
Platelet Mitochondrial Bioenergetics Reprogramming in Patients with Urothelial Carcinoma

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Blood Counts and Selected Metabolic Parameters of Subjects in Both Groups
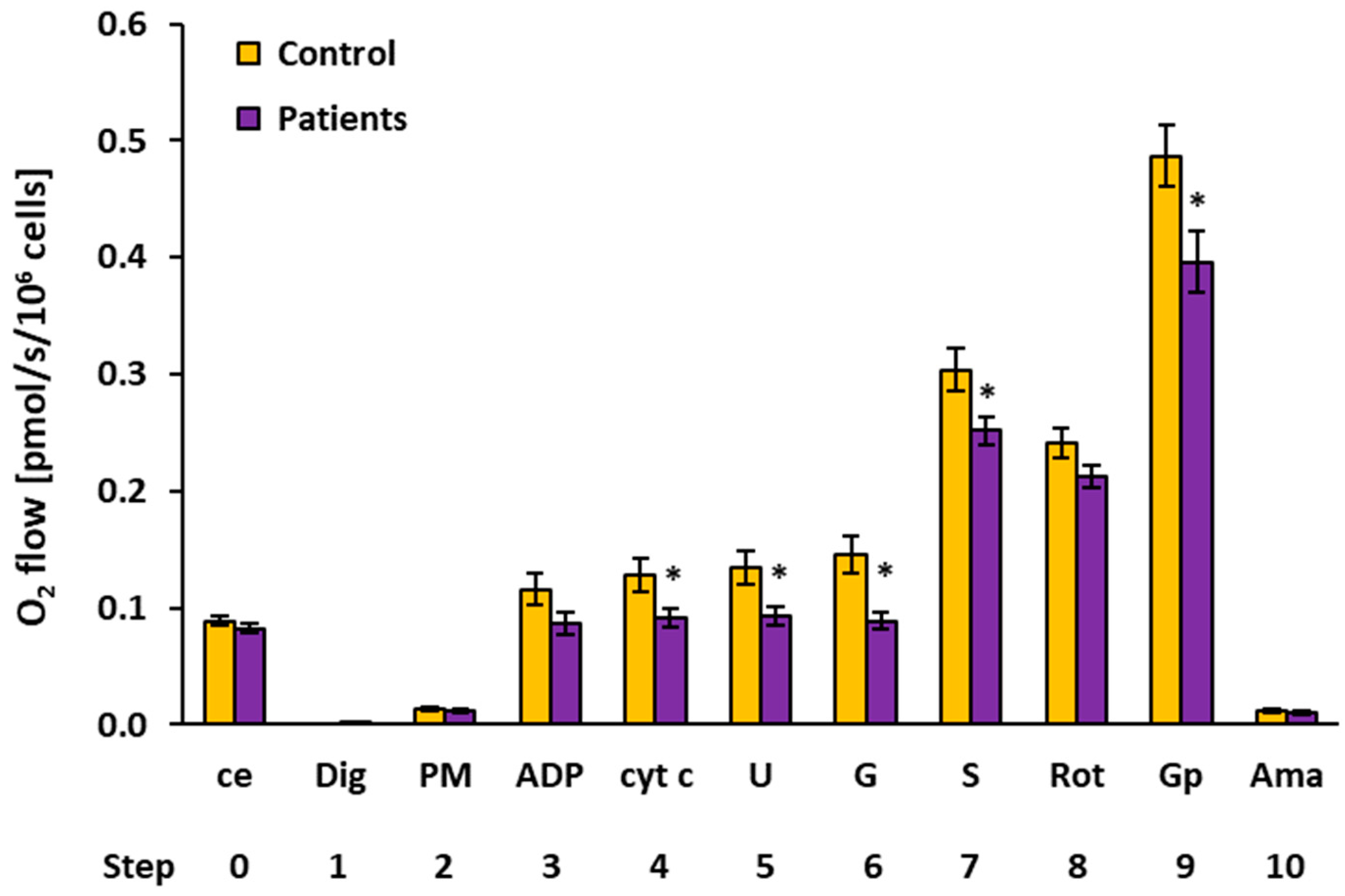
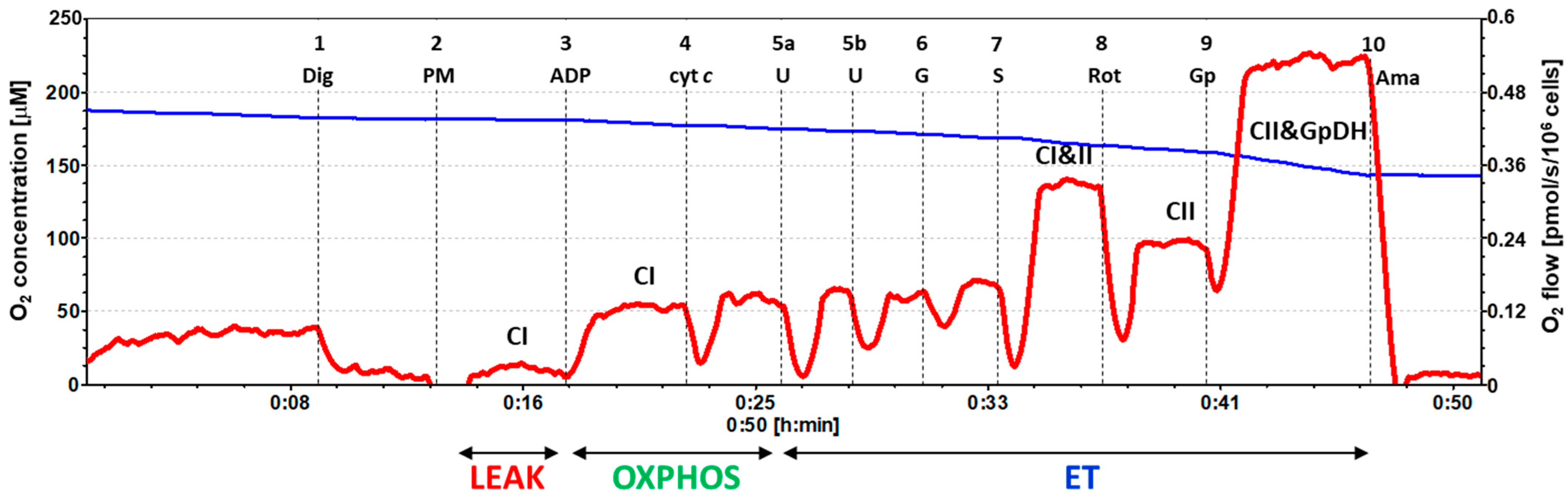
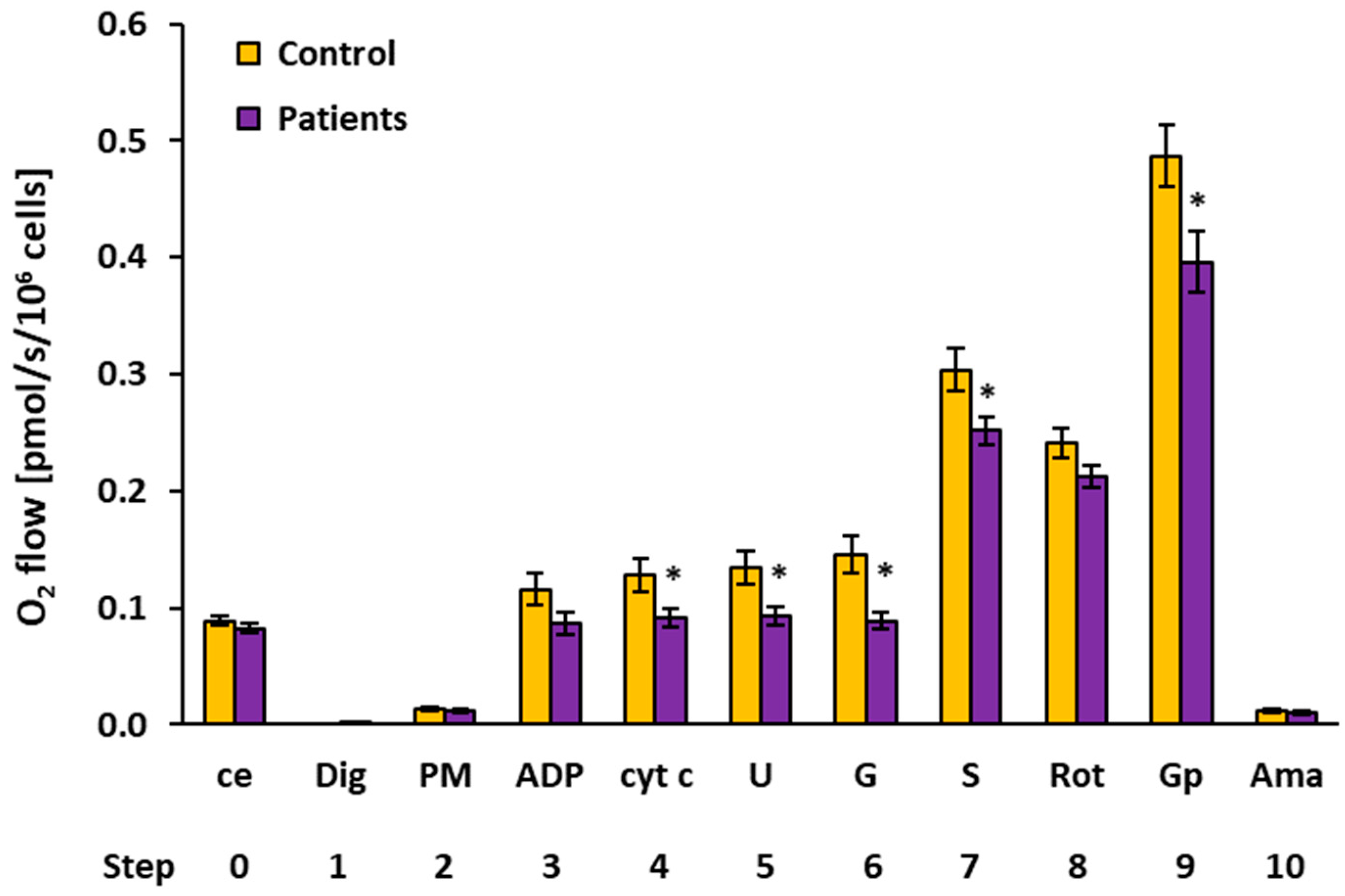
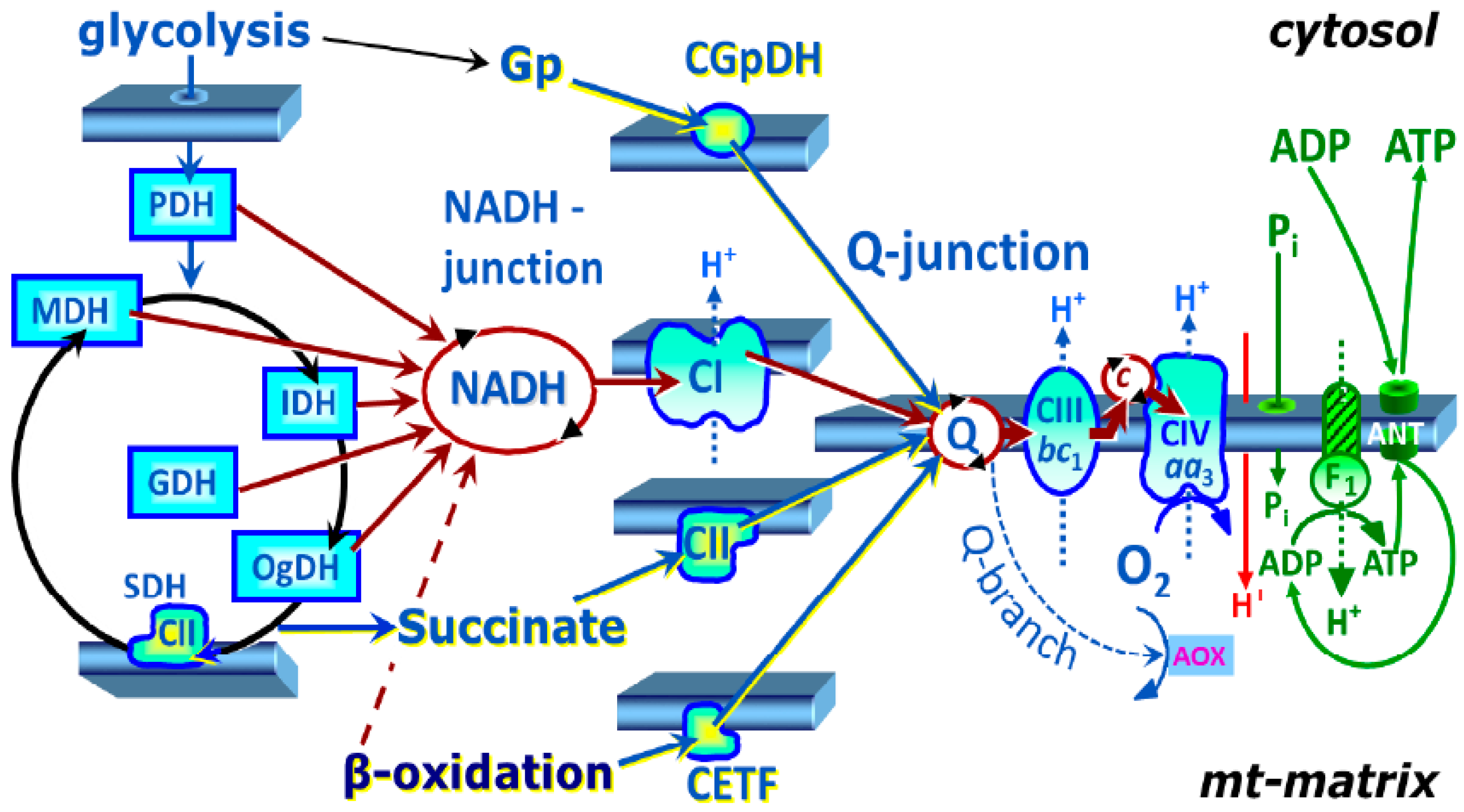
2.2. Platelet Mitochondrial Respiration by SUIT Protocol 1
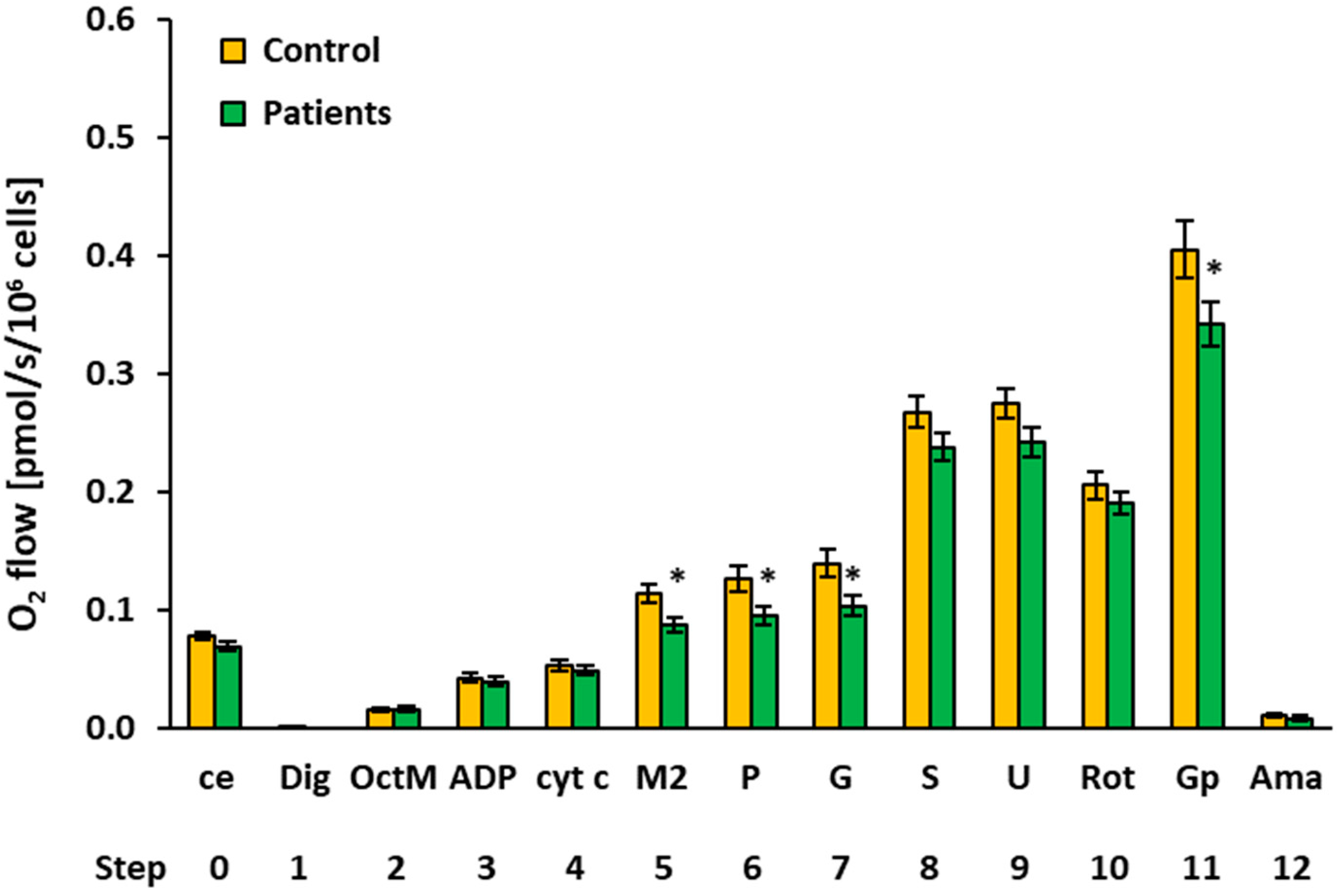
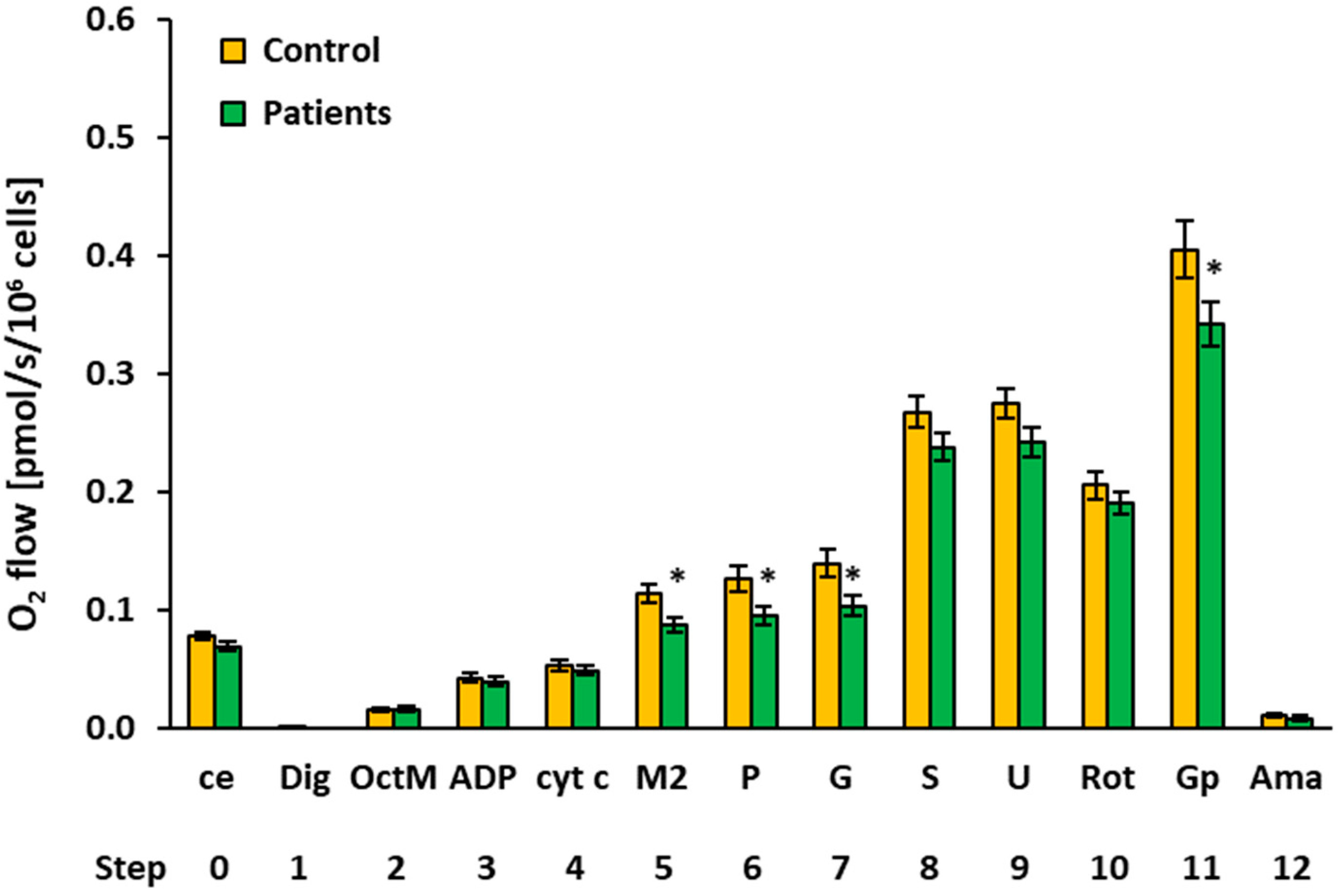
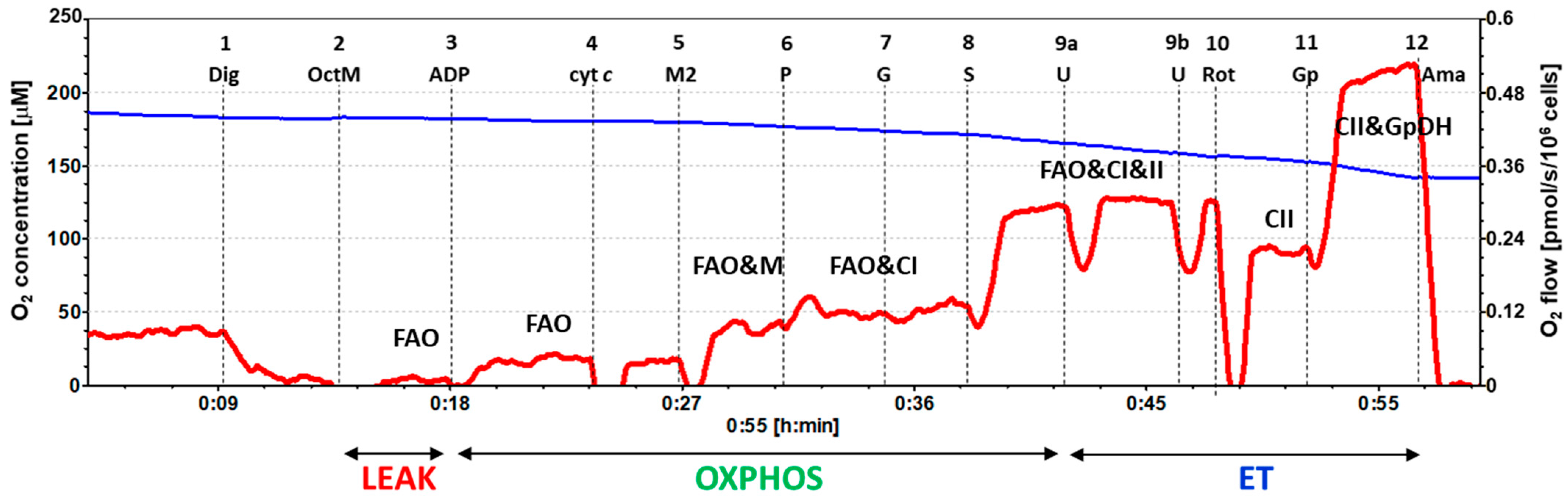
2.3. Platelet Mitochondrial Respiration and Fatty Acid Oxidation by SUIT Protocol 2
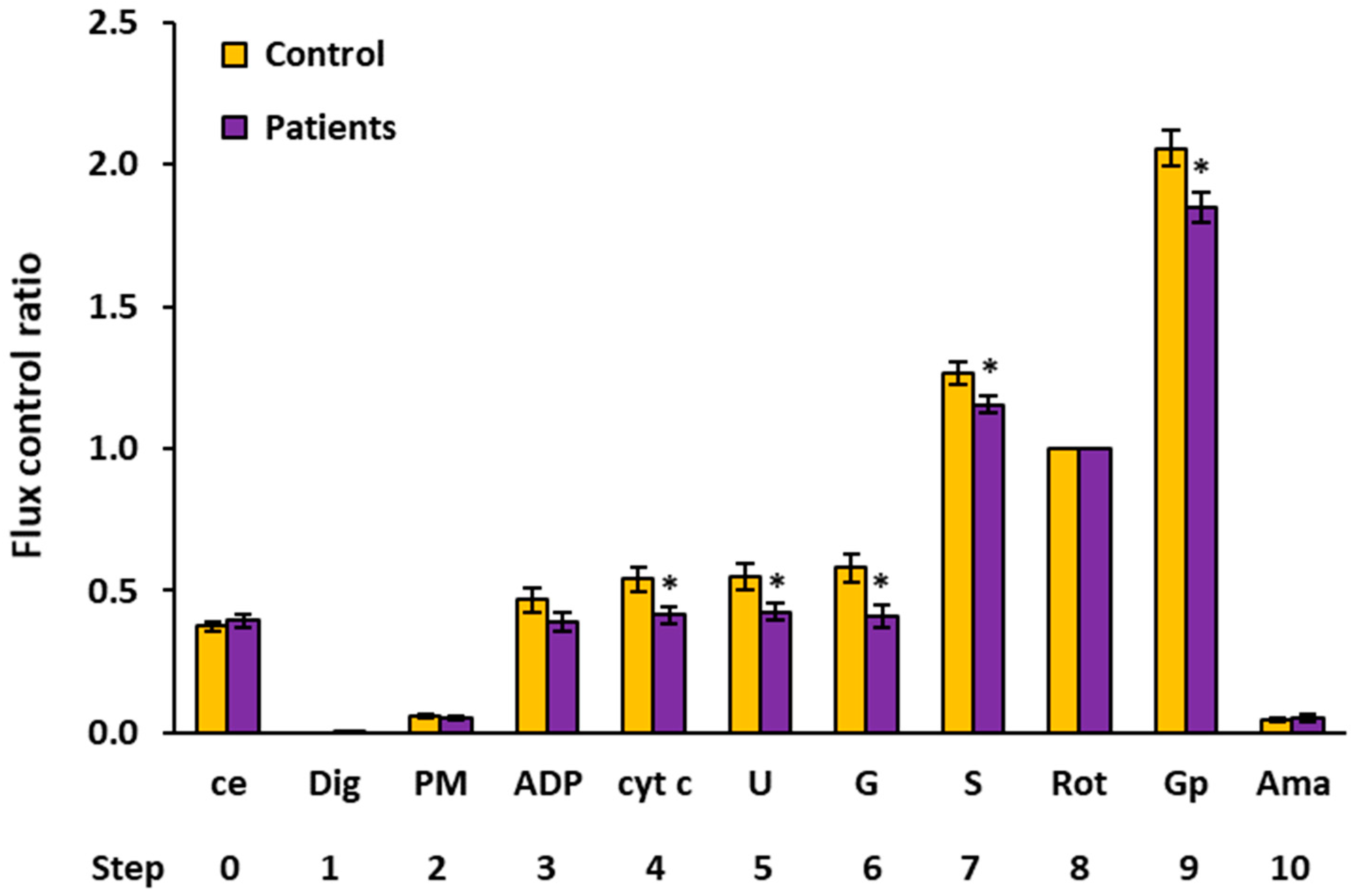
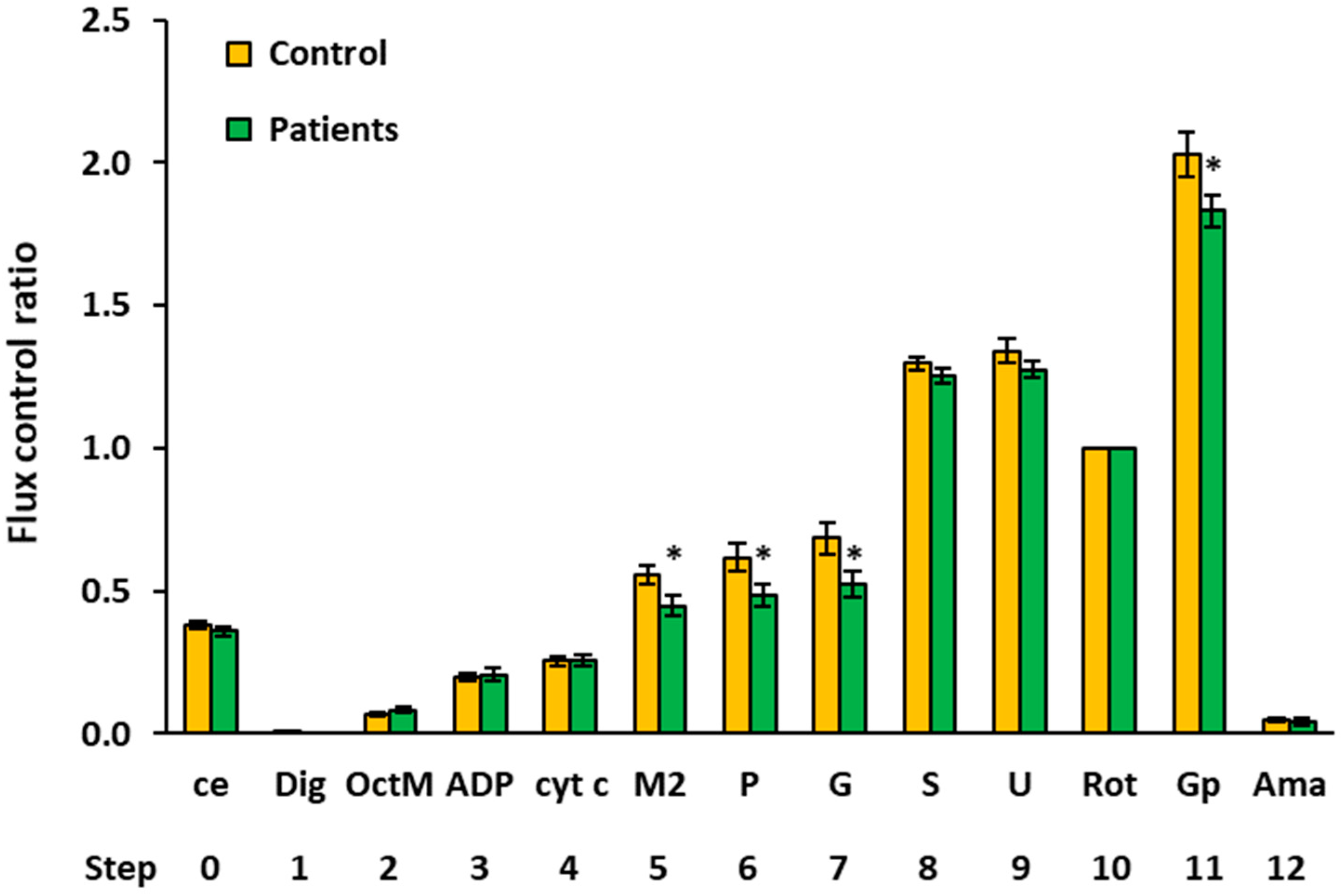
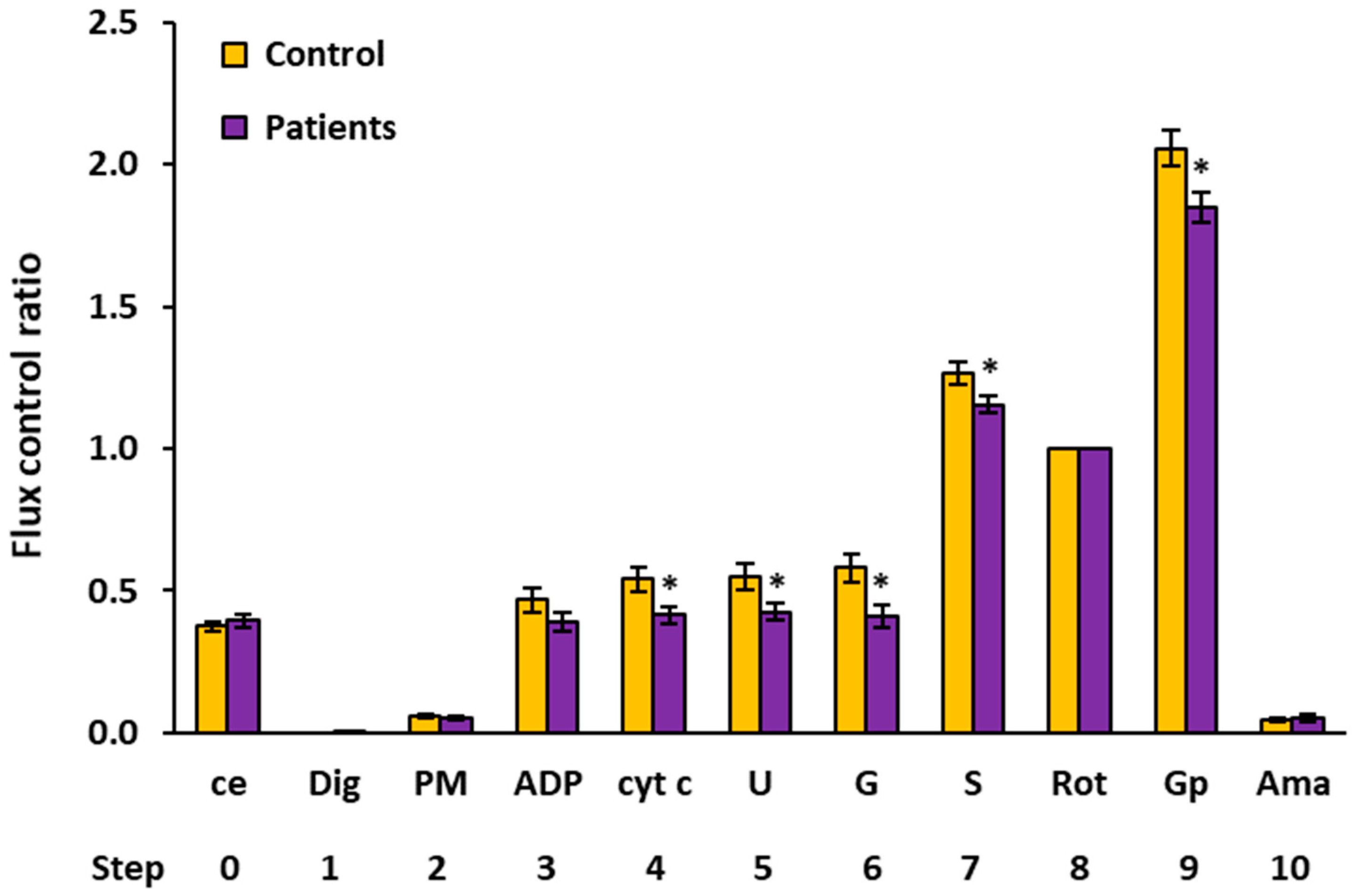
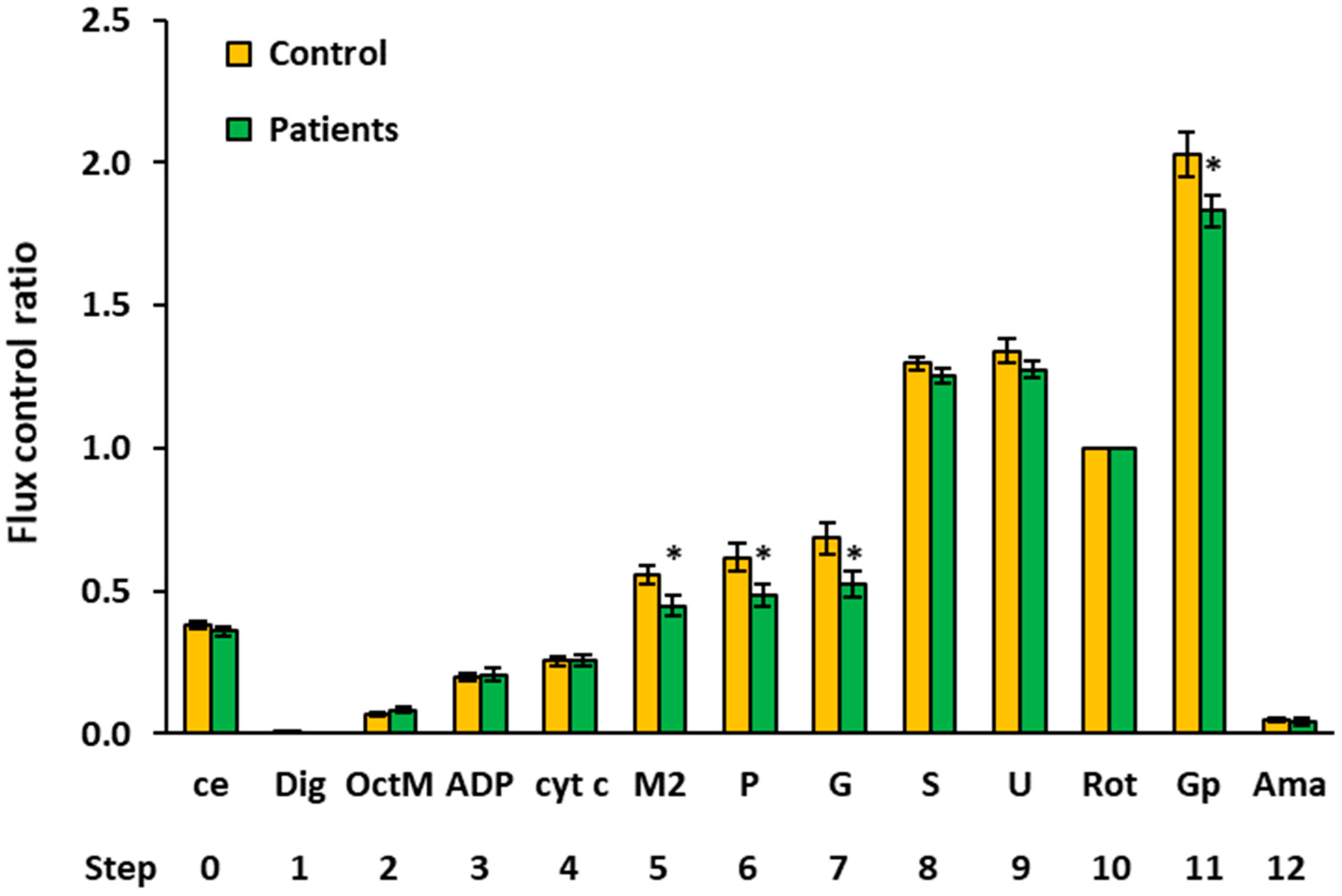
2.4. Flux Control Ratios of Platelet Mitochondrial Respiratory Capacities by SUIT Protocols 1 and 2
2.5. Citrate Synthase Activity in Patients with Urothelial Carcinoma
2.6. Endogenous Antioxidants and TBARS in Patients with Urothelial Carcinoma
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Baseline Characteristics of the Groups
4.3. Blood Count and Selected Metabolic Parameters of the Urothelial Carcinoma Patients and Control Subjects
4.4. Antioxidants and Oxidative Stress Determination
4.5. Platelet Preparation
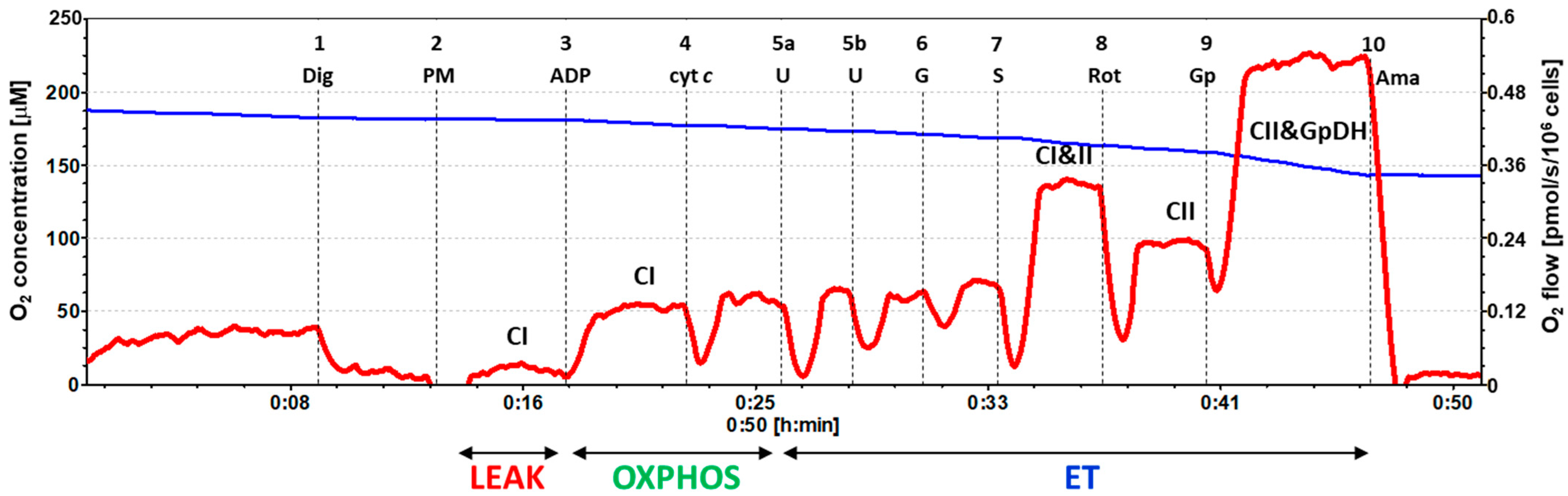
4.6. High-Resolution Respirometry Method
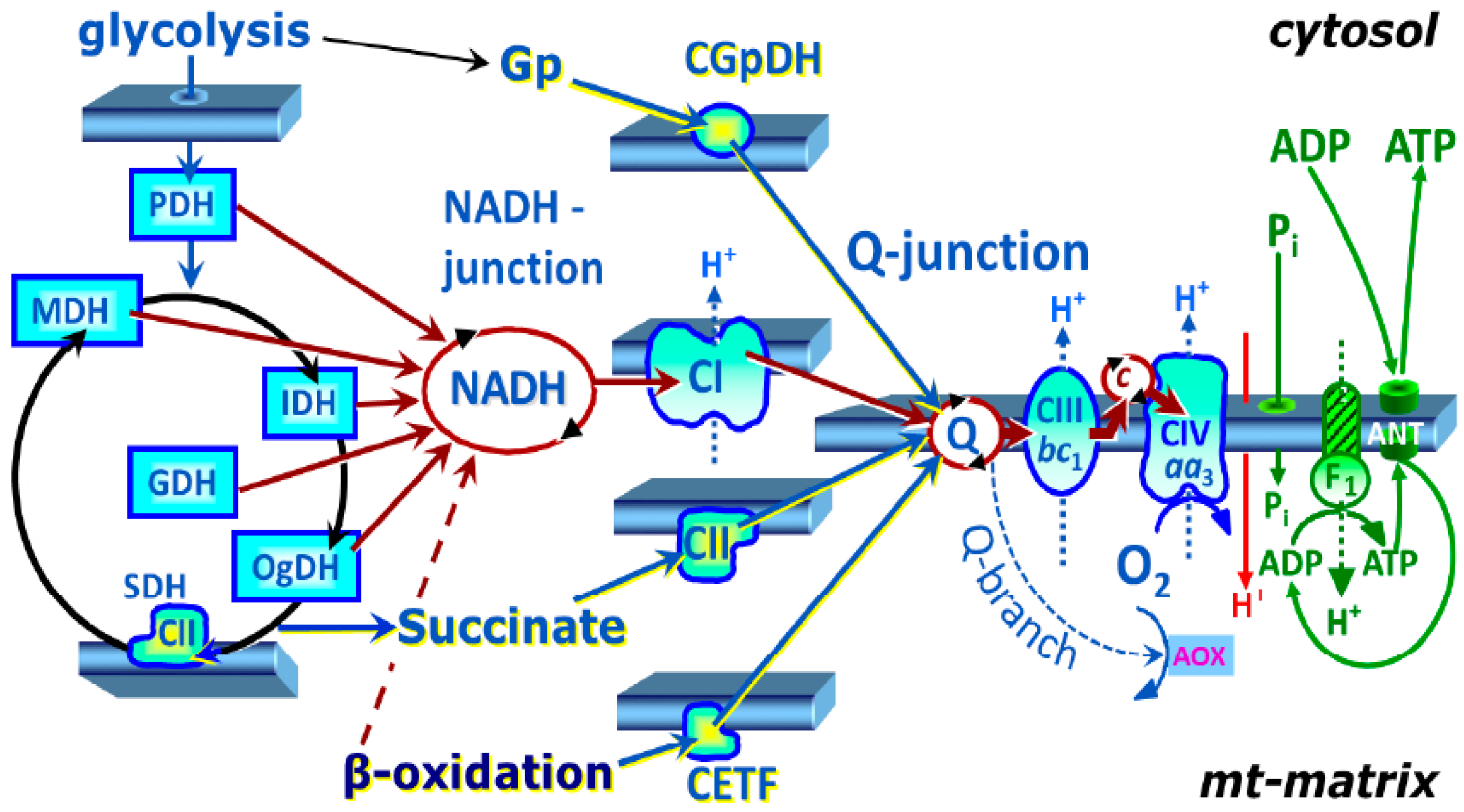
4.6.1. SUIT Protocol 1 for Evaluation of Platelet Mitochondrial Respiration
4.6.2. SUIT Protocol 2 for Evaluation of Platelet Mitochondrial Respiration and Fatty Acid Oxidation
4.6.3. Chemicals Used in SUIT Protocols for High-Resolution Respirometry
4.7. Citrate Synthase Activity Determination
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ama | Antimycin A |
| BMI | Body mass index |
| ce | Intact cells |
| CI | Complex I |
| CII | Complex II |
| CIII | Complex III |
| CoQ10 | Coenzyme Q10 |
| CoQ10-TOTAL | Ubiquinol plus ubiquinone |
| cyt c | Cytochrome c |
| Dig | Digitonin |
| ET | Electron transfer |
| FAO | Fatty acid oxidation |
| FCCP | Carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone |
| G | Glutamate |
| Gp | Glycerophosphate |
| M2 | Malate 2 mM |
| N | Number of patients |
| NADH | Nicotinamide adenine dinucleotide reduced |
| OctM | Octanoylcarnitine plus malate 0.1 mM |
| OXPHOS | Oxidative phosphorylation |
| P | Pyruvate |
| PDGF | Platelet-derived growth factor |
| PM | Pyruvate plus malate |
| Rot | Rotenone |
| S | Succinate |
| SUIT | Substrate–uncoupler–inhibitor titration |
| TBARS | Thiobarbituric acid reactive substances |
| TGF-β | Transforming growth factor β |
| U | Uncoupler |
| VEGF | Vascular endothelial growth factor |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, A.M.; Hegarty, P.K.; Gee, J.R.; Clark, P.E.; Svatek, R.S.; Hegarty, N.; Shariat, S.F.; Xylinas, E.; Schmitz-Dräger, B.J.; Lotan, Y.; et al. International consultation on urologic disease—European association of urology consultation on bladder cancer 2012. ICUD-EAU international consultation on bladder cancer 2012: Screening, diagnosis, and molecular markers. Eur Urol. 2013, 63, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A.; Moch, H.; Cubilla, A.L.; Ulbright, T.M.; Reuter, V.E. The 2016 WHO classification of tumours of the urinary system and male genital organs—Part B: Prostate and bladder tumours. Eur. Urol. 2016, 70, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, A.J.; Black, P.C. Variant histology in bladder cancer: Diagnostic and clinical implications. Transl. Cancer Res. 2020, 18, 6565–6575. [Google Scholar] [CrossRef]
- Gasic, G.J.; Gasic, T.B.; Galanti, N.; Johnson, T.; Murphy, S. Platelet-tumor-cell interactions in mice. The role of platelets in the spread of malignant disease. Int. J. Cancer 1973, 11, 704–718. [Google Scholar] [CrossRef]
- Schlesinger, M. The role of platelets and the platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.B.; Lee, S.K.; Bach, D.H. The role of platelets in the tumor-microenvironment and the drug resistance of cancer cells. Cancers 2019, 11, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Singh, K.; Costello, L. (Eds.) Mitochondria and Cancer; Springer: New York, NY, USA, 2009; p. 289. [Google Scholar]
- Ehinger, J.K.; Morota, S.; Hansson, M.J.; Paul, G.; Elmer, E. Mitochondrial dysfunction in blood cells from amyotrophic lateral sclerosis patients. J. Neurol. 2015, 262, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Sumbalova, Z.; Gvozdjakova, A.; Kucharska, J.; Rausova, Z.; Vancova, O.; Kuzmiakova, Z. Platelet mitochondrial function, coenzyme Q10, and oxidative stress in patients with chronic kidney diseases. MiP2019/MitoEAGLE 2019, 14, 35–36. [Google Scholar]
- Gvozdjakova, A.; Sumbalova, Z.; Kucharska, J.; Komlosi, M.; Rausova, Z.; Vancova, O.; Szamosova, M.; Mojto, V. Platelet mitochondrial respiration, endogenous coenzyme Q10 and oxidative stress in patients with chronic kidney disease. Diagnostics 2020, 10, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gvozdjakova, A.; Sumbalova, Z.; Kucharska, J.; Chladekova, A.; Rausova, Z.; Vancova, O.; Komlosi, M.; Ulicna, O.; Mojto, V. Platelet mitochondrial bioenergetic analysis in patients with nephropathies and non-communicable diseases: A new method. Bratisl. Lek. Listy 2019, 120, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Gvozdjakova, A.; Sumbalova, Z.; Kucharska, J.; Szamosova, M.; Capová, L.; Rausova, Z.; Vancova, O.; Mojto, V.; Langsjoen, P.; Palacka, P. Platelet mitochondrial respiration and coenzyme Q10 could be used as new diagnostic strategy for mitochondrial dysfunction in rheumatoid disease. PLoS ONE 2021, 16, e0256135. [Google Scholar] [CrossRef] [PubMed]
- Gvozdjakova, A.; Sumbalova, Z.; Kucharska, J.; Chladekova, A.; Rausova, Z.; Vancova, O.; Kubalova, M.; Kuzmiakova, Z.; Nemec, M.; Ulicna, O.; et al. Platelets mitochondrial function depends on coenzyme Q10 concentration in human young, not in elderly subjects. J. Nutr. Ther. 2018, 7, 67–76. [Google Scholar] [CrossRef]
- Sumbalova, Z.; Kucharska, J.; Palacka, P.; Rausova, Z.; Langsjoen, P.H.; Langsjoen, A.M.; Gvozdjakova, A. Platelet mitochondrial function and endogenous coenzyme Q10 levels are reduced in patients after COVID-19. Bratisl. Lek. Listy 2022, 123, 9–15. [Google Scholar]
- SUIT-001 O2 ce-pce D004. Available online: https://wiki.oroboros.at/index.php/SUIT-001_O2_ce-pce_D004 (accessed on 27 December 2021).
- SUIT-002 ce-pce D007a. Available online: https://wiki.oroboros.at/index.php/SUIT-002_O2_ce-pce_D007a (accessed on 27 December 2021).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sabrkhany, S.; Kuijpers, M.J.E.; Egbrink, M.G.A.; Griffioen, A.W. Platelet as messengers of early-stage cancer. Cancer Metastasis Rev. 2021, 40, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Baaten, C.C.C.F.M.J.; Swieringa, F.; Misztal, T.; Mastenbroek, T.G.; Feijge, M.A.H.; Bock, P.E.; Donners, M.M.P.C.; Collins, P.W.; Li, R.; Meijden, P.R.J.; et al. Platelet heterogeneity in activation-induced glycoprotein shedding: Functional effects. Blood Adv. 2018, 2, 2320–2331. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Stone, R.L.; Kaelber, J.T.; Rochat, R.H.; Nick, A.M.; Vijayan, K.V.; Sfshar-Kharghan, V.; Schmid, M.F.; Dong, J.F.; Sood, A.K.; et al. Electron cryotomography reveals ultrastructure alterations in platelets from patients with ovarian cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 14266–14271. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, K.; Bhattacharyya, M. Overview of platelet physiology: Its hemostatic and non-hemostatic role in disease pathogenesis. Sci. World J. 2014, 2014, 781857. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Olsson, A.K.; Cedervall, J. The pro-inflammatory role of platelets in cancer. Platelets 2018, 29, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Gakis, G.; Fritsche, H.M.; Hassan, F.; Kluth, L.; Miller, F.; Soave, A.; Otto, W.; Schwentner, C.; Todenhöfer, T.; Dahlem, R.; et al. Prognostic relevance of postoperative platelet count in upper tract urothelial carcinoma after radical nephroureterectomy. Eur. J. Cancer 2014, 50, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Q.; Fan, Z.; Xie, R.; Wang, Z.; Lu, Y. Platelet mitochondrial dysfunction and the correlation with human disease. Biochem. Soc. Transact. 2017, 45, 1213. [Google Scholar] [CrossRef]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of platelet mitochondria: Life in a nucleus-free zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Grasso, D.I.; Zampieri, L.X.; Capella, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria and cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Simonnet, H.; Demont, J.; Pfeiffer, K.; Guenaneche, L.; Bouvier, R.; Brandt, U.; Schagger, H.; Godinot, C. Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis 2003, 24, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, F.A.; Mayr, J.A.; Neureiter, D.; Feichtenger, R.; Alinger, B.; Jones, N.D.; Eder, W.; Sperl, W.; Kofler, B. Lack complex I is associated with oncocytic thyroid tumours. Br. J. Cancer. 2009, 100, 1434–1437. [Google Scholar] [CrossRef]
- Chai, W.; Cooney, R.V.; Franke, A.A.; Caberto, C.P.; Wilkens, L.R.; Le Marchand, L.; Goodman, M.T.; Henderson, B.E.; Kolonel, L.N. Plasma coenzyme Q10 levels and prostate cancer risk: The multiethnic cohort study. Cancer Epidemiol. Prev. Biomark. 2011, 20, 708–710. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Luo, P.; Zeng, Z.; Wang, H.; Malafa, M.; Suh, N. Vitamin E and cancer prevention: Studies with different forms of tocopherols and tocotrienols. Mol. Carcinog. 2020, 59, 365–389. [Google Scholar] [CrossRef]
- Slopovsky, J.; Kucharska, J.; Obertova, J.; Mego, M.; Kalavska, K.; Cingelova, S.; Svetlovska, D.; Gvozdjakova, A.; Furka, S.; Palacka, P. Plasma thiobarbituric acid reactive substances predict survival in chemotherapy naïve patients with metastatic urothelial carcinoma. Transl. Oncol. 2021, 14, 100890. [Google Scholar] [CrossRef]
- Enriquez, J.A.; Lenaz, G. Coenzyme q and the respiratory chain: Coenzyme q pool and mitochondrial supercomplexes. Mol. Syndromol. 2014, 5, 119–140. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channels VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Meierhofer, D.; Mayr, J.A.; Foetschi, U.; Berger, A.; Fink, K.; Schmeller, N.; Hacker, G.W.; Hauser-Kronberger, C.; Kofler, B.; Sperl, W. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis 2004, 25, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS complex III in breast cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [Green Version]
- World Medical Association. WMA declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 27 December 2021).
- International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use. Guideline for Good Clinical Practice E6(R2). Available online: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf (accessed on 27 December 2021).
- Sjovall, F.; Ehinger, J.K.; Marelsson, S.E.; Morota, S.; Frostner, E.A.; Uchino, H.; Lundgren, J.; Arnbjörnsson, E.; Hansson, M.J.; Fellman, V.; et al. Mitochondrial respiration in human viable platelets—methodology and ingluence gender, age and storage. Mitochondrion 2013, 13, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.K.; Gohil, K.; Packer, L. Simultaneous determination of tocopherols, ubiquinols, and ubiquinones in blood, plasma, tissue homogenates, and subcellular fractions. Anal. Biochem. 1986, 157, 106–116. [Google Scholar] [CrossRef]
- Kucharska, J.; Gvozdjakova, A.; Mizera, S.; Braunova, Z.; Schreinerova, Z.; Schramekova, E.; Pechan, I.; Fabian, J. Participation of coenzyme Q10 in the rejection development of the transplanted heart. Physiol. Res. 1998, 47, 399–404. [Google Scholar]
- Mosca, F.; Fattorini, D.; Bompadre, S.; Littarru, G.P. Assay of coenzyme Q10 in plasma by a single dilution step. Anal. Biochem. 2002, 305, 49–54. [Google Scholar] [CrossRef]
- Janero, D.R.; Bughardt, B. Thiobarbituric acid-reactive malondialdehyd formation during suproxide-dependent, iron-catalyzed lipid peroxidation: Influence of peroxidation conditions. Lipids 1989, 24, 125–131. [Google Scholar] [CrossRef]
- Sumbalova, Z.; Droescher, S.; Hiller, E.; Chang, S.C.; Garcia-Souza, L.F.; Calabria, E.; Volani, C.; Krumschnabel, G.; Gnaiger, E. 2k-Protocols: Isolation of peripheral blood mononuclear cells and platelets from human blood for HRFR. Mitochondrial Physiol. Netw. 2018, 17, 1–15. [Google Scholar]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [PubMed]
- Gnaiger, E. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. Bioenerg. Commun. 2020, 2, 112. [Google Scholar]
- Eigentler, A.; Draxl, A.; Gnaiger, E. Laboratory protocol: Citrate synthase a mitochondrial marker enzyme. Mitochondrial Physiol. Netw. 2020, 17, 1–12. [Google Scholar]
- NCSS Statistical Software, NCSS, LLC: Kaysville, UT, USA, 2020. Available online: https://www.ncss.com/(accessed on 27 December 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Mean | Median | SD | SEM | p | ||
|---|---|---|---|---|---|---|---|
| Parameter (unit) | |||||||
| White blood counts (109/L) | Controls | 15 | 6.23 | 5.90 | 1.87 | 1.09 | <0.0930 |
| Patients | 15 | 9.67 | 7.99 | 5.69 | 1.09 | ||
| Red blood counts (109/L) | Controls | 15 | 4.66 | 4.79 | 0.49 | 0.14 | <0.0062 * |
| Patients | 15 | 4.04 | 3.83 | 0.60 | 0.14 | ||
| Platelet counts (109/L) | Controls | 15 | 247.47 | 243.00 | 64.69 | 43.93 | <0.1710 |
| Patients | 15 | 366.47 | 283.00 | 231.75 | 43.93 | ||
| Hemoglobin level (g/L) | Controls | 15 | 140.67 | 139.00 | 13.30 | 4.08 | 0.0014 * |
| Patients | 15 | 117.07 | 110.00 | 17.97 | 4.08 | ||
| Creatinine (µmol/L) | Controls | 15 | 74.12 | 75.10 | 12.92 | 7.58 | <0.0002 * |
| Patients | 15 | 121.59 | 117.90 | 34.45 | 7.58 | ||
| Urea (mmol/L) | Controls | 15 | 5.55 | 5.20 | 1.47 | 0.60 | 0.0003 * |
| Patients | 15 | 9.36 | 8.50 | 2.96 | 0.60 | ||
| Uric acid (µmol/L) | Controls | 15 | 301.85 | 316.00 | 52.71 | 16.92 | <0.0107 * |
| Patients | 15 | 378.94 | 371.00 | 72.53 | 16.92 | ||
| γ-glutamyl-transferase (GMT) (µkat/L) | Controls | 14 | 0.38 | 0.31 | 0.22 | 0.08 | <0.0107 * |
| Patients | 15 | 0.63 | 0.49 | 0.38 | 0.08 | ||
| Alanine aminotransferase (ALT) (µmol/L) | Controls | 15 | 0.45 | 0.33 | 0.30 | 0.07 | <0.5753 |
| Patients | 15 | 0.40 | 0.32 | 0.23 | 0.07 | ||
| Aspartate transaminase (AST) (µmol/L) | Controls | 15 | 0.41 | 0.36 | 0.14 | 0.04 | <0.3718 |
| Patients | 15 | 0.37 | 0.36 | 0.16 | 0.04 | ||
| Alanine aminotransferase (ALP) (µmol/L) | Controls | 15 | 1.26 | 1.21 | 0.39 | 0.26 | <0.2058 |
| Patients | 15 | 1.82 | 1.58 | 1.36 | 0.26 | ||
| Total bilirubin (µmol/L) | Controls | 15 | 13.67 | 11.60 | 6.58 | 1.47 | <0.0326 * |
| Patients | 15 | 9.08 | 8.50 | 4.67 | 1.47 | ||
| Triacylglycerols (mmol/L) | Controls | 15 | 2.05 | 1.21 | 1.97 | 0.39 | <0.2627 |
| Patients | 15 | 1.87 | 1.73 | 0.82 | 0.39 | ||
| Cholesterol (mmol/L) | Controls | 15 | 5.32 | 5.53 | 1.06 | 0.29 | <0.0326 |
| Patients | 15 | 4.83 | 4.49 | 1.21 | 0.29 | ||
| High-density lipoprotein (HDL) cholesterol (mmol/L) | Controls | 15 | 1.41 | 1.30 | 0.52 | 0.13 | <0.7398 |
| Patients | 15 | 1.30 | 1.34 | 0.44 | 0.13 | ||
| Low-density lipoprotein (LDL) cholesterol (mmol/L) | Controls | 13 | 3.09 | 2.91 | 0.95 | 0.25 | <0.2222 |
| Patients | 15 | 2.68 | 2.30 | 0.87 | 0.23 | ||
| Very low-density lipoprotein (VLDL) cholesterol (mmol/L) | Controls | 13 | 0.62 | 0.55 | 0.23 | 0.09 | <0.0552 |
| Patients | 15 | 0.85 | 0.79 | 0.37 | 0.08 | ||
| Total protein (g/L) | Controls | 15 | 71.89 | 71.70 | 2.86 | 1.37 | <0.0042 * |
| Patients | 15 | 65.34 | 64.50 | 6.91 | 1.37 | ||
| Albumin (g/L) | Controls | 15 | 46.97 | 46.00 | 2.74 | 0.98 | <0.0083 * |
| Patients | 15 | 42.53 | 42.00 | 4.62 | 0.98 |
| O2 Flow (pmol/s/106 Cells) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Step | Titration | N | Mean | Median | Sd | Sem | p-Value | % of Control | |
| 0 | ce | Controls | 15 | 0.0885 | 0.0855 | 0.0160 | 0.0041 | ||
| Patients | 15 | 0.0820 | 0.0821 | 0.0156 | 0.0040 | 0.270 | 92.6% | ||
| 1 | Dig | Controls | 15 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | ||
| Patients | 15 | 0.0003 | 0.0000 | 0.0007 | 0.0002 | ||||
| 2 | PM | Controls | 13 | 0.0142 | 0.0113 | 0.0060 | 0.0017 | ||
| Patients | 14 | 0.0120 | 0.0110 | 0.0044 | 0.0012 | 0.298 | 84.9% | ||
| 3 | ADP | Controls | 12 | 0.1160 | 0.1169 | 0.0477 | 0.0138 | ||
| Patients | 11 | 0.0865 | 0.0866 | 0.0322 | 0.0097 | 0.099 | 74.5% | ||
| 4 | cyt c | Controls | 12 | 0.1285 | 0.1442 | 0.0500 | 0.0144 | ||
| Patients | 11 | 0.0916 | 0.0913 | 0.0288 | 0.0087 | 0.044 * | 71.2% | ||
| 5 | U | Controls | 12 | 0.1347 | 0.1473 | 0.0503 | 0.0145 | ||
| Patients | 11 | 0.0933 | 0.0962 | 0.0250 | 0.0075 | 0.023 * | 69.3% | ||
| 6 | G | Controls | 10 | 0.1456 | 0.1414 | 0.0522 | 0.0165 | ||
| Patients | 11 | 0.0888 | 0.0874 | 0.0242 | 0.0073 | 0.004 * | 61.0% | ||
| 7 | S | Controls | 15 | 0.3040 | 0.2803 | 0.0728 | 0.0188 | ||
| Patients | 13 | 0.2517 | 0.2547 | 0.0422 | 0.0117 | 0.031 * | 82.8% | ||
| 8 | Rot | Controls | 15 | 0.2408 | 0.2301 | 0.0488 | 0.0126 | ||
| Patients | 15 | 0.2124 | 0.2236 | 0.0378 | 0.0097 | 0.085 | 88.2% | ||
| 9 | Gp 1 | Controls | 15 | 0.4865 | 0.4509 | 0.1009 | 0.0260 | ||
| Patients | 15 | 0.3961 | 0.3735 | 0.1006 | 0.0260 | 0.020 * | 81.4% | ||
| 10 | Ama | Controls | 15 | 0.0113 | 0.0129 | 0.0060 | 0.0016 | ||
| Patients | 15 | 0.0102 | 0.0101 | 0.0079 | 0.0020 | 0.679 | 90.5% | ||
| O2 Flow (pmol/s/106 Cells) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Step | Titration | N | Mean | Median | Sd | Sem | p-Value | % of Control | |
| 0 | ce | Controls | 15 | 0.0775 | 0.0797 | 0.0116 | 0.0030 | ||
| Patients | 13 | 0.0689 | 0.0702 | 0.0117 | 0.0032 | 0.062 | 88.9% | ||
| 1 | Dig | Controls | 15 | 0.0004 | 0.0000 | 0.0014 | 0.0004 | ||
| Patients | 14 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.343 | |||
| 2 | OctM | Controls | 15 | 0.0145 | 0.0131 | 0.0071 | 0.0018 | ||
| Patients | 12 | 0.0155 | 0.0153 | 0.0063 | 0.0018 | 0.718 | 106.5% | ||
| 3 | ADP | Controls | 15 | 0.0421 | 0.0426 | 0.0150 | 0.0039 | ||
| Patients | 13 | 0.0390 | 0.0380 | 0.0142 | 0.0039 | 0.584 | 92.7% | ||
| 4 | cyt c | Controls | 15 | 0.0528 | 0.0563 | 0.0184 | 0.0047 | ||
| Patients | 14 | 0.0481 | 0.0493 | 0.0148 | 0.0040 | 0.463 | 91.2% | ||
| 5 | M2 | Controls | 11 | 0.1129 | 0.1183 | 0.0261 | 0.0079 | ||
| Patients | 14 | 0.0865 | 0.0929 | 0.0230 | 0.0062 | 0.013* | 76.6% | ||
| 6 | P | Controls | 12 | 0.1257 | 0.1382 | 0.0360 | 0.0104 | ||
| Patients | 14 | 0.0947 | 0.0990 | 0.0278 | 0.0074 | 0.021 * | 75.3% | ||
| 7 | G | Controls | 12 | 0.1392 | 0.1440 | 0.0423 | 0.0122 | ||
| Patients | 14 | 0.1030 | 0.1049 | 0.0334 | 0.0089 | 0.023 * | 74.0% | ||
| 8 | S | Controls | 15 | 0.2671 | 0.2535 | 0.0517 | 0.0134 | ||
| Patients | 14 | 0.2374 | 0.2308 | 0.0446 | 0.0119 | 0.110 | 88.9% | ||
| 9 | U | Controls | 15 | 0.2744 | 0.2703 | 0.0489 | 0.0126 | ||
| Patients | 14 | 0.2418 | 0.2370 | 0.0474 | 0.0127 | 0.080 | 88.1% | ||
| 10 | Rot | Controls | 11 | 0.2055 | 0.2044 | 0.0389 | 0.0117 | ||
| Patients | 14 | 0.1899 | 0.1792 | 0.0352 | 0.0094 | 0.304 | 92.4% | ||
| 11 | Gp 1 | Controls | 11 | 0.4047 | 0.3827 | 0.0811 | 0.0245 | ||
| Patients | 13 | 0.3415 | 0.3155 | 0.0677 | 0.0188 | 0.049 * | 84.4% | ||
| 12 | Ama | Controls | 13 | 0.0107 | 0.0116 | 0.0065 | 0.0018 | ||
| Patients | 14 | 0.0079 | 0.0048 | 0.0076 | 0.0020 | 0.303 | 73.2% | ||
| Flux Control Ratio (Relative Units) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Step | Titration | N | Mean | Median | Sd | Sem | p-Value | % of Control | |
| 0 | ce | Controls | 15 | 0.3741 | 0.3668 | 0.0721 | 0.0186 | ||
| Patients | 15 | 0.3934 | 0.3767 | 0.0880 | 0.0227 | 0.517 | 105.1% | ||
| 1 | Dig | Controls | 15 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | ||
| Patients | 15 | 0.0014 | 0.0000 | 0.0037 | 0.0010 | 0.156 | |||
| 2 | PM | Controls | 13 | 0.0574 | 0.0517 | 0.0197 | 0.0055 | ||
| Patients | 11 | 0.0513 | 0.0500 | 0.0158 | 0.0048 | 0.423 | 89.5% | ||
| 3 | ADP | Controls | 12 | 0.4665 | 0.4773 | 0.1465 | 0.0423 | ||
| Patients | 11 | 0.3894 | 0.3963 | 0.1140 | 0.0344 | 0.176 | 83.5% | ||
| 4 | cyt c | Controls | 11 | 0.5393 | 0.5868 | 0.1359 | 0.0410 | ||
| Patients | 11 | 0.4141 | 0.4453 | 0.1057 | 0.0319 | 0.026 * | 76.8% | ||
| 5 | U | Controls | 12 | 0.5466 | 0.5699 | 0.1620 | 0.0468 | ||
| Patients | 11 | 0.4250 | 0.4602 | 0.0998 | 0.0301 | 0.044 * | 77.8% | ||
| 6 | G | Controls | 10 | 0.5799 | 0.5639 | 0.1507 | 0.0476 | ||
| Patients | 11 | 0.4115 | 0.3682 | 0.1285 | 0.0388 | 0.012 * | 71.0% | ||
| 7 | S | Controls | 15 | 1.2635 | 1.2688 | 0.1537 | 0.0397 | ||
| Patients | 13 | 1.1550 | 1.1352 | 0.1136 | 0.0315 | 0.046 * | 91.4% | ||
| 8 | Rot | Controls | 15 | 1.0000 | 1.0000 | 0.0000 | 0.0000 | ||
| Patients | 15 | 1.0000 | 1.0000 | 0.0000 | 0.0000 | ||||
| 9 | Gp 1 | Controls | 14 | 2.0569 | 2.0941 | 0.2302 | 0.0615 | ||
| Patients | 15 | 1.8476 | 1.9119 | 0.2053 | 0.0530 | 0.015 * | 89.8% | ||
| 10 | Ama | Controls | 15 | 0.0474 | 0.0439 | 0.0266 | 0.0069 | ||
| Patients | 15 | 0.0505 | 0.0398 | 0.0445 | 0.0115 | 0.822 | 106.4% | ||
| Flux Control Ratio (Relative Units) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Step | Titration | N | Mean | Median | Sd | Sem | p-Value | % of Control | |
| 0 | ce | Controls | 15 | 0.3811 | 0.3867 | 0.0546 | 0.0141 | ||
| Patients | 14 | 0.3584 | 0.3484 | 0.0589 | 0.0157 | 0.291 | 94.0% | ||
| 1 | Dig | Controls | 15 | 0.0019 | 0.0000 | 0.0075 | 0.0019 | ||
| Patients | 14 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.343 | |||
| 2 | OctM | Controls | 15 | 0.0695 | 0.0668 | 0.0302 | 0.0078 | ||
| Patients | 12 | 0.0832 | 0.0772 | 0.0371 | 0.0107 | 0.300 | 119.7% | ||
| 3 | ADP | Controls | 15 | 0.2015 | 0.2119 | 0.0511 | 0.0132 | ||
| Patients | 13 | 0.2082 | 0.2114 | 0.0782 | 0.0217 | 0.788 | 103.3% | ||
| 4 | cyt c | Controls | 15 | 0.2539 | 0.2439 | 0.0673 | 0.0174 | ||
| Patients | 14 | 0.2565 | 0.2579 | 0.0763 | 0.0204 | 0.925 | 101.0% | ||
| 5 | M2 | Controls | 11 | 0.5558 | 0.5781 | 0.1084 | 0.0327 | ||
| Patients | 13 | 0.4479 | 0.4264 | 0.1303 | 0.0361 | 0.040 * | 80.6% | ||
| 6 | P | Controls | 12 | 0.6185 | 0.6716 | 0.1590 | 0.0459 | ||
| Patients | 13 | 0.4873 | 0.4853 | 0.1380 | 0.0383 | 0.037 * | 78.8% | ||
| 7 | G | Controls | 12 | 0.6844 | 0.7221 | 0.1882 | 0.0543 | ||
| Patients | 13 | 0.5247 | 0.5347 | 0.1593 | 0.0442 | 0.031 * | 76.7% | ||
| 8 | S | Controls | 15 | 1.2968 | 1.3176 | 0.0847 | 0.0219 | ||
| Patients | 14 | 1.2522 | 1.2478 | 0.1025 | 0.0274 | 0.211 | 96.6% | ||
| 9 | U | Controls | 15 | 1.3416 | 1.3182 | 0.1591 | 0.0411 | ||
| Patients | 14 | 1.2753 | 1.2335 | 0.1169 | 0.0313 | 0.215 | 95.1% | ||
| 10 | Rot | Controls | 15 | 1.0000 | 1.0000 | 0.0000 | 0.0000 | ||
| Patients | 14 | 1.0000 | 1.0000 | 0.0000 | 0.0000 | ||||
| 11 | Gp 1 | Controls | 10 | 2.0291 | 2.0845 | 0.2561 | 0.0810 | ||
| Patients | 13 | 1.8305 | 1.8184 | 0.1913 | 0.0531 | 0.045 * | 90.2% | ||
| 12 | Ama | Controls | 15 | 0.0496 | 0.0516 | 0.0305 | 0.0079 | ||
| Patients | 14 | 0.0427 | 0.0271 | 0.0416 | 0.0111 | 0.612 | 86.1% | ||
| N | Mean | Median | Sd | Sem | p-Value | % of Control | ||
|---|---|---|---|---|---|---|---|---|
| Whole blood (µmol/L) | ||||||||
| Coenzyme Q10-TOTAL | Controls | 14 | 0.313 | 0.321 | 0.077 | 0.026 | ||
| Patients | 15 | 0.333 | 0.351 | 0.111 | 0.025 | 0.5126 | 106.4% | |
| α-tocopherol | Controls | 15 | 20.82 | 20.20 | 6.481 | 1.549 | ||
| Patients | 15 | 23.33 | 22.60 | 5.477 | 1.549 | 0.1198 | 112.1% | |
| γ-tocopherol | Controls | 15 | 1.387 | 1.400 | 0.926 | 0.186 | ||
| Patients | 15 | 1.184 | 1.260 | 0.426 | 0.186 | 0.8195 | 85.4% | |
| β-carotene | Controls | 14 | 0.249 | 0.249 | 0.171 | 0.042 | ||
| Patients | 15 | 0.144 | 0.144 | 0.142 | 0.040 | 0.0701 | 57.8% | |
| Platelets (pmol/109 cells) | ||||||||
| Coenzyme Q10-TOTAL | Controls | 12 | 84.14 | 79.5 | 19.26 | 5.39 | ||
| Patients | 14 | 63.56 | 69.65 | 18.14 | 4.99 | 0.0372 * | 75.5% | |
| α-tocopherol | Controls | 13 | 2546.5 | 2091.9 | 1482.0 | 321.1 | ||
| Patients | 15 | 1392.9 | 1174.9 | 778.5 | 298.9 | 0.0200 * | 54.7% | |
| γ-tocopherol | Controls | 14 | 325.6 | 292.2 | 157.9 | 72.3 | ||
| Patients | 15 | 231.0 | 106.0 | 343.2 | 69.8 | 0.0019 * | 71.0% | |
| Plasma (µmol/L) | ||||||||
| Coenzyme Q10-TOTAL | Controls | 13 | 0.516 | 0.498 | 0.114 | 0.037 | ||
| Patients | 15 | 0.466 | 0.462 | 0.15 | 0.035 | 0.3449 | 90.3% | |
| α-tocopherol | Controls | 15 | 31.14 | 31.40 | 8.78 | 1.946 | ||
| Patients | 15 | 31.40 | 32.30 | 6.04 | 1.946 | 0.3614 | 100.8% | |
| γ-tocopherol | Controls | 15 | 2.086 | 1.46 | 1.352 | 0.263 | ||
| Patients | 15 | 1.542 | 1.55 | 0.498 | 0.263 | 0.4806 | 73.9% | |
| β-carotene | Controls | 14 | 0.370 | 0.291 | 0.223 | 0.050 | ||
| Patients | 14 | 0.217 | 0.152 | 0.143 | 0.050 | 0.0387 * | 58.6% | |
| TBARS | Controls | 14 | 5.035 | 4.905 | 0.829 | 0.301 | ||
| Patients | 15 | 6.545 | 6.160 | 1.342 | 0.290 | 0.0022 * | 130.0% |
| Median (Range) | N (%) | p | ||
|---|---|---|---|---|
| Age (years) | Control subjects | 53 (35–67) | 15 (100) | |
| UC patients | 73 (58–83) | 15 (100) | <0.0001 | |
| Male/female | Control subjects | 6/9 (40.0/60.0) | ||
| UC patients | 10/5 (66.7/33.3) | |||
| BMI (kg/m2) | Control subjects | 25.5 (17.9–32.9) | ||
| UC patients | 25.2 (17.2–40.4) | 0.885 | ||
| UC patients | ||||
| Primary tumor site | Bladder/renal pelvis | 13/2 (86.7/13.3) | ||
| Histology type | Urothelial carcinoma | 15 (100.0) | ||
| Histology variants | Plasmacytoid/sarcomatoid | 1/1 (6.7/6.7) | ||
| ECOG performance status | ≤1 | 10 (66.7) | ||
| >1 | 5 (33.3) | |||
| TNM classification | Local disease (T2N0M0) | 2 (13.3) | ||
| Locally advanced disease (T3-4N0-3M0) | 7 (46.7) | |||
| Metastatic disease (M1) | 6 (40.0) | |||
| Metastasis localization * | Distant lymph nodes | 2 (33.3) | ||
| Bones | 3 (50.0) | |||
| Lungs | 4 (66.7) | |||
| Liver | 2 (33.3) | |||
| Peritoneum | 2 (33.3) | |||
| Other ** | 1 (16.7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palacka, P.; Gvozdjáková, A.; Rausová, Z.; Kucharská, J.; Slopovský, J.; Obertová, J.; Furka, D.; Furka, S.; Singh, K.K.; Sumbalová, Z. Platelet Mitochondrial Bioenergetics Reprogramming in Patients with Urothelial Carcinoma. Int. J. Mol. Sci. 2022, 23, 388. https://doi.org/10.3390/ijms23010388
Palacka P, Gvozdjáková A, Rausová Z, Kucharská J, Slopovský J, Obertová J, Furka D, Furka S, Singh KK, Sumbalová Z. Platelet Mitochondrial Bioenergetics Reprogramming in Patients with Urothelial Carcinoma. International Journal of Molecular Sciences. 2022; 23(1):388. https://doi.org/10.3390/ijms23010388
Chicago/Turabian StylePalacka, Patrik, Anna Gvozdjáková, Zuzana Rausová, Jarmila Kucharská, Ján Slopovský, Jana Obertová, Daniel Furka, Samuel Furka, Keshav K. Singh, and Zuzana Sumbalová. 2022. "Platelet Mitochondrial Bioenergetics Reprogramming in Patients with Urothelial Carcinoma" International Journal of Molecular Sciences 23, no. 1: 388. https://doi.org/10.3390/ijms23010388
APA StylePalacka, P., Gvozdjáková, A., Rausová, Z., Kucharská, J., Slopovský, J., Obertová, J., Furka, D., Furka, S., Singh, K. K., & Sumbalová, Z. (2022). Platelet Mitochondrial Bioenergetics Reprogramming in Patients with Urothelial Carcinoma. International Journal of Molecular Sciences, 23(1), 388. https://doi.org/10.3390/ijms23010388