
Loss of αA or αB-Crystallin Accelerates Photoreceptor Cell Death in a Mouse Model of P23H Autosomal Dominant Retinitis Pigmentosa


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
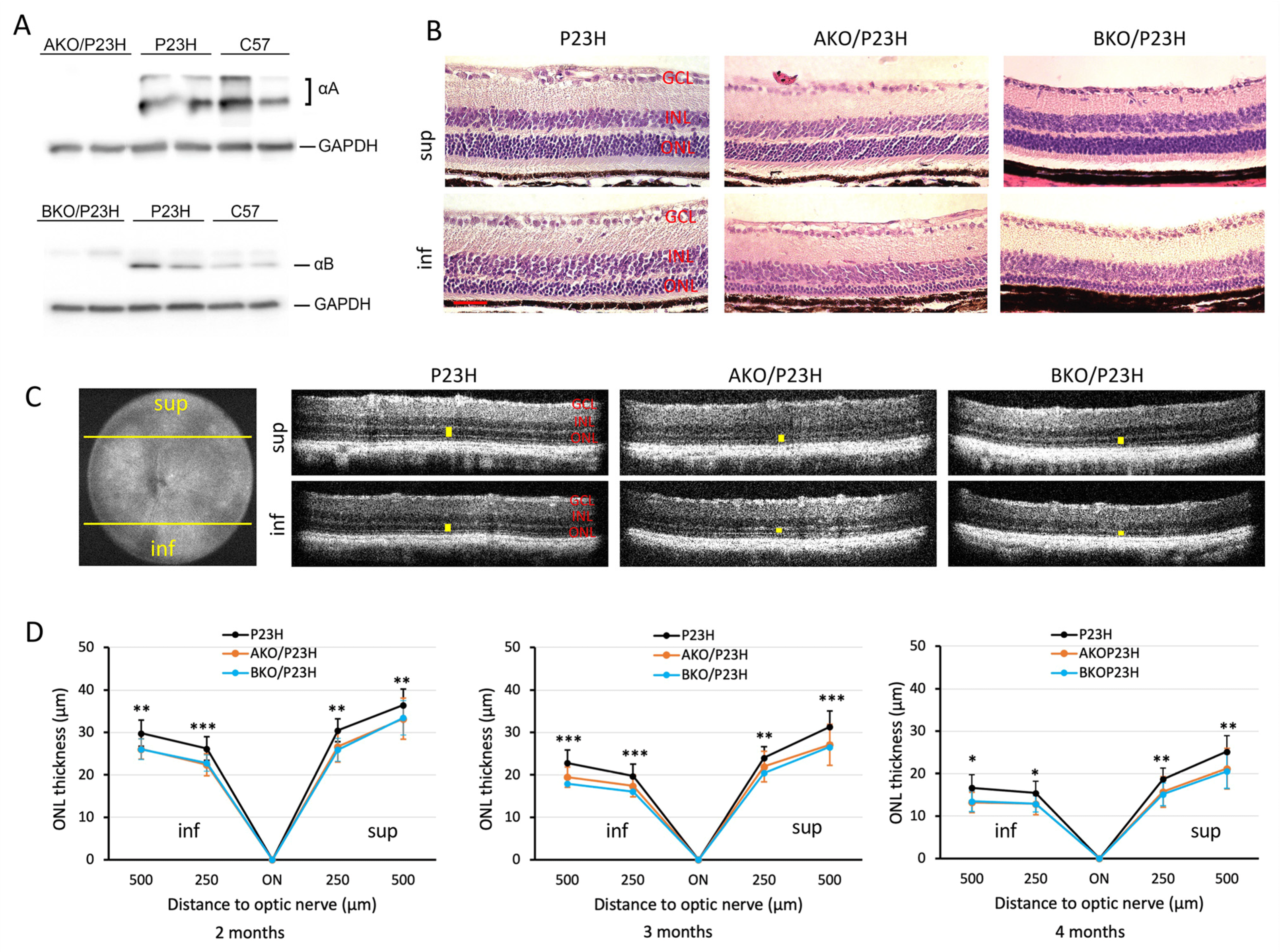
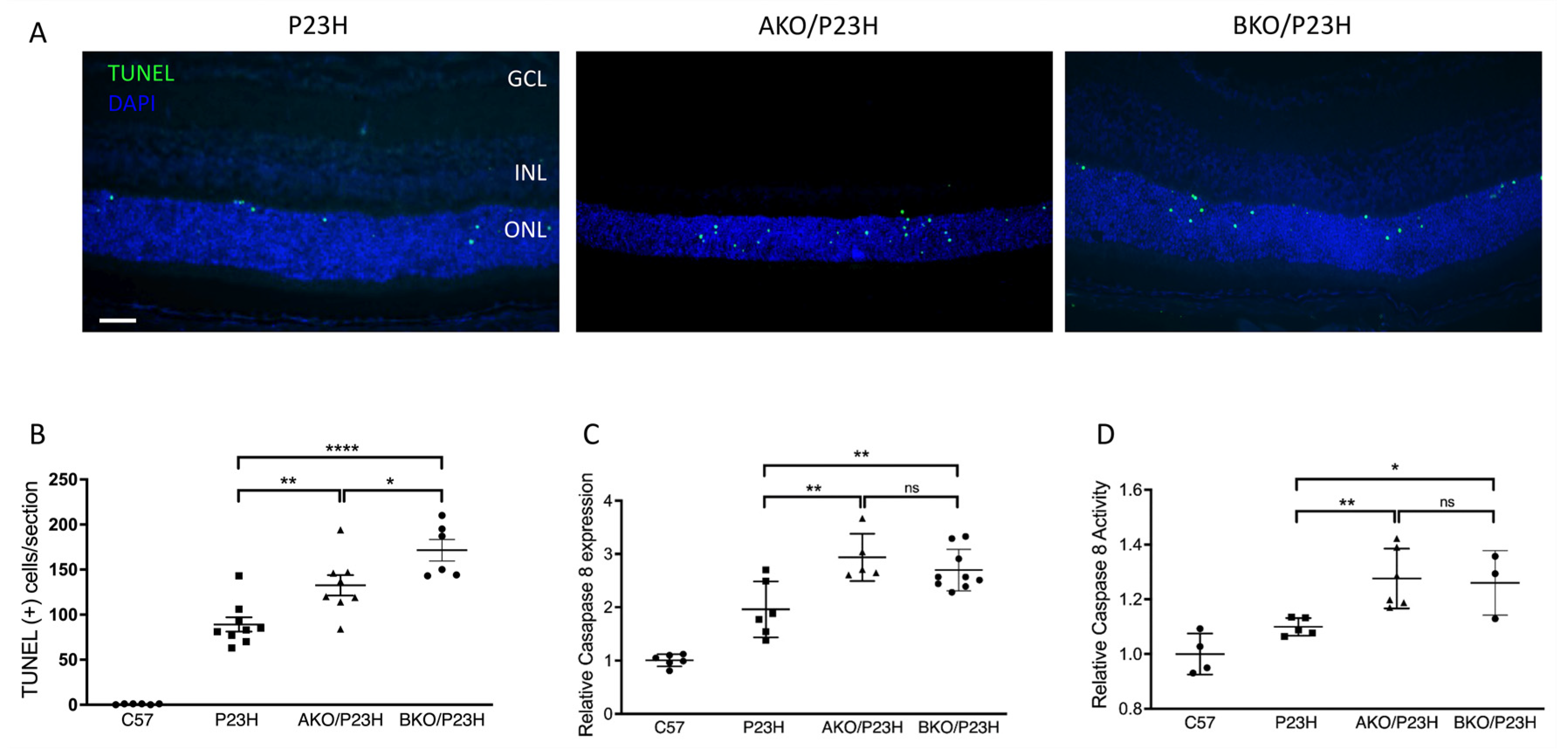
2.1. Increased Photoreceptor Cell Death in AKO/P23H and BKO/P23H Mouse Retina
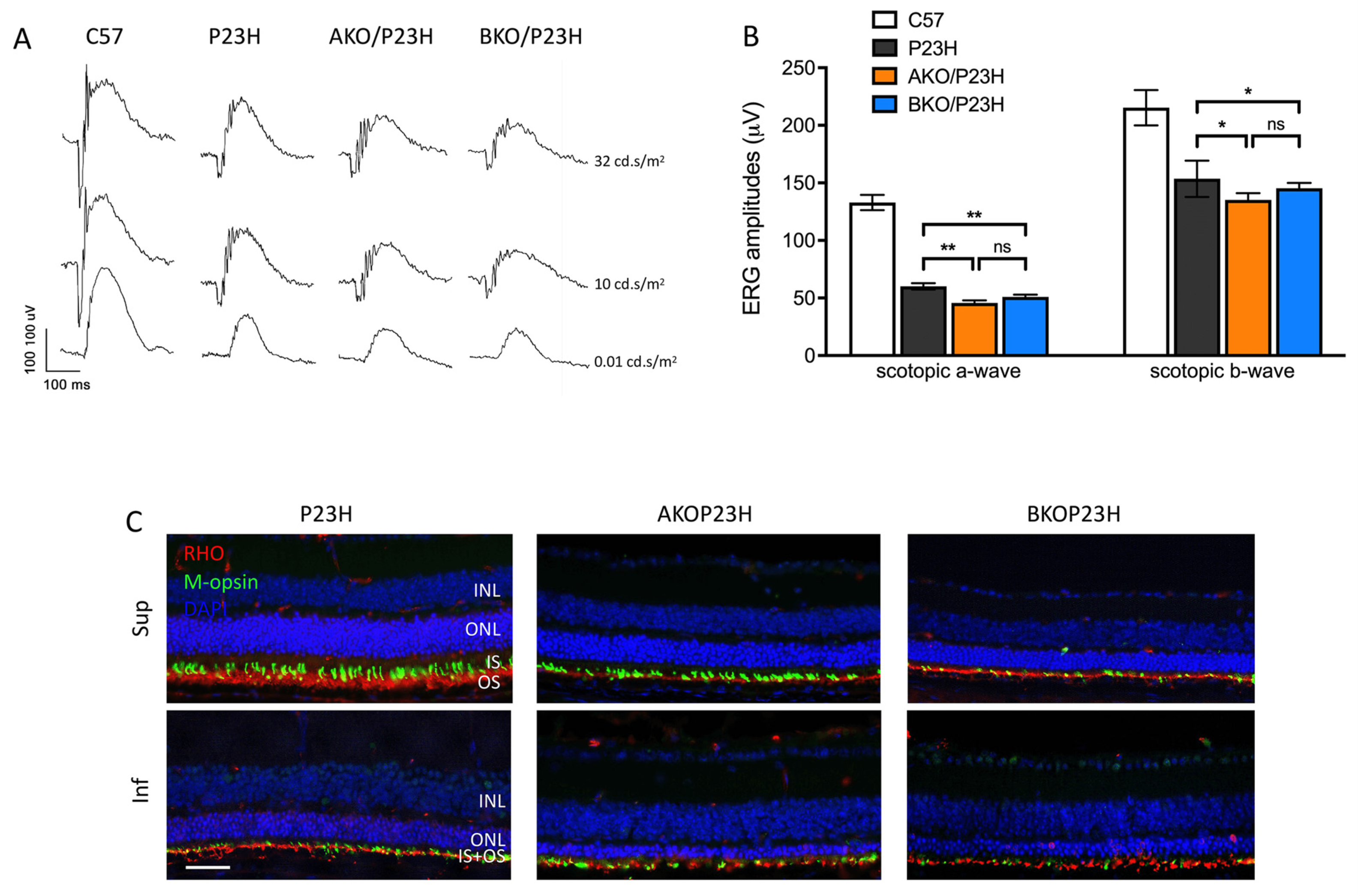
2.2. Reduced Rod-Photoreceptor Function in AKO/P23H and BKO/P23H Mouse Retina
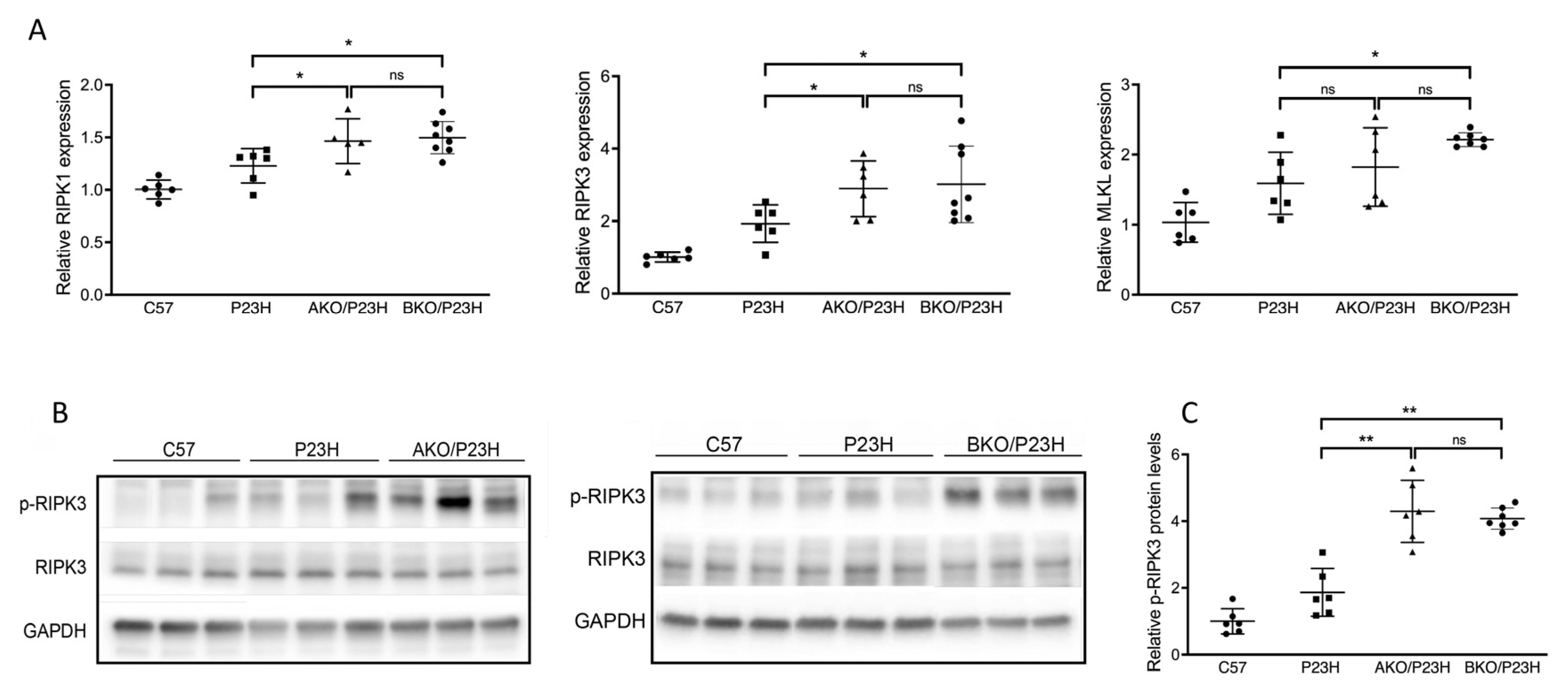
2.3. Increased Activation of Cell Death Pathways in AKO/P23H and BKO/P23H Mouse Retina
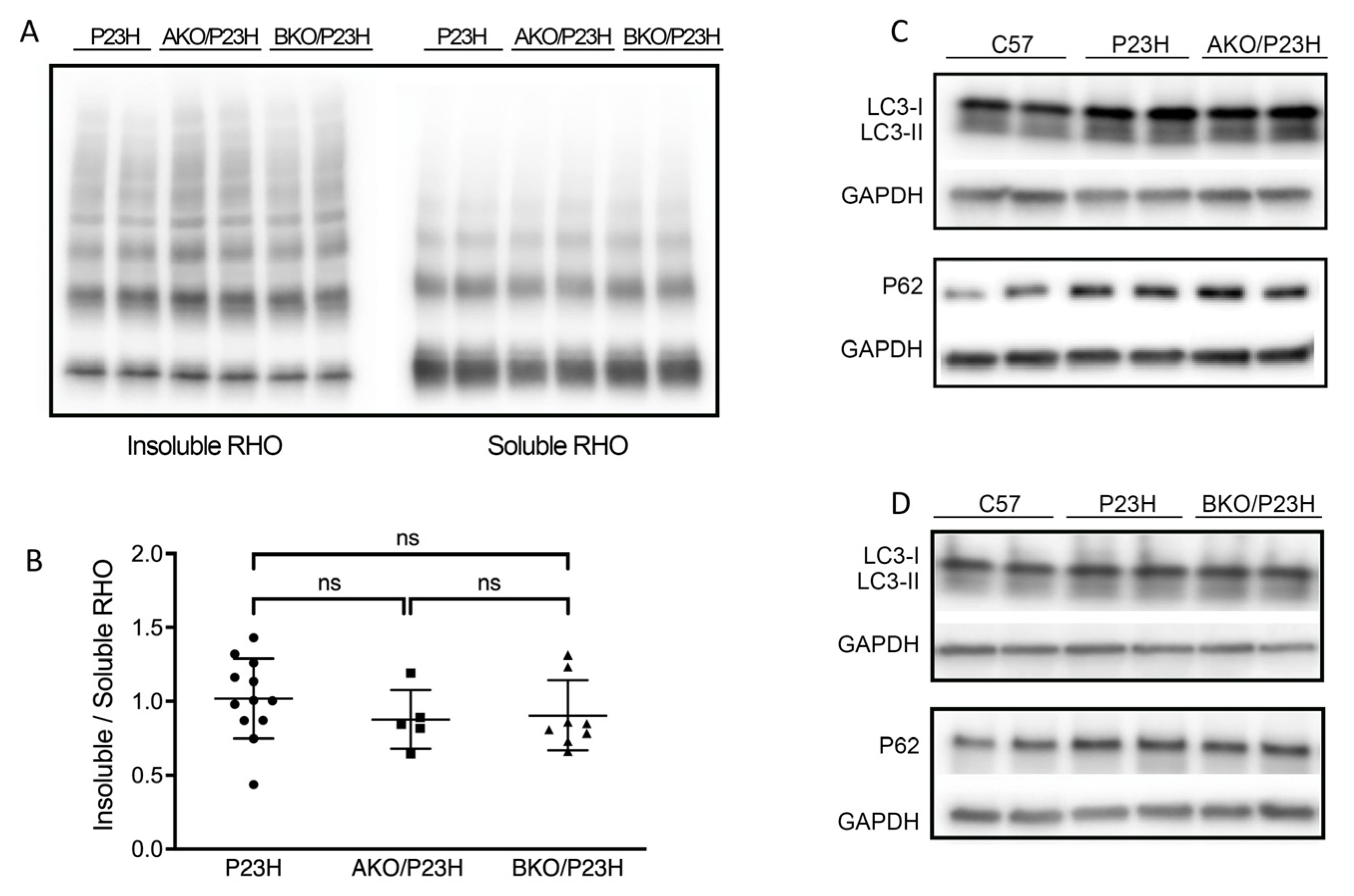
2.4. Autophagy Activation Unchanged in the AKO/P23H and BKO/P23H Mouse Retina
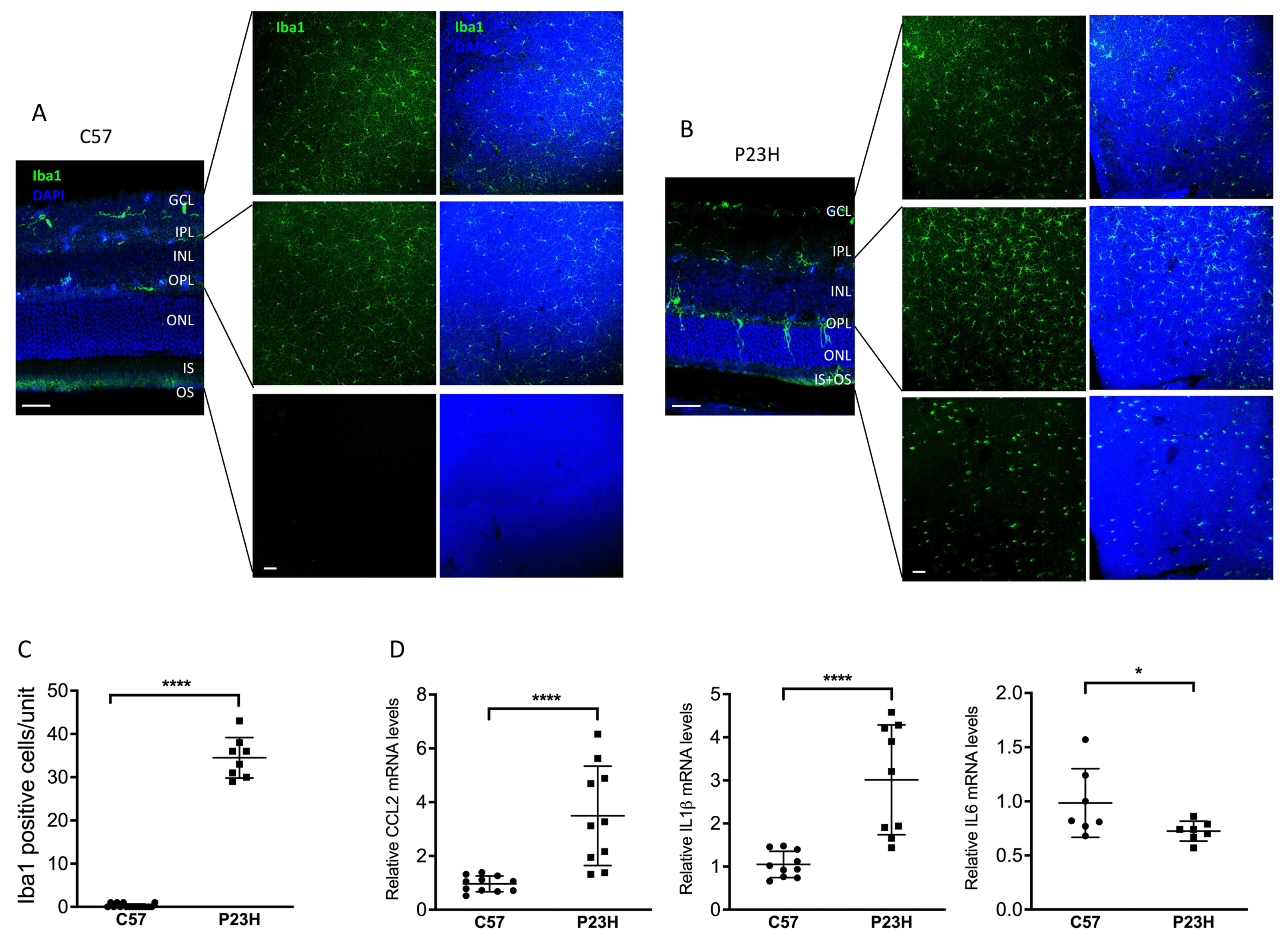
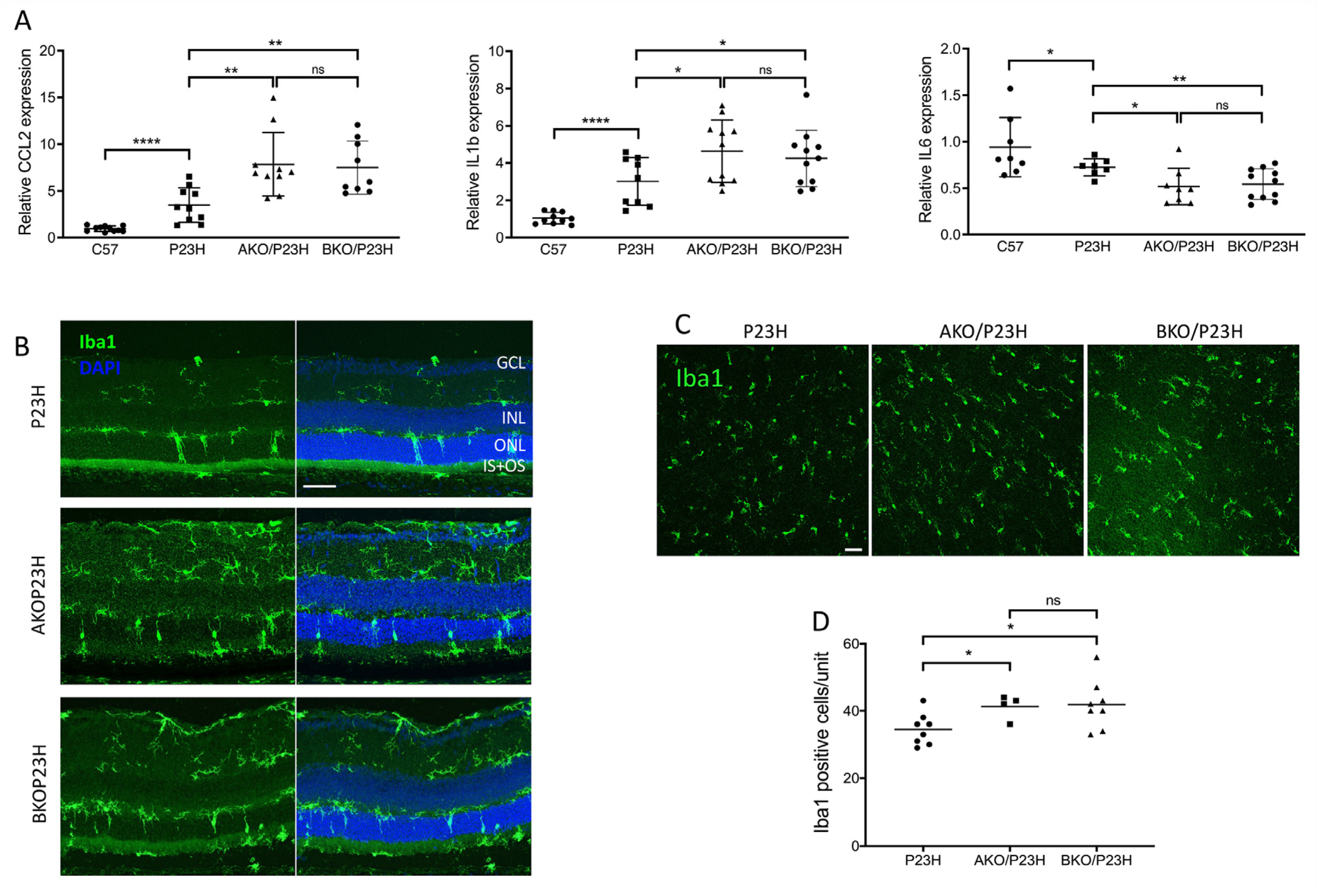
2.5. Cytokine and Microglia Activation in the P23H, AKO/P23H, and BKO/P23H Retina
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Spectral Domain Optical Coherence Tomography
4.3. Electroretinography
4.4. Histology and Immunohistochemistry
4.5. Western Blot Analysis
4.6. Soluble and Insoluble Rhodopsin Fractionation Assay
4.7. TUNEL Staining
4.8. Caspase 8 Activity Assay
4.9. Real-Time Polymerase Chain Reaction (RT-PCR)
4.10. Immunohistochemistry and Cell Count on Retinal Whole Mount
4.11. Statistical Analyses
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- RetNet–Retinal Information Network Home Page. Available online: https://sph.uth.edu/retnet/ (accessed on 30 September 2018).
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef]
- Sahel, J.A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef] [PubMed]
- Arbabi, A.; Liu, A.; Ameri, H. Gene Therapy for Inherited Retinal Degeneration. J. Ocul. Pharmacol. Ther. 2019, 35, 79–97. [Google Scholar] [CrossRef]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its role in photoreceptor physiology, human disease, and future therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef]
- He, Y.; Zhang, Y.; Su, G. Recent advances in treatment of retinitis pigmentosa. Curr. Stem Cell Res. Ther. 2015, 10, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 2007, 125, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R. Differential effects of antipsychotic drugs on contrast response functions of retinal ganglion cells in wild-type Sprague-Dawley rats and P23H retinitis pigmentosa rats. PLoS ONE 2019, 14, e0218200. [Google Scholar] [CrossRef] [PubMed]
- Perdices, L.; Fuentes-Broto, L.; Segura, F.; Ben Gdara, N.; Sánchez-Cano, A.I.; Insa, G.; Orduna, E.; Pinilla, I. Hepatic oxidative stress in pigmented P23H rhodopsin transgenic rats with progressive retinal degeneration. Free Radic. Biol. Med. 2018, 124, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.J.; Daniello, K.M.; Duncan, J.L.; Yang, H.; Yasumura, D.; Matthes, M.T.; LaVail, M.M. Influence of eye pigmentation on retinal degeneration in P23H and S334ter mutant rhodopsin transgenic rats. Exp. Eye Res. 2019, 187, 107755. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef]
- Ross, J.W.; de Fernandez Castro, J.P.; Zhao, J.; Samuel, M.; Walters, E.; Rios, C.; Bray-Ward, P.; Jones, B.W.; Marc, R.E.; Wang, W.; et al. Generation of an inbred miniature pig model of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2012, 53, 501–507. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Kanuga, N.; Adamson, P.; Cheetham, M.E. The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2017, 26, 4896–4905. [Google Scholar] [CrossRef] [PubMed]
- Comitato, A.; Di Salvo, M.T.; Turchiano, G.; Montanari, M.; Sakami, S.; Palczewski, K.; Marigo, V. Dominant and recessive mutations in rhodopsin activate different cell death pathways. Hum. Mol. Genet. 2016, 25, 2801–2812. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wen, R.H.; Stanar, P.; Tam, B.; Moritz, O.L. Autophagy in Xenopus laevis rod photoreceptors is independently regulated by phototransduction and misfolded RHOP23H. Autophagy 2019, 15, 1970–1989. [Google Scholar] [CrossRef] [PubMed]
- Comitato, A.; Sanges, D.; Rossi, A.; Humphries, M.M.; Marigo, V. Activation of Bax in three models of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Comitato, A.; Schiroli, D.; Montanari, M.; Marigo, V. Calpain Activation Is the Major Cause of Cell Death in Photoreceptors Expressing a Rhodopsin Misfolding Mutation. Mol. Neurobiol. 2020, 57, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Arango-Gonzalez, B.; Trifunović, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef]
- Sizova, O.S.; Shinde, V.M.; Lenox, A.R.; Gorbatyuk, M.S. Modulation of cellular signaling pathways in P23H rhodopsin photoreceptors. Cell Signal. 2014, 26, 665–672. [Google Scholar] [CrossRef]
- Yao, J.; Qiu, Y.; Frontera, E.; Jia, L.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Thompson, D.A.; Zacks, D.N. Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 2018, 14, 1226–1238. [Google Scholar] [CrossRef]
- Qiu, Y.; Yao, J.; Jia, L.; Thompson, D.A.; Zacks, D.N. Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: A novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 2019, 10, 547. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J. Alpha-crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. USA 1992, 89, 10449–10453. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J. Proctor Lecture. The function of alpha-crystallin. Investig. Ophthalmol. Vis. Sci. 1993, 34, 10–22. [Google Scholar]
- Narberhaus, F. Alpha-crystallin-type heat shock proteins: Socializing minichaperones in the context of a multichaperone network. Microbiol. Mol. Biol. Rev. 2002, 66, 64–93. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.P.; Garland, D.; Duglas-Tabor, Y.; Robison, W.G., Jr.; Groome, A.; Wawrousek, E.F. Targeted disruption of the mouse alpha A-crystallin gene induces cataract and cytoplasmic inclusion bodies containing the small heat shock protein alpha B-crystallin. Proc. Natl. Acad. Sci. USA 1997, 94, 884–889. [Google Scholar] [CrossRef]
- Ruebsam, A.; Dulle, J.E.; Myers, A.M.; Sakrikar, D.; Green, K.M.; Khan, N.W.; Schey, K.; Fort, P.E. A specific phosphorylation regulates the protective role of αA-crystallin in diabetes. JCI Insight 2018, 3, e97919. [Google Scholar] [CrossRef]
- Dong, Y.; Dong, Z.; Kase, S.; Ando, R.; Fukuhara, J.; Kinoshita, S.; Inafuku, S.; Tagawa, Y.; Ishizuka, E.T.; Saito, W.; et al. Phosphorylation of alphaB-crystallin in epiretinal membrane of human proliferative diabetic retinopathy. Int. J. Ophthalmol. 2016, 9, 1100–1105. [Google Scholar] [CrossRef]
- Andley, U.P. Crystallins in the eye: Function and pathology. Prog. Retin. Eye Res. 2007, 26, 78–98. [Google Scholar] [CrossRef]
- Wu, N.; Yu, J.; Chen, S.; Xu, J.; Ying, X.; Ye, M.; Li, Y.; Wang, Y. α-Crystallin protects RGC survival and inhibits microglial activation after optic nerve crush. Life Sci. 2014, 94, 17–23. [Google Scholar] [CrossRef]
- Lu, S.Z.; Guo, Y.S.; Liang, P.Z.; Zhang, S.Z.; Yin, S.; Yin, Y.Q.; Wang, X.M.; Ding, F.; Gu, X.S.; Zhou, J.W. Suppression of astrocytic autophagy by αB-crystallin contributes to α-synuclein inclusion formation. Transl. Neurodegener. 2019, 8, 3. [Google Scholar] [CrossRef]
- Pasupuleti, N.; Matsuyama, S.; Voss, O.; Doseff, A.I.; Song, K.; Danielpour, D.; Nagaraj, R.H. The anti-apoptotic function of human αA-crystallin is directly related to its chaperone activity. Cell Death Dis. 2010, 1, e31. [Google Scholar] [CrossRef]
- Tao, Y.; Ding, L.; Yao, A.; Yang, Z.; Yang, Q.; Qin, L.; Yu, L.; Gao, Y.; Huang, Y.F.; Li, Z.; et al. Intravitreous Delivery of Ab-Crystallin Ameliorates N-Methyl-N-Nitrosourea Induced Photoreceptor Degeneration in Mice: An in vivo and ex vivo Study. Cell. Physiol. Biochem. 2018, 48, 2147–2160. [Google Scholar] [CrossRef]
- Kase, S.; Meghpara, B.B.; Ishida, S.; Rao, N.A. Expression of α-crystallin in the retina of human sympathetic ophthalmia. Mol. Med. Rep. 2012, 5, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jiang, Z.; Lou, B.; Duan, F.; Qiu, S.; Cheng, Z.; Ma, X.; Yang, Y.; Lin, X. αB-Crystallin Alleviates Endotoxin-Induced Retinal Inflammation and Inhibits Microglial Activation and Autophagy. Front. Immunol. 2021, 12, 641999. [Google Scholar] [CrossRef] [PubMed]
- Sakami, S.; Maeda, T.; Bereta, G.; Okano, K.; Golczak, M.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G.; Palczewski, K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 2011, 286, 10551–10567. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Jürgensmeier, J.M.; Shin, H.; Deveraux, Q.; Wolf, B.B.; Yang, X.; Zhou, Q.; Ellerby, H.M.; Ellerby, L.M.; Bredesen, D.; et al. Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 1998, 273, 27084–27090. [Google Scholar] [CrossRef]
- Degrugillier, F.; Aissat, A.; Prulière-Escabasse, V.; Bizard, L.; Simonneau, B.; Decrouy, X.; Jiang, C.; Rotin, D.; Fanen, P.; Simon, S. Phosphorylation of the Chaperone-Like HspB5 Rescues Trafficking and Function of F508del-CFTR. Int. J. Mol. Sci. 2020, 21, 4844. [Google Scholar] [CrossRef]
- Cox, D.; Ecroyd, H. The small heat shock proteins αB-crystallin (HSPB5) and Hsp27 (HSPB1) inhibit the intracellular aggregation of α-synuclein. Cell Stress Chaperones 2017, 22, 589–600. [Google Scholar] [CrossRef]
- Parcellier, A.; Schmitt, E.; Brunet, M.; Hammann, A.; Solary, E.; Garrido, C. Small heat shock proteins HSP27 and alphaB-crystallin: Cytoprotective and oncogenic functions. Antioxid. Redox Signal. 2005, 7, 404–413. [Google Scholar] [CrossRef]
- Illing, M.E.; Rajan, R.S.; Bence, N.F.; Kopito, R.R. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 2002, 277, 34150–34160. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. Autophagy in polyglutamine disease: Imposing order on disorder or contributing to the chaos? Mol. Cell Neurosci. 2015, 66, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Fei, F.; Jones, A.; Busto, P.; Marshak-Rothstein, A.; Ksander, B.R.; Gregory-Ksander, M. Overexpression of soluble Fas ligand following Adeno-associated virus gene therapy prevents retinal ganglion cell death in chronic and acute murine models of glauoma. J. Immunol. 2016, 197, 4626–4638. [Google Scholar] [CrossRef]
- Kiang, L.; Ross, B.X.; Yao, J.; Shanmugam, S.; Andrews, C.A.; Hansen, S.; Besirli, C.G.; Zacks, D.N.; Abcouwer, S.F. Vitreous Cytokine Expression and a Murine Model Suggest a Key Role of Microglia in the Inflammatory Response to Retinal Detachment. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3767–3778. [Google Scholar] [CrossRef] [PubMed]
- Noailles, A.; Maneu, V.; Campello, L.; Gómez-Vicente, V.; Lax, P.; Cuenca, N. Persistent inflammatory state after photoreceptor loss in an animal model of retinal degeneration. Sci. Rep. 2016, 6, 33356. [Google Scholar] [CrossRef]
- Chong, D.Y.; Boehlke, C.S.; Zhang, L.; Han, Y.; Zacks, D.N. Interleukin-6 as a photoreceptor neuroprotectant in an experimental model of retinal detachment. Investig. Opthalmol. Vis. Sci. 2008, 49, 3193–3200. [Google Scholar] [CrossRef]
- Nath, M.; Shan, Y.; Myers, A.M.; Fort, P.E. HspB4/αA-Crystallin Modulates Neuroinflammation in the Retina via the Stress-Specific Inflammatory Pathways. J. Clin. Med. 2021, 10, 2384. [Google Scholar] [CrossRef]
- Thanos, S.; Böhm, M.R.; Meyer zu Hörste, M.; Prokosch-Willing, V.; Hennig, M.; Bauer, D.; Heiligenhaus, A. Role of crystallins in ocular neuroprotection and axonal regeneration. Prog. Retin. Eye Res. 2014, 42, 145–161. [Google Scholar] [CrossRef]
- Phadte, A.S.; Sluzala, Z.B.; Fort, P.E. Therapeutic Potential of α-Crystallins in Retinal Neurodegenerative Diseases. Antioxidants 2021, 10, 1001. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.A.; Saraswathy, S.; Wu, G.S.; Katselis, G.S.; Wawrousek, E.F.; Bhat, S. Elevated retina-specific expression of the small heat shock protein, alphaA-crystallin, is associated with photoreceptor protection in experimental uveitis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Whiston, E.A.; Sugi, N.; Kamradt, M.C.; Sack, C.; Heimer, S.R.; Engelbert, M.; Wawrousek, E.F.; Gilmore, M.S.; Ksander, B.R.; Gregory, M.S. Alphab-crystallin protects retinal tissue during Staphylococcus aureus-induced endophthalmitis. Infect. Immun. 2008, 76, 1781–1790. [Google Scholar] [CrossRef]
- Arac, A.; Brownell, S.E.; Rothbard, J.B.; Chen, C.; Ko, R.M.; Pereira, M.P.; Albers, G.W.; Steinman, L.; Steinberg, G.K. Systemic augmentation of alphaB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 13287–13292. [Google Scholar] [CrossRef]
- Anders, F.; Liu, A.; Mann, C.; Teister, J.; Lauzi, J.; Thanos, S.; Grus, F.H.; Pfeiffer, N.; Prokosch, V. The Small Heat Shock Protein α-Crystallin B Shows Neuroprotective Properties in a Glaucoma Animal Model. Int. J. Mol. Sci. 2017, 18, 2418. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Notomi, S.; Hisatomi, T.; Nakazawa, T.; Ishibashi, T.; Miller, J.W.; Vavvas, D.G. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog. Retin. Eye Res. 2013, 37, 114–140. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Jun, S.; Datta, S.; Wang, L.; Pegany, R.; Cano, M.; Handa, J.T. The impact of lipids, lipid oxidation, and inflammation on AMD, and the potential role of miRNAs on lipid metabolism in the RPE. Exp. Eye Res. 2019, 181, 346–355. [Google Scholar] [CrossRef]
- Arroba, A.I.; Campos-Caro, A.; Aguilar-Diosdado, M.; Valverde, Á.M. IGF-1, Inflammation and Retinal Degeneration: A Close Network. Front. Aging Neurosci. 2018, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Thanos, S. Sick photoreceptors attract activated microglia from the ganglion cell layer: A model to study the inflammatory cascades in rats with inherited retinal dystrophy. Brain Res. 1992, 588, 21–28. [Google Scholar] [CrossRef]
- Noailles, A.; Maneu, V.; Campello, L.; Lax, P.; Cuenca, N. Systemic inflammation induced by lipopolysaccharide aggravates inherited retinal dystrophy. Cell Death Dis. 2018, 9, 350. [Google Scholar] [CrossRef]
- Massengill, M.T.; Ash, N.F.; Young, B.M.; Ildefonso, C.J.; Lewin, A.S. Sectoral activation of glia in an inducible mouse model of autosomal dominant retinitis pigmentosa. Sci. Rep. 2020, 10, 16967. [Google Scholar] [CrossRef]
- Zhu, C.L.; Ji, Y.; Lee, E.J.; Grzywacz, N.M. Spatiotemporal pattern of rod degeneration in the S334ter-line-3 rat model of retinitis pigmentosa. Cell Tissue Res. 2013, 351, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, T.J.; Hubbard, M.G.; Levi, H.J.; White, W.; Wang, X.; Simpson, R.; Jablonski, M.M.; Gross, A.K. Proinflammatory Pathways Are Activated in the Human Q344X Rhodopsin Knock-In Mouse Model of Retinitis Pigmentosa. Biomolecules 2021, 11, 1163. [Google Scholar] [CrossRef]
- Noailles, A.; Fernández-Sánchez, L.; Lax, P.; Cuenca, N. Microglia activation in a model of retinal degeneration and TUDCA neuroprotective effects. J. Neuroinflamm. 2014, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jia, L.; Shelby, S.J.; Ganios, A.M.; Feathers, K.; Thompson, D.A.; Zacks, D.N. Circadian and noncircadian modulation of autophagy in photoreceptors and retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.X.; Choi, J.; Yao, J.; Hager, H.M.; Abcouwer, S.F.; Zacks, D.N. Loss of High-Mobility Group Box 1 (HMGB1) Protein in Rods Accelerates Rod Photoreceptor Degeneration After Retinal Detachment. Investig. Ophthalmol. Vis. Sci. 2020, 61, 50. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Yao, J.; Jia, L.; Fort, P.E.; Zacks, D.N. Loss of αA or αB-Crystallin Accelerates Photoreceptor Cell Death in a Mouse Model of P23H Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2022, 23, 70. https://doi.org/10.3390/ijms23010070
Wang T, Yao J, Jia L, Fort PE, Zacks DN. Loss of αA or αB-Crystallin Accelerates Photoreceptor Cell Death in a Mouse Model of P23H Autosomal Dominant Retinitis Pigmentosa. International Journal of Molecular Sciences. 2022; 23(1):70. https://doi.org/10.3390/ijms23010070
Chicago/Turabian StyleWang, Tiantian, Jingyu Yao, Lin Jia, Patrice E. Fort, and David N. Zacks. 2022. "Loss of αA or αB-Crystallin Accelerates Photoreceptor Cell Death in a Mouse Model of P23H Autosomal Dominant Retinitis Pigmentosa" International Journal of Molecular Sciences 23, no. 1: 70. https://doi.org/10.3390/ijms23010070
APA StyleWang, T., Yao, J., Jia, L., Fort, P. E., & Zacks, D. N. (2022). Loss of αA or αB-Crystallin Accelerates Photoreceptor Cell Death in a Mouse Model of P23H Autosomal Dominant Retinitis Pigmentosa. International Journal of Molecular Sciences, 23(1), 70. https://doi.org/10.3390/ijms23010070