
Homologous Recombination Deficiencies and Hereditary Tumors


Abstract
1. Introduction
2. Molecular Components and Cascade Overview of HR
2.1. Key Initial Step for HR: Sensing DSBs
2.2. ATM, CHEK2, BRIP1, and BARD1: Activators and Partners of BRCA-1 and Their Roles in Cancer Susceptibility
2.3. PALB2: Bridging Mediator of BRCA1 and BRCA2 and the Roles in Cancer Susceptibility
2.4. RAD51, RAD51C, RAD51D: The Final Effector of HR and Paralogs
3. HR-Based Precision Oncology for Hereditary Tumor Clinics
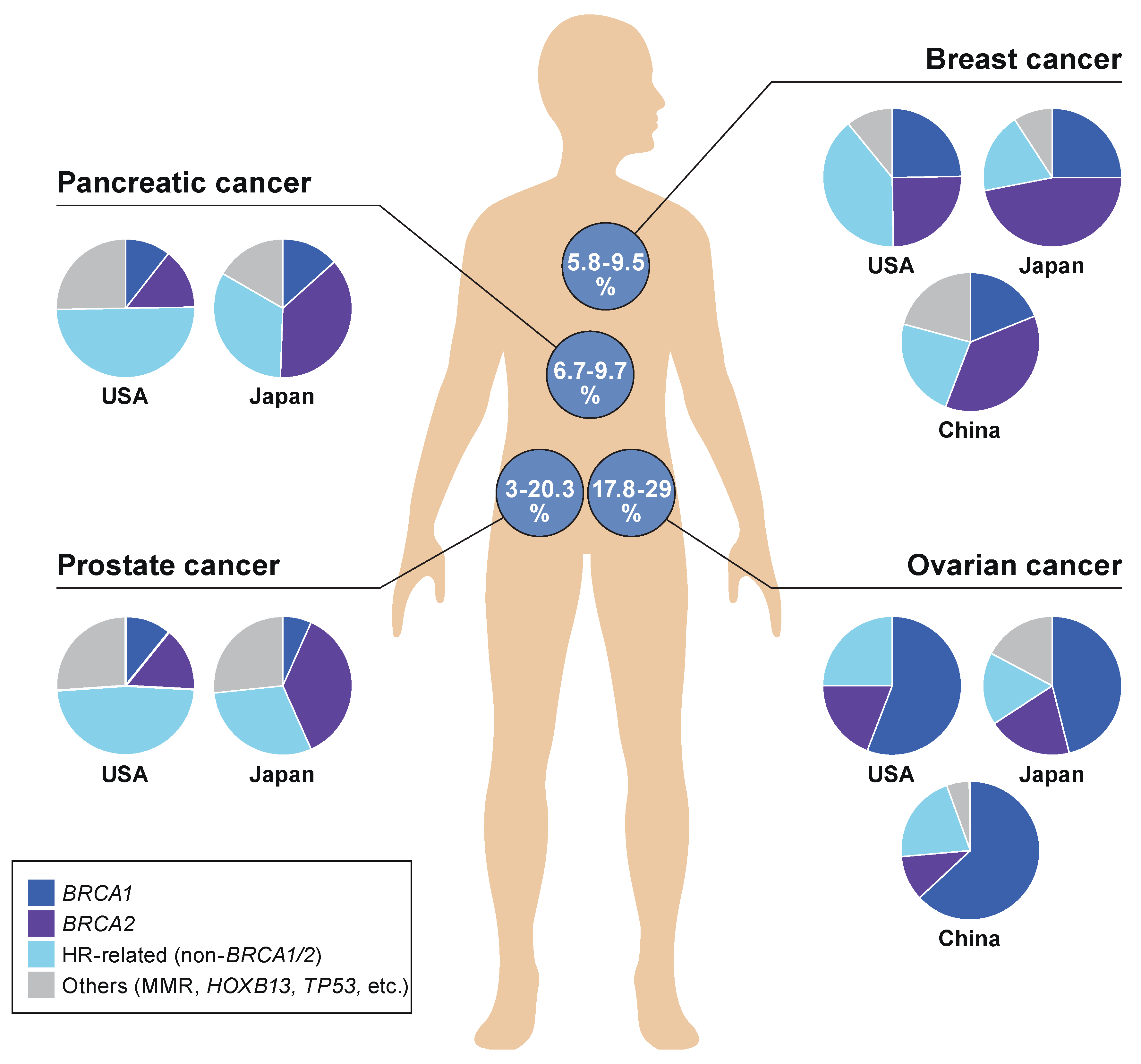
3.1. HR Deficiency (HRD) and HRD-Associated Genes
3.2. Differential Features of BRCA1-Type and BRCA2-Type
3.3. Reversion of BRCA1/2 Variants and Other Resistance Mechanisms to PARP Inhibitors or Platinum Chemotherapy
4. Rational Strategies for Diagnosis of Hereditary Tumors Using Multi-Gene Panel Testing (MGPT)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | ataxia telangiectasia mutated |
| ATR | ATM- and Rad3-related |
| BACH1 | BRCA1-associated C-terminal helicase 1 |
| BARD1 | BRCA1 Associated RING domain 1 |
| BRIP1 | BRCA1 interacting helicase 1 |
| CAP | College of American Pathologist |
| CHEK2 | checkpoint kinase 2 |
| CHK2 | CHEK2 gene coding checkpoint kinase 2 protein |
| CLIA | Clinical Laboratory Improvement Amendments |
| CtIP | CtBP-interacting protein |
| DSB | double strand break |
| GPV | germline pathogenic variant |
| HBOC | hereditary breast and ovarian cancer syndrome |
| HER2 | human epidermal growth factor |
| HGSC | high-grade serous carcinoma |
| HR | homologous recombination |
| HRD | homologous recombination deficiency |
| MGPT | multi-gene panel testing |
| NGS | next-generation sequencing |
| MMEJ | microhomology-mediated end-joining |
| NHEJ | non-homologous end-joining |
| MRN | MRE11-RAD50-NBS1 (or NBN) |
| NES | nuclear exporting signal |
| PALB2 | partner and localizer of BRCA2 |
| PARP | poly (ADP-ribose) polymerase |
| PARPi | poly (ADP-ribose) polymerase inhibitors |
| RPA | replication protein A |
| RRSO | risk-reducing salpingo-oophorectomy |
| TNBC | triple-negative breast cancer |
| VUS | variant of unknown significance |
References
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef]
- Ueki, A.; Hirasawa, A. Molecular Features and Clinical Management of Hereditary Gynecological Cancers. Int. J. Mol. Sci. 2020, 21, 9504. [Google Scholar] [CrossRef] [PubMed]
- Piombino, C.; Cortesi, L.; Lambertini, M.; Punie, K.; Grandi, G.; Toss, A. Secondary Prevention in Hereditary Breast and/or Ovarian Cancer Syndromes Other Than BRCA. J. Oncol. 2020, 2020, 6384190. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Eckert-Boulet, N.; Rothstein, R.; Lisby, M. Cell biology of homologous recombination in yeast. Methods Mol. Biol. 2011, 745, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hurov, K.; Hofmann, K.; Elledge, S.J. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009, 23, 729–739. [Google Scholar] [CrossRef]
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680. [Google Scholar] [CrossRef]
- Yoshida, R. Hereditary breast and ovarian cancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2020, 28, 1167–1180. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanova, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Lee, J.H.; Paull, T.T. Rad50 ATPase activity is regulated by DNA ends and requires coordination of both active sites. Nucleic Acids Res. 2017, 45, 5255–5268. [Google Scholar] [CrossRef]
- Paull, T.T.; Gellert, M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar] [CrossRef]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef]
- Deng, C.X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Hirasawa, A. Hereditary Gynecological Malignancy and Molecular Features. In Molecular Diagnosis and Targeting for Gynecologic Malignancy; Springer Nature Singapore Pte Ltd.: Singapore, 2021; pp. 145–165. [Google Scholar]
- Toss, A.; Tenedini, E.; Piombino, C.; Venturelli, M.; Marchi, I.; Gasparini, E.; Barbieri, E.; Razzaboni, E.; Domati, F.; Caggia, F.; et al. Clinicopathologic Profile of Breast Cancer in Germline ATM and CHEK2 Mutation Carriers. Genes 2021, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Nevanlinna, H.; Bartek, J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006, 25, 5912–5919. [Google Scholar] [CrossRef]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; Rooimans, M.A.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Gupta, I.; Shaikh, Z. BRIP1, a potential candidate gene in development of non-BRCA1/2 breast cancer. Front. Biosci. 2016, 8, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.B.; Wu, H.T.; Zhang, M.L.; Liu, J.; Zhang, G.J. Fanconi Anemia Pathway: Mechanisms of Breast Cancer Predisposition Development and Potential Therapeutic Targets. Front. Cell Dev. Biol. 2020, 8, 160. [Google Scholar] [CrossRef]
- Guenard, F.; Labrie, Y.; Ouellette, G.; Beauparlant, C.J.; Simard, J.; BRCAs, I.; Durocher, F. Mutational analysis of the breast cancer susceptibility gene BRIP1 /BACH1/FANCJ in high-risk non-BRCA1/BRCA2 breast cancer families. J. Hum. Genet. 2008, 53, 579. [Google Scholar] [CrossRef][Green Version]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Ali, M.; Delozier, C.D.; Chaudhary, U. BRIP-1 germline mutation and its role in colon cancer: Presentation of two case reports and review of literature. BMC Med. Genet. 2019, 20, 75. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horvath, J.; Niederacher, D.; Arnold, N.; Kaulfuss, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Slavin, T.P.; Maxwell, K.N.; Lilyquist, J.; Vijai, J.; Neuhausen, S.L.; Hart, S.N.; Ravichandran, V.; Thomas, T.; Maria, A.; Villano, D.; et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017, 3, 22. [Google Scholar] [CrossRef]
- Abe, K.; Ueki, A.; Urakawa, Y.; Kitago, M.; Yoshihama, T.; Nanki, Y.; Kitagawa, Y.; Aoki, D.; Kosaki, K.; Hirasawa, A. Familial pancreatic cancer with PALB2 and NBN pathogenic variants: A case report. Hered Cancer Clin. Pr. 2021, 19, 5. [Google Scholar] [CrossRef]
- Canto, M.I.; Harinck, F.; Hruban, R.H.; Offerhaus, G.J.; Poley, J.W.; Kamel, I.; Nio, Y.; Schulick, R.S.; Bassi, C.; Kluijt, I.; et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013, 62, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kitago, M.; Kitagawa, Y.; Hirasawa, A. Hereditary pancreatic cancer. Int. J. Clin. Oncol. 2021, 26, 1784–1792. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu. Rev. Pathol. 2009, 4, 461–487. [Google Scholar] [CrossRef] [PubMed]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005, 434, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Hirasawa, A.; Imoto, I.; Naruto, T.; Akahane, T.; Yamagami, W.; Nomura, H.; Masuda, K.; Susumu, N.; Tsuda, H.; Aoki, D. Prevalence of pathogenic germline variants detected by multigene sequencing in unselected Japanese patients with ovarian cancer. Oncotarget 2017, 8, 112258–112267. [Google Scholar] [CrossRef]
- Futagawa, M.; Yamamoto, H.; Kochi, M.; Urakawa, Y.; Sogawa, R.; Kato, F.; Okazawa-Sakai, M.; Ennishi, D.; Shinozaki, K.; Inoue, H.; et al. Retroperitoneal leiomyosarcoma in a female patient with a germline splicing variant RAD51D c.904-2A > T: A case report. Hered Cancer Clin. Pr. 2021, 19, 48. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 2014, 343, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Pharoah, P.D.; McMullan, G.; Day, N.E.; Ponder, B.A.; Easton, D. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet. Epidemiol. 2001, 21, 1–18. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Ho, B.N.; Singh, A.T.K. The Evolution, Functions and Applications of the Breast Cancer Genes BRCA1 and BRCA2. Cancer Genom. Proteom. 2017, 14, 293–298. [Google Scholar] [CrossRef][Green Version]
- Momozawa, Y.; Iwasaki, Y.; Hirata, M.; Liu, X.; Kamatani, Y.; Takahashi, A.; Sugano, K.; Yoshida, T.; Murakami, Y.; Matsuda, K.; et al. Germline Pathogenic Variants in 7636 Japanese Patients With Prostate Cancer and 12,366 Controls. J. Natl. Cancer Inst. 2020, 112, 369–376. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Hu, C.; LaDuca, H.; Shimelis, H.; Polley, E.C.; Lilyquist, J.; Hart, S.N.; Na, J.; Thomas, A.; Lee, K.Y.; Davis, B.T.; et al. Multigene Hereditary Cancer Panels Reveal High-Risk Pancreatic Cancer Susceptibility Genes. JCO Precis. Oncol. 2018, 2, 1–28. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Giannakis, M.; Moore, N.D.; Han, G.C.; Reardon, B.; Hamada, T.; Mu, X.J.; Nishihara, R.; Qian, Z.; Liu, L.; et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am. J. Hum. Genet. 2018, 102, 401–414. [Google Scholar] [CrossRef]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived From Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef]
- Brose, M.S.; Rebbeck, T.R.; Calzone, K.A.; Stopfer, J.E.; Nathanson, K.L.; Weber, B.L. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J. Natl. Cancer Inst. 2002, 94, 1365–1372. [Google Scholar] [CrossRef]
- Finch, A.; Beiner, M.; Lubinski, J.; Lynch, H.T.; Moller, P.; Rosen, B.; Murphy, J.; Ghadirian, P.; Friedman, E.; Foulkes, W.D.; et al. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 Mutation. JAMA 2006, 296, 185–192. [Google Scholar] [CrossRef]
- Levine, D.A.; Argenta, P.A.; Yee, C.J.; Marshall, D.S.; Olvera, N.; Bogomolniy, F.; Rahaman, J.A.; Robson, M.E.; Offit, K.; Barakat, R.R.; et al. Fallopian tube and primary peritoneal carcinomas associated with BRCA mutations. J. Clin. Oncol. 2003, 21, 4222–4227. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Friedman, E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Brit. J. Cancer 2007, 96, 11–15. [Google Scholar] [CrossRef]
- Oh, M.; Alkhushaym, N.; Fallatah, S.; Althagafi, A.; Aljadeed, R.; Alsowaida, Y.; Jeter, J.; Martin, J.R.; Babiker, H.M.; McBride, A.; et al. The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: A meta-analysis. Prostate 2019, 79, 880–895. [Google Scholar] [CrossRef]
- Nyberg, T.; Frost, D.; Barrowdale, D.; Evans, D.G.; Bancroft, E.; Adlard, J.; Ahmed, M.; Barwell, J.; Brady, A.F.; Brewer, C.; et al. Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur. Urol. 2020, 77, 24–35. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Levine, D.A.; Tang, L.H.; Allen, P.J.; Jarnagin, W.; Brennan, M.F.; Offit, K.; Robson, M.E. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2009, 27, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, H.; Rogers, K.M. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin. Pr. 2015, 13, 16. [Google Scholar] [CrossRef]
- Rafnar, T.; Gudbjartsson, D.F.; Sulem, P.; Jonasdottir, A.; Sigurdsson, A.; Jonasdottir, A.; Besenbacher, S.; Lundin, P.; Stacey, S.N.; Gudmundsson, J.; et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011, 43, 1104–1107. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokolorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojc, B.; Deebniak, T.; Gorski, B.; Blecharz, P.; et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef] [PubMed]
- Brandao, A.; Paulo, P.; Maia, S.; Pinheiro, M.; Peixoto, A.; Cardoso, M.; Silva, M.P.; Santos, C.; Eeles, R.A.; Kote-Jarai, Z.; et al. The CHEK2 Variant C.349A > G Is Associated with Prostate Cancer Risk and Carriers Share a Common Ancestor. Cancers 2020, 12, 3254. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Gorski, B.; Huzarski, T.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef]
- Thompson, E.R.; Rowley, S.M.; Li, N.; McInerny, S.; Devereux, L.; Wong-Brown, M.W.; Trainer, A.H.; Mitchell, G.; Scott, R.J.; James, P.A.; et al. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J. Clin. Oncol. 2016, 34, 1455–1459. [Google Scholar] [CrossRef]
- Steffen, J.; Nowakowska, D.; Niwinska, A.; Czapczak, D.; Kluska, A.; Piatkowska, M.; Wisniewska, A.; Paszko, Z. Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int. J. Cancer 2006, 119, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Gorski, B.; Debniak, T.; Gliniewicz, B.; Mierzejewski, M.; Masojc, B.; Jakubowska, A.; Matyjasik, J.; Zlowocka, E.; Sikorski, A.; et al. NBS1 is a prostate cancer susceptibility gene. Cancer Res. 2004, 64, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkas, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef]
- Cortesi, L.; Toss, A.; Cucinotto, I. PARP Inhibitors for the Treatment of Ovarian Cancer. Curr. Cancer Drug Targets 2018, 18, 877–893. [Google Scholar] [CrossRef]
- Loizzi, V.; Ranieri, G.; Laforgia, M.; Gadaleta, C.D.; Gargano, G.; Kardhashi, A.; De Liso, M.; Naglieri, E.; Del Vecchio, V.; Cicinelli, E.; et al. PARP inhibitors and epithelial ovarian cancer: Molecular mechanisms, clinical development and future prospective. Oncol. Lett. 2020, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Burstein, H.J.; Schwartz, R.S. Molecular origins of cancer. N. Engl. J. Med. 2008, 358, 527. [Google Scholar] [CrossRef]
- Cortesi, L.; Piombino, C.; Toss, A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? J. Pers. Med. 2021, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Kasi, P.M.; Bamlet, W.R.; Ho, T.P.; Polley, E.C.; Hu, C.; Hart, S.N.; Rabe, K.G.; Boddicker, N.J.; Gnanaolivu, R.D.; et al. Effect of Germline Mutations in Homologous Recombination Repair Genes on Overall Survival of Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2020, 26, 6505–6512. [Google Scholar] [CrossRef]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Casolino, R.; Paiella, S.; Azzolina, D.; Beer, P.A.; Corbo, V.; Lorenzoni, G.; Gregori, D.; Golan, T.; Braconi, C.; Froeling, F.E.M.; et al. Homologous Recombination Deficiency in Pancreatic Cancer: A Systematic Review and Prevalence Meta-Analysis. J. Clin. Oncol. 2021, 39, 2617–2631. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Norquist, B.; Lacchetti, C.; Armstrong, D.; Grisham, R.N.; Goodfellow, P.J.; Kohn, E.C.; Levine, D.A.; Liu, J.F.; Lu, K.H.; et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1222–1245. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef]
- Miao, K.; Lei, J.H.; Valecha, M.V.; Zhang, A.; Xu, J.; Wang, L.; Lyu, X.; Chen, S.; Miao, Z.; Zhang, X.; et al. NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. Nat. Commun. 2020, 11, 3256. [Google Scholar] [CrossRef] [PubMed]
- Sonderstrup, I.M.H.; Jensen, M.R.; Ejlertsen, B.; Eriksen, J.O.; Gerdes, A.M.; Kruse, T.A.; Larsen, M.J.; Thomassen, M.; Laenkholm, A.V. Subtypes in BRCA-mutated breast cancer. Hum. Pathol. 2019, 84, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Couch, F.J.; Parsons, M.T.; McGuffog, L.; Barrowdale, D.; Bolla, M.K.; Wang, Q.; Healey, S.; Schmutzler, R.; Wappenschmidt, B.; et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: A large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014, 16, 3419. [Google Scholar] [CrossRef]
- Limaiem, F.; Mlika, M. Medullary Breast Carcinoma. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/31194432/ (accessed on 23 November 2021).
- Taylor, R.A.; Fraser, M.; Rebello, R.J.; Boutros, P.C.; Murphy, D.G.; Bristow, R.G.; Risbridger, G.P. The influence of BRCA2 mutation on localized prostate cancer. Nat. Rev. Urol. 2019, 16, 281–290. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Cook-Deegan, R.; Niehaus, A. After Myriad: Genetic Testing in the Wake of Recent Supreme Court Decisions about Gene Patents. Curr. Genet. Med. Rep. 2014, 2, 223–241. [Google Scholar] [CrossRef]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shao, D.; Li, L.; Wu, M.; Ma, S.; Tan, X.; Zhong, S.; Guo, F.; Wang, Z.; Ye, M. Germline and somatic mutations of multi-gene panel in Chinese patients with epithelial ovarian cancer: A prospective cohort study. J. Ovarian Res. 2019, 12, 80. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef]
- Mizukami, K.; Iwasaki, Y.; Kawakami, E.; Hirata, M.; Kamatani, Y.; Matsuda, K.; Endo, M.; Sugano, K.; Yoshida, T.; Murakami, Y.; et al. Genetic characterization of pancreatic cancer patients and prediction of carrier status of germline pathogenic variants in cancer-predisposing genes. EBioMedicine 2020, 60, 103033. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, F.; Easton, D.F.; Renault, A.L.; Tavtigian, S.V.; Bernstein, J.L.; Kote-Jarai, Z.; Eeles, R.A.; Plaseska-Karanfia, D.; Feliubadalo, L.; Spanish ATM Working Group; et al. First international workshop of the ATM and cancer risk group (4–5 December 2019). Fam. Cancer 2021, 1–17. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| HR-Related Genes | Molecular Functions | Functions in HR | Susceptible Organs | Risk Estimates | References | |
|---|---|---|---|---|---|---|
| ATM | serine-threonine kinase/checkpoint activation (G2-M and G1-S checkpoints)/p53 activation | BRCA1 phosphorylation | Breast (luminal B-like/HER2-negative), Ovary, Pancreas Prostate, Colon | Breast cancer (2.8–3.3), Ovarian cancer (0.9–2.4), Pancreatic cancer (5.7–9.0), Prostate cancer (2.9), Colorectal cancer (2.8) | [40,52,53,54,55,56,57,58,59] | |
| BRCA1 (FANCS) | mediator/adaptor/enzymatic/cell cycle regulator (G2-M checkpoint control) | BRCT domain-mediated phospho-protein interactions | Breast (TNBC), Ovary (HGSC), Pancreas, Prostate | Breast cancer # (46–87% vs. 12%), Ovarian cancer # (39–63% vs. 1–2%), Pancreatic cancer # (1–3% vs. 0.50%), Prostate cancer # (8.6% by age 65 vs. 6% through age 69) | [54,60,61,62,63,64,65,66,67,68,69,70] | |
| BARD1 | constitutive RING-mediated heterodimerization with BRCA1 | forming BRCA1-BARD1 E3 ubiquitin ligase/interacting with BRCA1 through RING domain | Breast(early-age onset), Ovary | Breast cancer (1.9–3.2), Ovarian cancer (4.2) | [36,40,53,57,59] | |
| BRCA2 (FANCD1) | mediator of RAD51 | recombination mediator/RAD51 binding | Breast, Ovary (HGSC), Pancreas, Prostate, Skin (potential risk for Malignant Melanoma) | Breast cancer # (38–84% vs. 12%), Ovarian cancer # (16.5–27% vs. 1–2%), Pancreatic cancer # ( 2–7% vs. 0.50%), Prostate cancer # (15% by age 65; 20% lifetime vs. 6% through age 69) | [54,60,61,62,64,65,66,67,68,69,70] | |
| BRIP1 (FANCJ) | 5′ to 3′ DNA helicase/binds to BRCA1/phosphorylated following DNA damage | interacting with BRCA1 through BRCT domain/genome integrity/tumor suppressive | Breast, Ovary, Pancreas, Prostate | Breast cancer (1.6), Ovarian cancer (2.4–8.1), Pancreatic cancer (2.7) | [53,57,58,71] | |
| CHEK2 | serine/threonine kinase (ATM-mediated) | BRCA1 phosphorylation | Breast (luminal A-like), Prostate, Colon | Breast cancer (2.68), Prostate cancer (ethnic differences in CHEK2 variant frequencies) | [21,72,73,74] | |
| MRN complex | MRE11 (MRE11A) | enzymatic/nuclease | MRE11, endonuclease; 3′–5′ exonuclease; cooperate with CtIP to initiate DSB resection | Breast | Breast cancer (0.9–9.0) | [40,53,75] |
| RAD50 | structural/ATPase | RAD50, structural maintenance of chromosomes SMC-like protein | – | – | [53,58,59,75] | |
| NBS1 (NBN) | adaptor/checkpoint | NBS1 (NBN), phospho-protein and ATM kinase interactions | Breast, Ovary, Prostate | Breast cancer (3.2), Ovarian cancer (1.9), Prostate cancer (3.9) | [57,76,77] | |
| PALB2 (FANCN) | scaffold/mediator/partner for BRCA2 stability/nuclear localization | complex formation/linking BRCA1 and BRCA2 | Breast, Ovary, Pancreas, Prostate, Colon, Kidney (Wilm’s tumor), CNS (Medulloblastoma) | Breast cancer (2.3–9.5), Ovarian cancer (2.9–4.4), Pancreatic cancer (2.3–14.8), Prostate cancer (0.4), Colorectal cancer (4.9) | [6,53,54,55,56,57,58,59,75,78,79] | |
| RAD51C | RAD51 paralog | interacting with RAD51/forming subcomplex (BCDX2 complex) | Breast, Ovary, Pancreas | Breast cancer (0.4–1.4), Ovarian cancer (3.4–5.1) | [40,53,57,58,59,80] | |
| RAD51D | RAD51 paralog | interacting with RAD51/forming subcomplex (BCDX2 complex) | Breast, Ovary, Pancreas | Breast cancer (3.1–8.3), Ovarian cancer(4.8–10.9) | [40,53,57,58,59] | |
| RAD51 | recombinase | final effector of HR/nucleofilament formation/strand invasion | Not specified | – | – | |
| #: Estimated lifetime cumulative risks for the indicated malignancy due to pathogenic germline variants versus general population risks are shown as risk estimates by referring to GeneReviews® [Internet] https://www.ncbi.nlm.nih.gov/books/NBK1247/ (accessed on 19 November 2021) and [81]. | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, H.; Hirasawa, A. Homologous Recombination Deficiencies and Hereditary Tumors. Int. J. Mol. Sci. 2022, 23, 348. https://doi.org/10.3390/ijms23010348
Yamamoto H, Hirasawa A. Homologous Recombination Deficiencies and Hereditary Tumors. International Journal of Molecular Sciences. 2022; 23(1):348. https://doi.org/10.3390/ijms23010348
Chicago/Turabian StyleYamamoto, Hideki, and Akira Hirasawa. 2022. "Homologous Recombination Deficiencies and Hereditary Tumors" International Journal of Molecular Sciences 23, no. 1: 348. https://doi.org/10.3390/ijms23010348
APA StyleYamamoto, H., & Hirasawa, A. (2022). Homologous Recombination Deficiencies and Hereditary Tumors. International Journal of Molecular Sciences, 23(1), 348. https://doi.org/10.3390/ijms23010348