on Caco2/TC7 Colon Tumor Cell Cultures. Biol. Pharm. Bull. 2002, 25, 1600–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Flow Cytometry-Based Apoptosis Detection. In Apoptosis; Humana Press: Totowa, NJ, USA, 2009; Volume 559, pp. 19–32. [Google Scholar] [CrossRef] [Green Version]
- Kontek, R.; Jakubczak, M.; Matlawska-Wasowska, K. The antioxidants, vitamin A and E but not vitamin C and melatonin enhance the proapoptotic effects of irinotecan in cancer cells in vitro. Toxicol. Vitr. 2014, 28, 282–291. [Google Scholar] [CrossRef]
- Xiao, B.; Deng, X.; Zhou, W.; Tan, E.-K. Flow Cytometry-Based Assessment of Mitophagy Using MitoTracker. Front. Cell. Neurosci. 2016, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Mujwar, S. Computational repurposing of tamibarotene against triple mutant variant of SARS-CoV-2. Comput. Biol. Med. 2021, 136, 104748. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. Application of the SwissDrugDesign Online Resources in Virtual Screening. Int. J. Mol. Sci. 2019, 20, 4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MM-Compounds | Cell Lines | ||||
|---|---|---|---|---|---|
| Mean IC50 [µM] | |||||
| BxPC-3 | HCT-116 | PC-3 | L929 | WI-38 | |
| MM134 | 0.32 ± 0.1 | 0.38 ± 0.03 | 0.16 ± 0.02 | 0.58 ± 0.005 | 0.65 ± 0.07 |
| MM136 | 0.25 ± 0.08 | 0.25 ± 0.07 | 0.13 ± 0.01 | 0.22 ± 0.08 | 0.48 ± 0.09 |
| MM137 | 0.16 ± 0.04 | 0.14 ± 0.02 | 0.11 ± 0.007 | 0.18 ± 0.0008 | 0.27 ± 0.04 |
| MM139 | 0.33 ± 0.14 | 0.35 ± 0.05 | 0.17 ± 0.003 | 0.26 ± 0.02 | 0.54 ± 0.06 |
| BxPC-3 | |||
|---|---|---|---|
| 24 h incubation | |||
| Compound/Concentration | 0.5IC50 | IC50 | 2xIC50 |
| MM134 | 88.19 ± 13.62 | 81.23 ± 11.94 | 78.31 ± 14.34 |
| MM136 | 82.83 ± 13.02 | 65.68 ± 4.58 | 57.67 ± 5.56 |
| MM137 | 89.34 ± 5.91 | 79.5 ± 3.34 | 79.22 ± 2.22 |
| MM139 | 81.28 ± 14.44 | 66.73 ± 3.72 | 64.23 ± 6.08 |
| 48 h incubation | |||
| MM134 | 79.98 ± 11.47 | 66.04 ± 16.7 | 68.95 ± 7.78 |
| MM136 | 69.08 ± 4.40 | 62.37 ± 0.05 | 61.83 ± 0.46 |
| MM137 | 84.62 ± 9.01 | 79.8 ± 6.76 | 73.8 ± 2.58 |
| MM139 | 76.9 ± 6.40 | 60.35 ± 0.91 | 65.95 ± 12.53 |
| PC-3 | |||
| 24 h incubation | |||
| MM134 | 100.35 ± 1.57 | 82.5 ± 2.21 | 82.34 ± 2.13 |
| MM136 | 96.3 ± 5.74 | 92.78 ± 0.9 | 86.9 ± 7.22 |
| MM137 | 96.26 ± 2.83 | 87.9 ± 2.71 | 82.5 ± 7.99 |
| MM139 | 89.2 ± 6.28 | 87.28 ± 15.8 | 78.14 ± 6.55 |
| 48 h incubation | |||
| MM134 | 82.1 ± 14.45 | 80.26 ± 13.44 | 64.56 ± 3.33 |
| MM136 | 81.34 ± 0.4 | 61.24 ± 3.62 | 47.38 ± 2.74 |
| MM137 | 78.26 ± 2.62 | 75.49 ± 5.55 | 64.19 ± 8.72 |
| MM139 | 78.91 ± 15.21 | 65.93 ± 19.56 | 61.28 ± 9.16 |
| Drug Target | PDB Code | x-D | y-D | z-D | Spacing (Ả) | x Center | y Center | z Center |
|---|---|---|---|---|---|---|---|---|
| ABL1 | 4wa9 | 40 | 40 | 40 | 0.431 | 23.95 | 127.557 | 14.335 |
| ABL2 | 2xyn | 40 | 40 | 40 | 0.536 | −54.025 | 48.668 | −7.666 |
| AKT1 | 3mvh | 40 | 40 | 40 | 0.453 | 17.948 | −1.885 | 27.56 |
| AKT2 | 3d0e | 40 | 40 | 40 | 0.475 | 22.521 | −19.611 | 7.41 |
| AKT3 | 2x18 | 40 | 40 | 40 | 0.458 | 25.066 | 66.608 | −19.071 |
| BCR | 5n7e | 40 | 40 | 40 | 0.636 | 22.531 | −2.925 | 111.276 |
| BTK | 3gen | 40 | 40 | 40 | 0.492 | −16.796 | 6.794 | −14.126 |
| CA-IX | 6qn5 | 40 | 40 | 40 | 0.375 | −29.939 | −3.656 | 0.027 |
| CA-XII | 6qnl | 40 | 40 | 40 | 0.375 | −41.046 | 6.288 | 31.866 |
| CDK2 | 3bhu | 40 | 40 | 40 | 0.375 | −7.638 | 20.962 | −21.4 |
| CDK4 | 2w9z | 50 | 50 | 50 | 0.453 | 20.281 | 25.506 | 8.713 |
| CDK6 | 5l2s | 50 | 50 | 50 | 0.403 | 22.452 | 38.566 | −8.687 |
| CDK7 | 1ua2 | 40 | 40 | 40 | 0.375 | 41.304 | −4.892 | 23.033 |
| ICAM-1 | 5mza | 40 | 40 | 40 | 0.531 | 6.393 | −16.531 | 9.728 |
| mTOR1 | 6bcx | 40 | 40 | 40 | 0.625 | 247.93 | 204.864 | 264.981 |
| mTOR2 | 6zwo | 40 | 40 | 40 | 0.625 | 249.818 | 191.487 | 206.7 |
| PD-L1 | 7bea | 40 | 40 | 40 | 0.492 | −5.537 | −10.417 | 19.104 |
| S. No. | Ligand | Binding Energy ABL1 (4wa9) | Binding Energy ABL2 (2xyn) | Binding Energy AKT1 (3mvh) | Binding Energy AKT2 (3d0e) | Binding Energy AKT3 (2x18) | Binding Energy BCR (5n7e) | Binding Energy BTK (3gen) | Binding Energy CA-IX (6qn5) | Binding Energy CA-XII (6qnl) | Binding Energy CDK2 (3bhu) | Binding Energy CDK4 (2w9z) | Binding Energy CDK6 (5ls2) | Binding Energy CDK7 (1ua2) | Binding Energy ICAM-1 (5mza) | Binding Energy mTOR1 (6bcx) | Binding Energy mTOR2 (6zwo) | Binding Energy PD-L1 (7bea) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Reference Ligand | −9.35 (AXI) | −9.28 (VX6) | −10.51 (WFE) | −9.54 (G93) | −4.4 (EPE) | - | −9.31 (B43) | −8.18 (J8N) | −7.63 (J92) | −7.67 (MHR) | −5.7 (NAG) | −9.4 (6ZV) | −7.64 (ATP) | - | −3.26 (ATP) | −4.71 (AGS) | −10.09 (TK2) |
| 3 | MM129 | −9.71 | −9.17 | −10.70 | −9.52 | −6.68 | −8.63 | −8.52 | −8.44 | −8.21 | −9.61 | −8.97 | −10.83 | −10.04 | −6.86 | −8.31 | −8.12 | −9.86 |
| 4 | MM131 | −9.48 | −9.23 | −9.92 | −9.62 | −6.74 | −8.62 | −8.76 | −7.90 | −7.87 | −8.80 | −8.52 | −9.01 | −8.74 | −6.51 | −7.19 | −7.44 | −9.88 |
| 5 | MM134 | −9.88 | −10.08 | −11.35 | −11.81 | −6.94 | −9.51 | −10.11 | −8.01 | −8.04 | −9.63 | −8.08 | −9.69 | −9.14 | −7.20 | −8.45 | −7.64 | −10.30 |
| 6 | MM136 | −9.53 | −9.81 | −12.42 | −9.93 | −6.63 | −9.04 | −9.55 | −8.39 | −8.54 | −8.60 | −8.97 | −9.26 | −9.40 | −7.68 | −9.08 | −8.79 | −12.33 |
| 7 | MM137 | −10.11 | −10.46 | −11.51 | −10.79 | −6.84 | −8.49 | −10.06 | −7.86 | −8.52 | −8.31 | −8.78 | −8.31 | −9.78 | −7.19 | −8.57 | −7.95 | −11.54 |
| 8 | MM139 | −10.42 | −9.84 | −12.16 | −10.55 | −7.24 | −8.52 | −10.51 | −9.34 | −9.06 | −10.02 | −9.62 | −9.68 | −9.88 | −7.68 | −8.46 | −8.29 | −11.43 |
| Property | Parameter | MM129 | MM131 | MM134 | MM136 | MM137 | MM139 |
|---|---|---|---|---|---|---|---|
| Flexibility | Num. rotatable bonds | 5 | 5 | 6 | 3 | 3 | 3 |
| Lipophilicity | XLOGP3 | −0.31 | −0.21 | −0.30 | −0.15 | 0.04 | 1.07 |
| Molecular weight | MW | 459.44 g/mol | 389.39 g/mol | 444.47 g/mol | 401.40 g/mol | 414.44 g/mol | 399.43 g/mol |
| Polarity | TPSA | 178.97 Å2 | 161.46 Å2 | 153.70 Å2 | 141.67 Å2 | 135.68 Å2 | 132.44 Å2 |
| Saturation | Fraction Csp3 | 0.35 | 0.29 | 0.41 | 0.33 | 0.38 | 0.38 |
| Compound | MM129 | MM131 | MM134 | MM136 | MM137 | MM139 |
|---|---|---|---|---|---|---|
| GI absorption | Low | Low | Low | Low | High | High |
| BBB permeant | No | No | No | No | No | No |
| P-gp substrate | Yes | Yes | Yes | Yes | Yes | Yes |
| CYP2D6 Inhibitor | No | No | No | No | No | No |
| CYP3A4 Inhibitor | No | No | Yes | No | No | No |
| Compound | MM129 | MM131 | MM134 | MM136 | MM137 | MM139 |
|---|---|---|---|---|---|---|
| Molecular weight | 459.44 g/mol | 389.39 g/mol | 444.47 g/mol | 401.40 g/mol | 414.44 g/mol | 399.43 g/mol |
| Num. H-bond acceptors | 12 | 10 | 11 | 10 | 10 | 9 |
| Num. H-bond donors | 1 | 2 | 1 | 0 | 0 | 0 |
| Consensus Log Po/w | 0.07 | 0.28 | 0.35 | 0.51 | 0.41 | 1.31 |
| Lipinski violation | 1 | 1 | 1 | 1 | 1 | 1 |
| Type of Cells | Medium | Supplements |
|---|---|---|
| BxPC-3 HCT-116 | RPMI-1640 | 10% (v/v) FBS, L-Glutamine, 25 mM Hepes, 1% penicillin-streptomycin |
| PC-3 | DMEM-F12 | 10% (v/v) FBS, L-Glutamine, 15 mM Hepes 1% penicillin-streptomycin |
| L929 | RPMI-1640 | 10% (v/v) FBS, L-Glutamine, 25 mM Hepes, 1% penicillin-streptomycin 1% β-mercaptoethanol |
| WI-38 | MEM | 10% (v/v) FBS, L-Glutamine, 25 mM Hepes 1% penicillin-streptomycin |
| Cancer Cell Lines | Cell Density/2 mL Culture Medium Annexin V-FITC | |
|---|---|---|
| 24 h Incubation | 48 h Incubation | |
| BxPC-3 | 6 × 105 | 5 × 105 |
| PC-3 | 7 × 105 | 5 × 105 |
| Cell density/200 µL culture medium MitoTracker Red | ||
| 24 h incubation | 48 h incubation | |
| BxPC-3 | 2 × 104 | 1.5 × 104 |
| PC-3 | 1.2 × 104 | 1.5 × 104 |
| Cell density/mL culture medium AO/EB | ||
| 24 h incubation | 48 h incubation | |
| BxPC-3 | 1.5 × 105 | 1 × 105 |
| PC-3 | 1.5 × 105 | 1 × 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Mujwar, S.; Szymanowska, A.; Marciniak, B.; Bukowski, K.; Mojzych, M.; Kontek, R. Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies. Int. J. Mol. Sci. 2022, 23, 5892. https://doi.org/10.3390/ijms23115892
Kciuk M, Mujwar S, Szymanowska A, Marciniak B, Bukowski K, Mojzych M, Kontek R. Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies. International Journal of Molecular Sciences. 2022; 23(11):5892. https://doi.org/10.3390/ijms23115892
Chicago/Turabian StyleKciuk, Mateusz, Somdutt Mujwar, Anna Szymanowska, Beata Marciniak, Karol Bukowski, Mariusz Mojzych, and Renata Kontek. 2022. "Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies" International Journal of Molecular Sciences 23, no. 11: 5892. https://doi.org/10.3390/ijms23115892
APA StyleKciuk, M., Mujwar, S., Szymanowska, A., Marciniak, B., Bukowski, K., Mojzych, M., & Kontek, R. (2022). Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies. International Journal of Molecular Sciences, 23(11), 5892. https://doi.org/10.3390/ijms23115892