
Exploring the Catalytic Mechanism of the RNA Cap Modification by nsp16-nsp10 Complex of SARS-CoV-2 through a QM/MM Approach

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
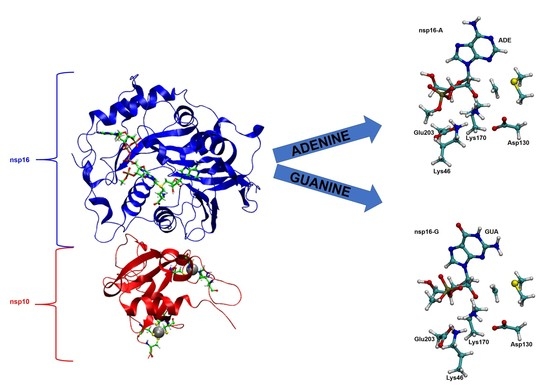
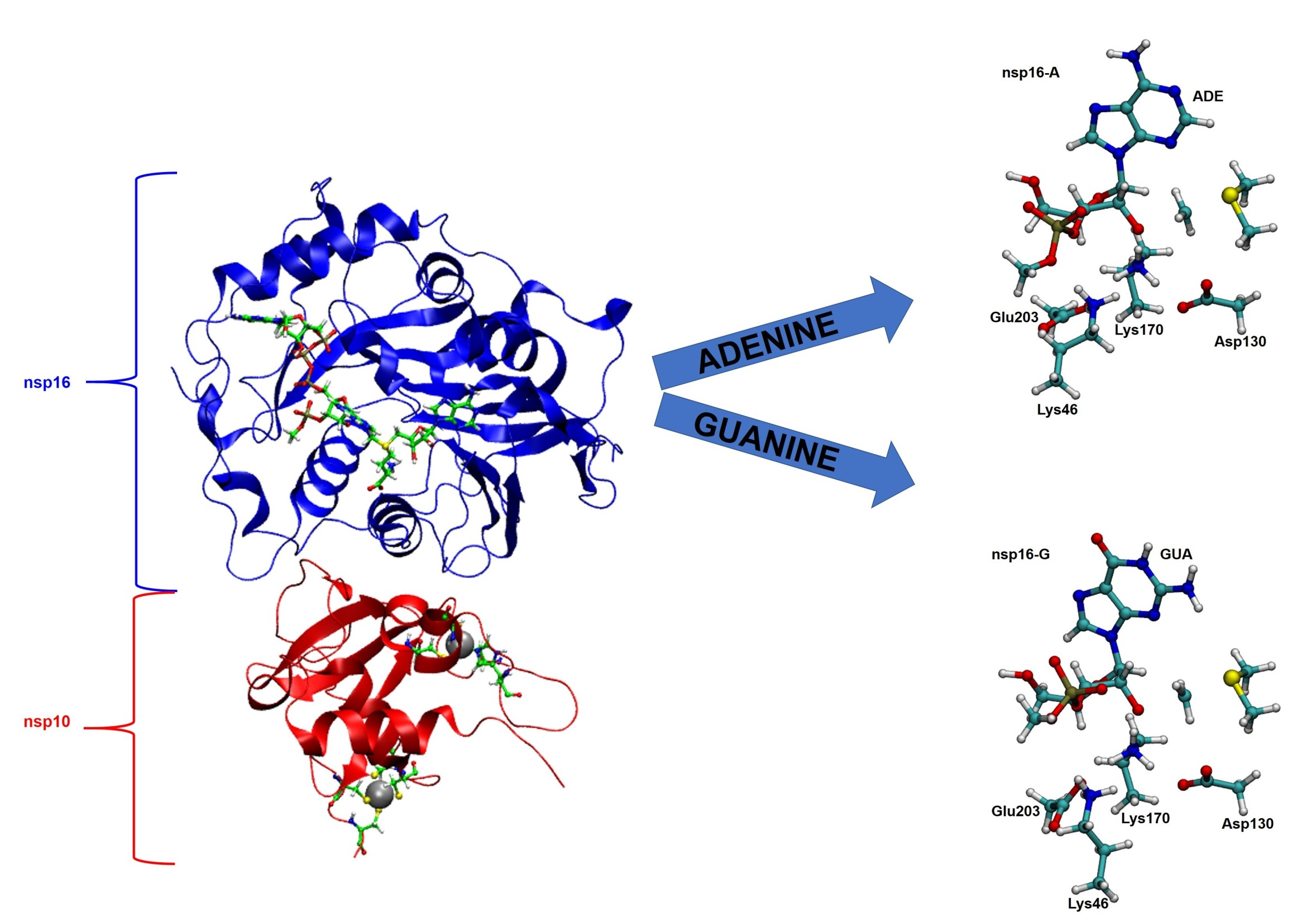
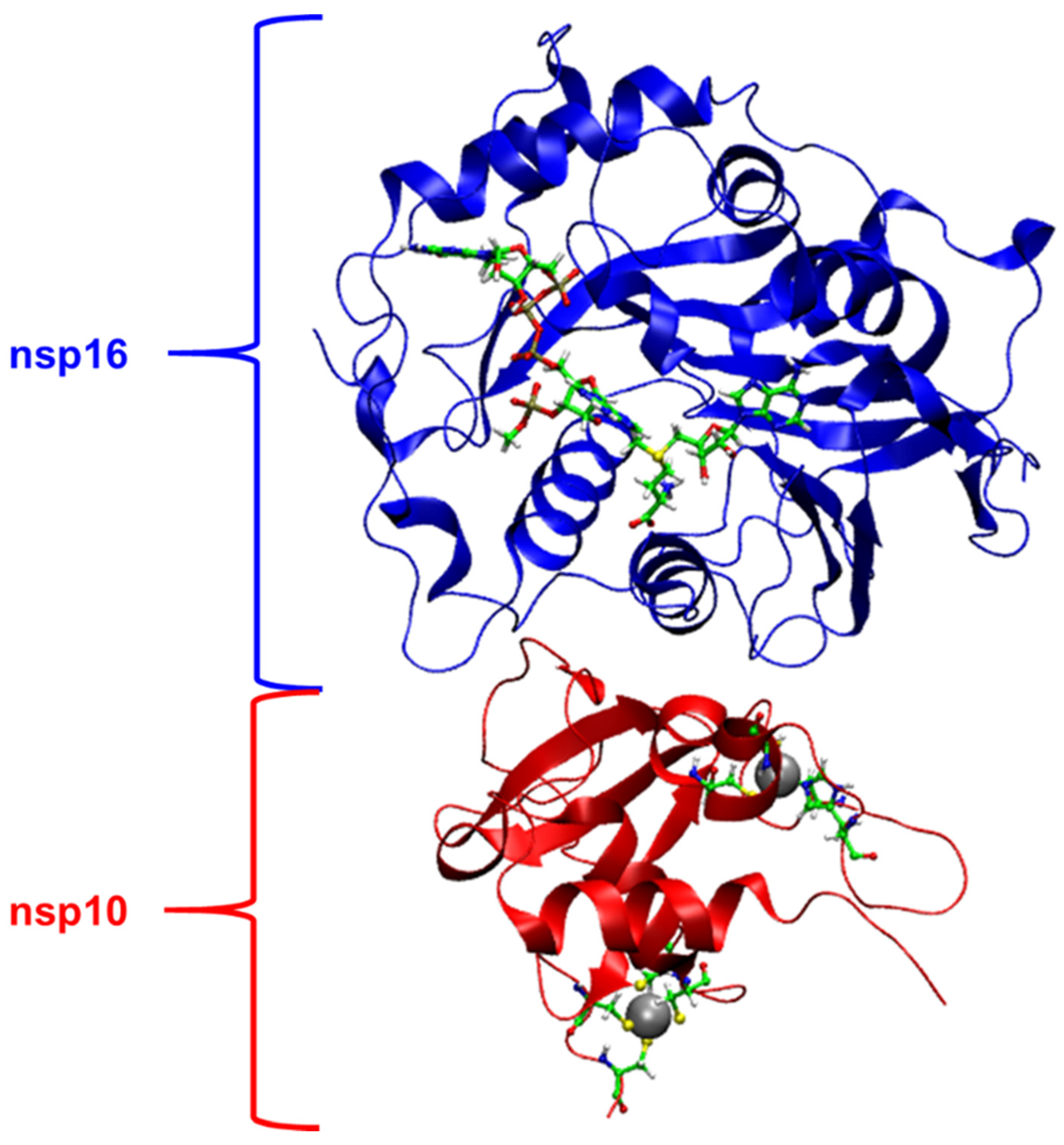
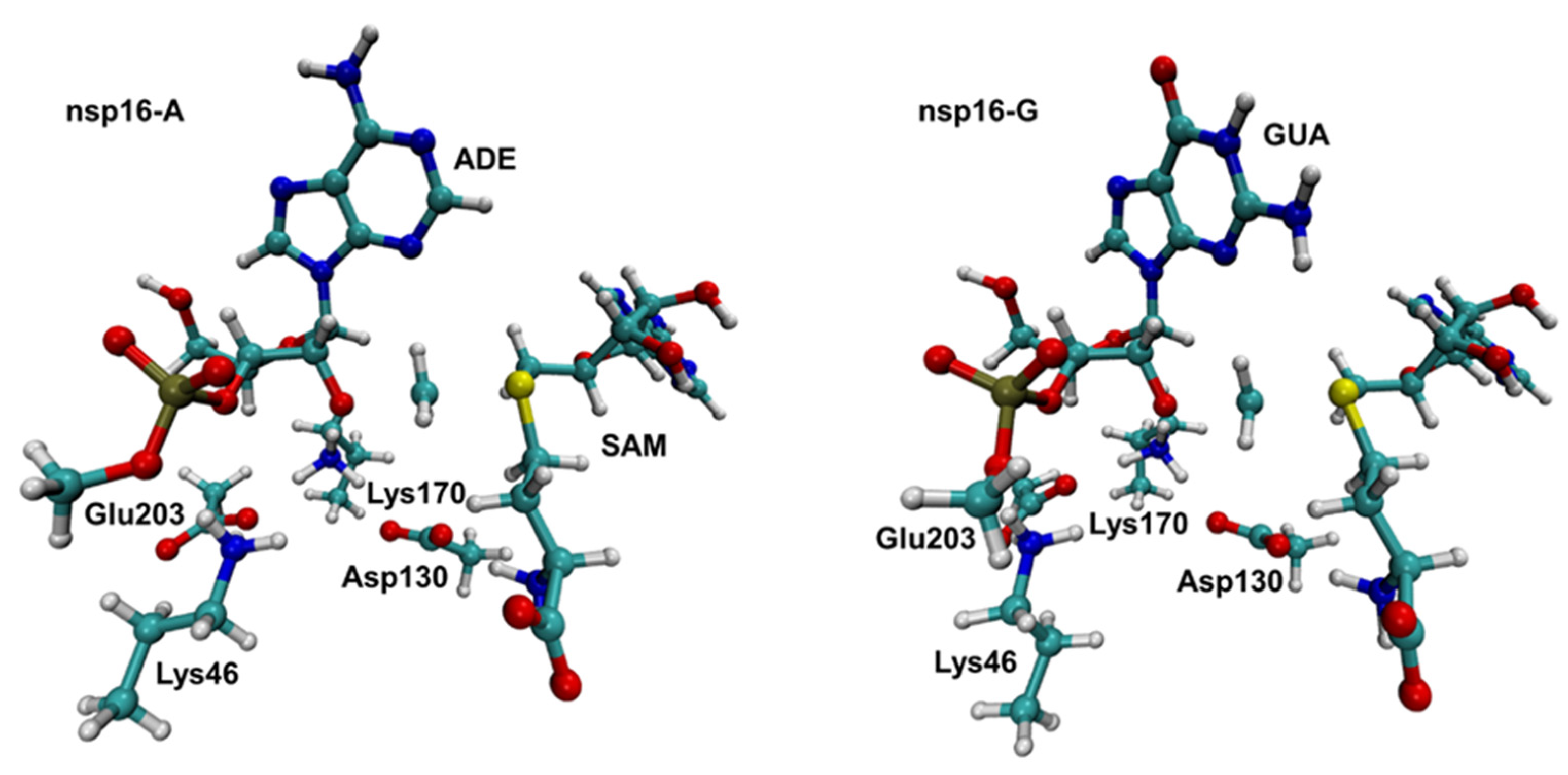
2.1. Structural Analysis of MD Simulations
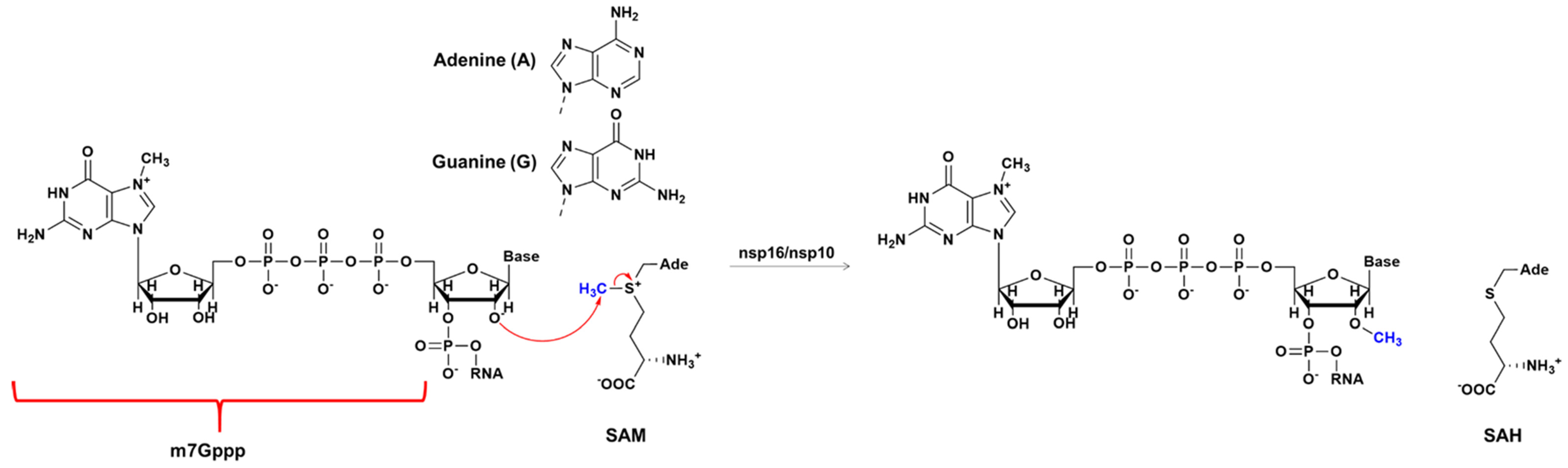
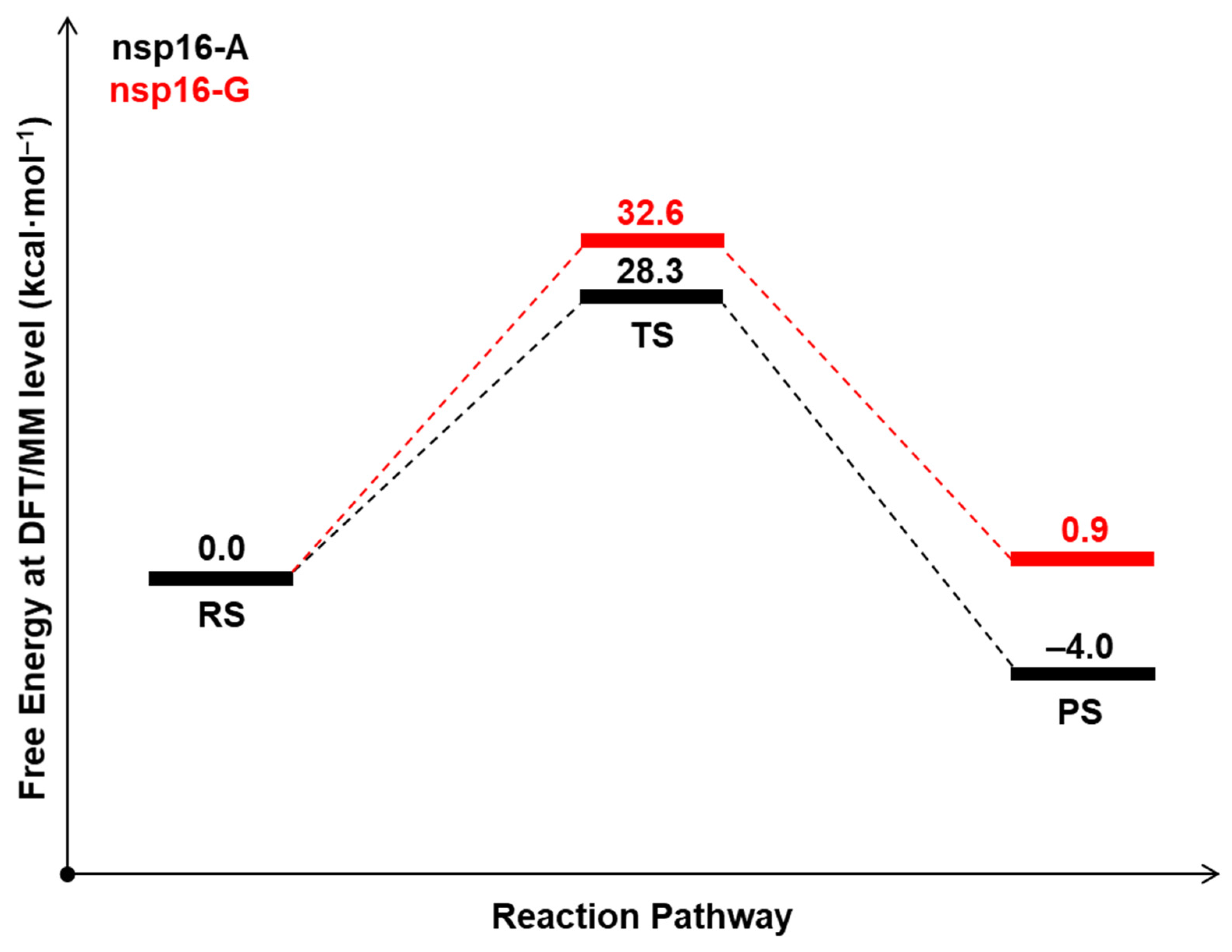
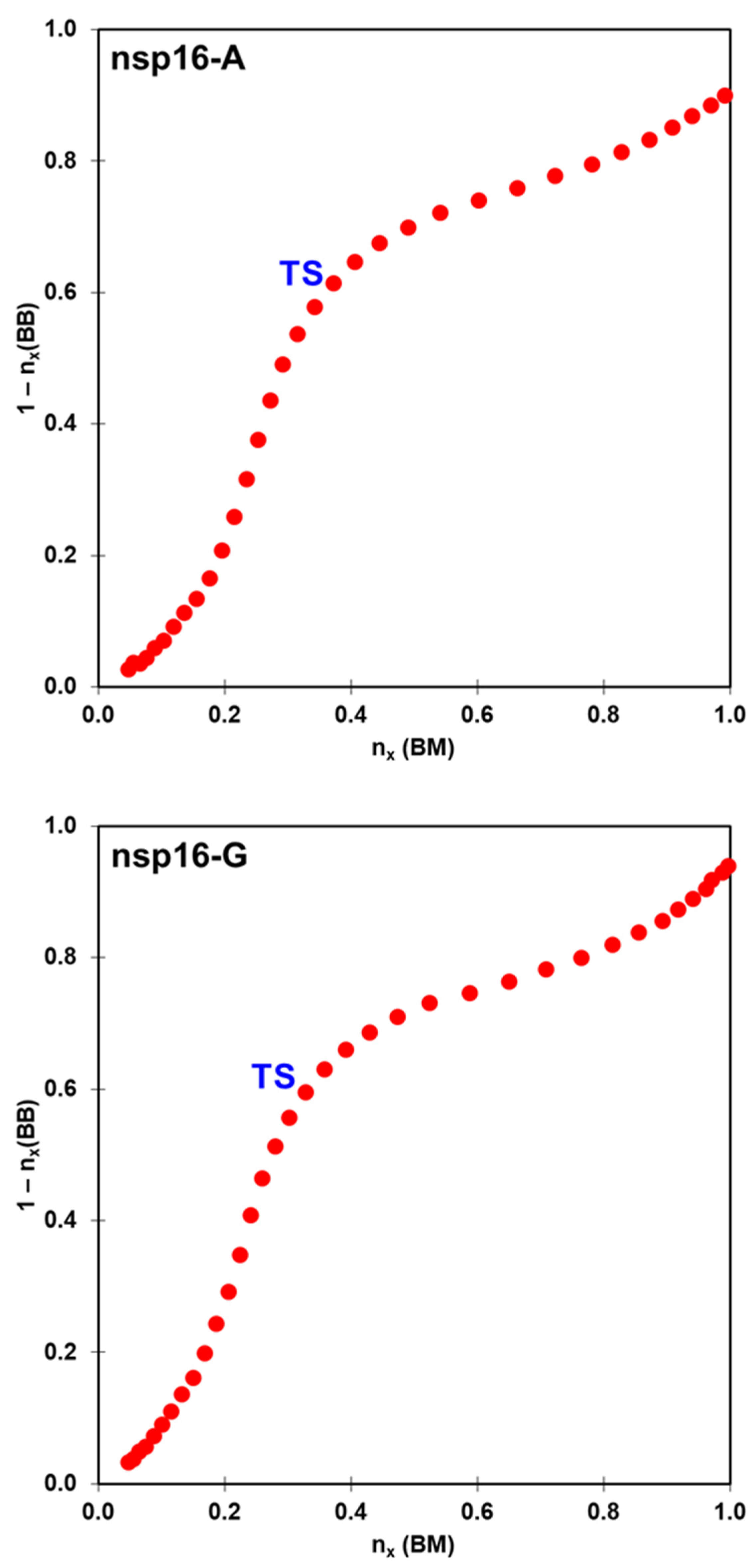
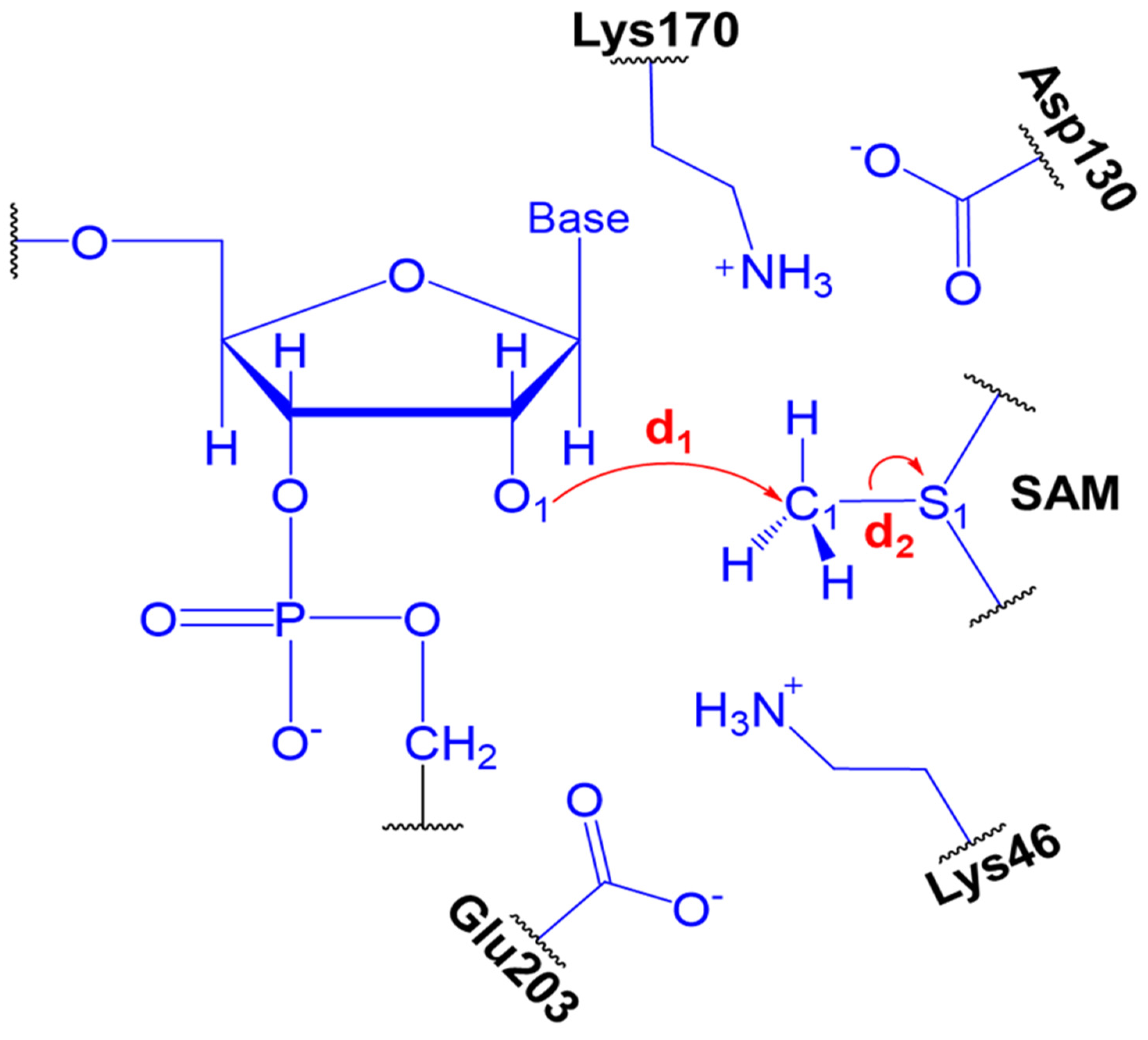
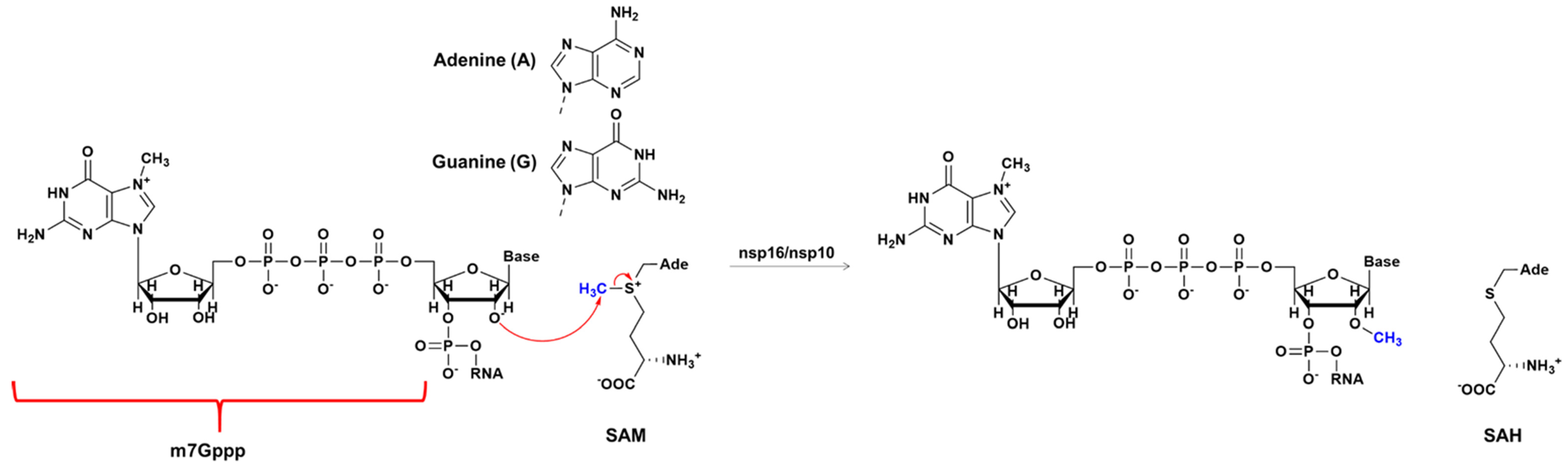
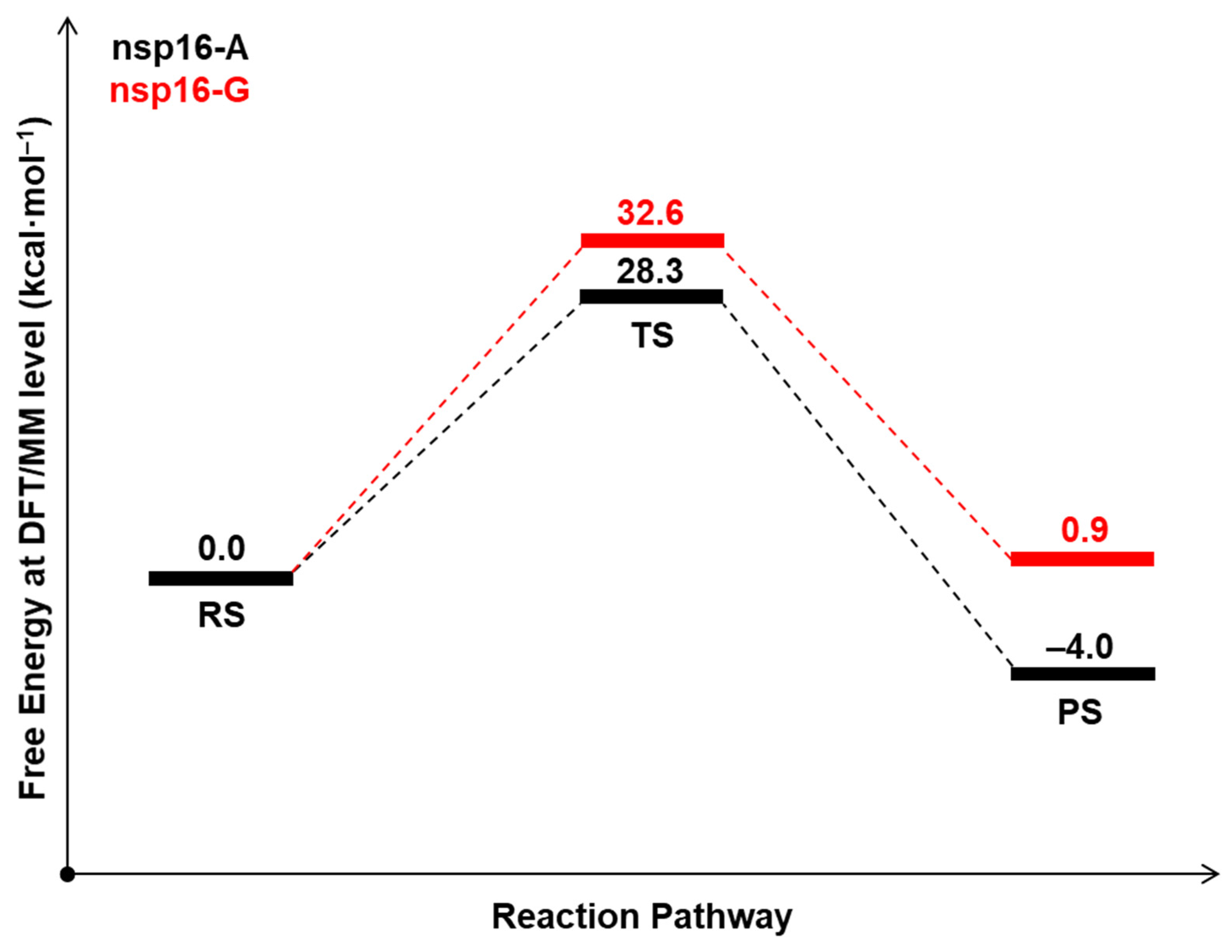
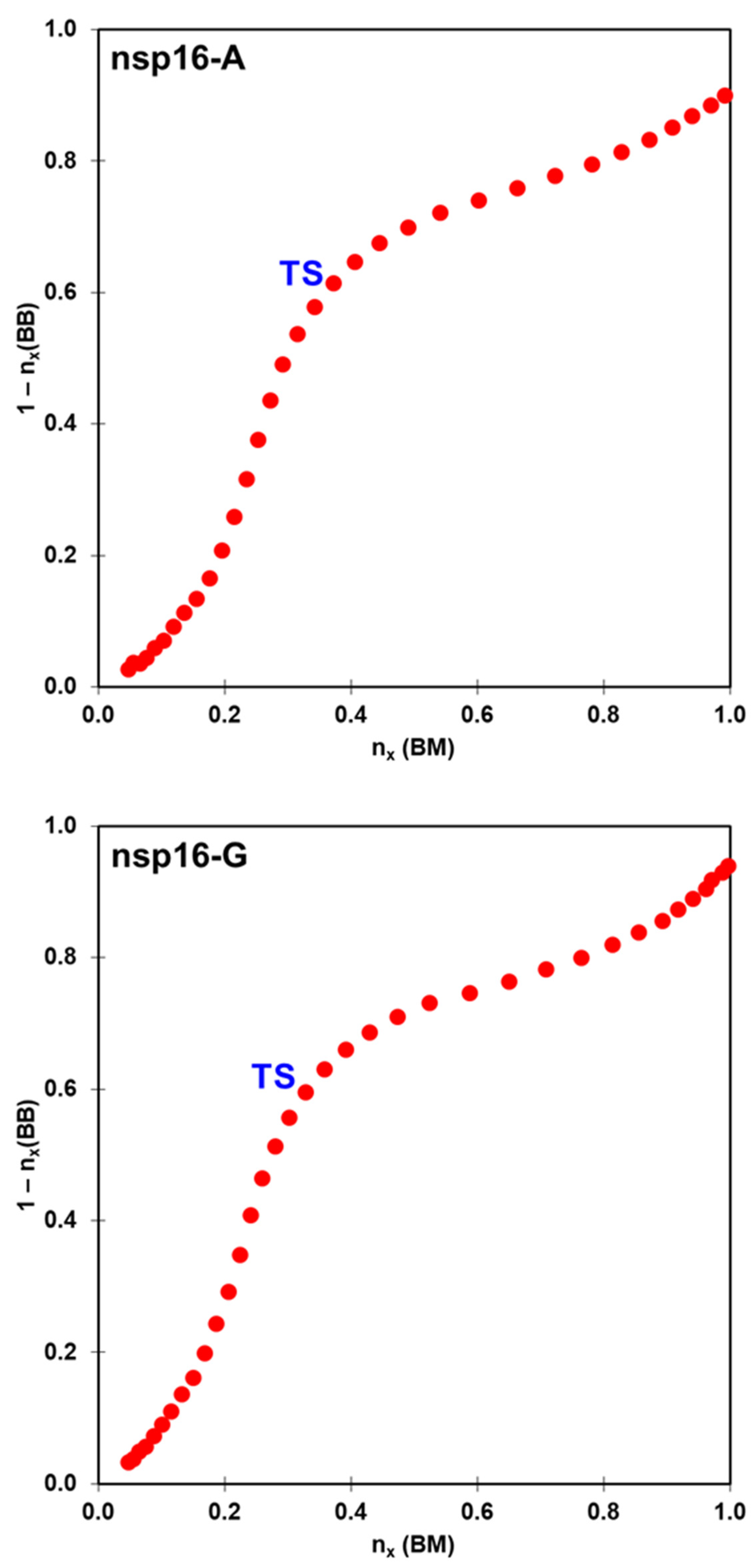
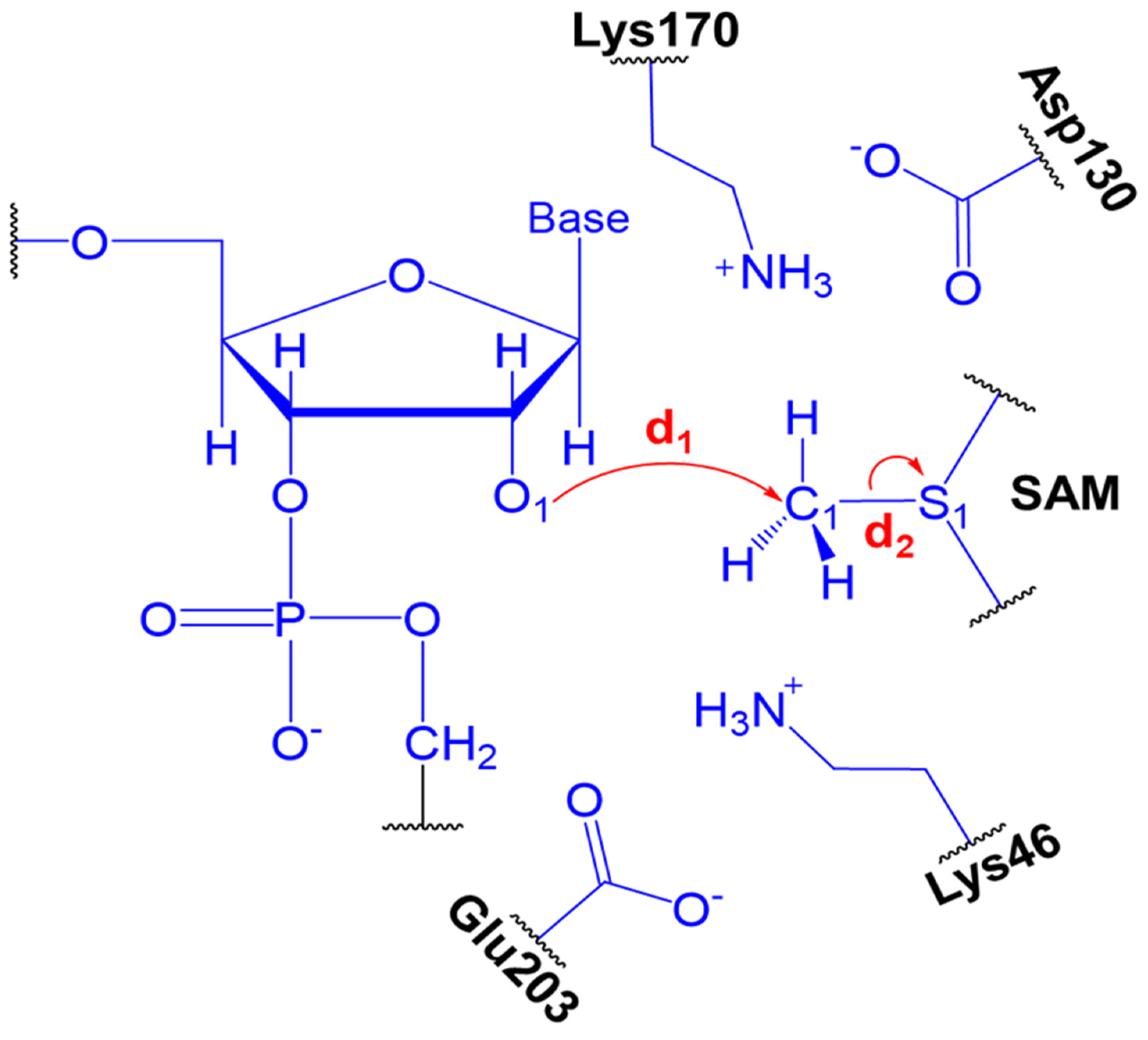
2.2. The Methyl Transfer from SAM to Ribose
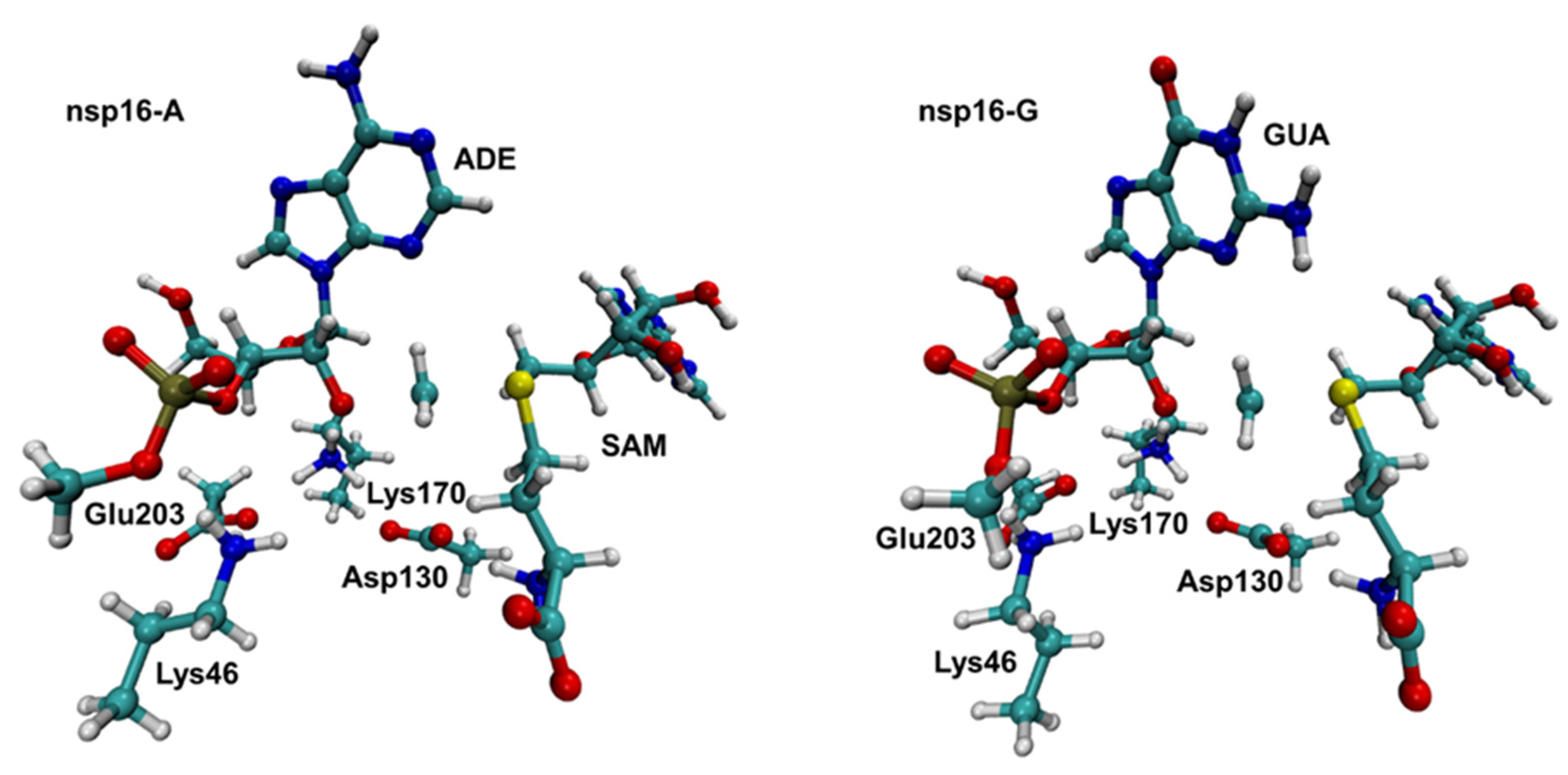
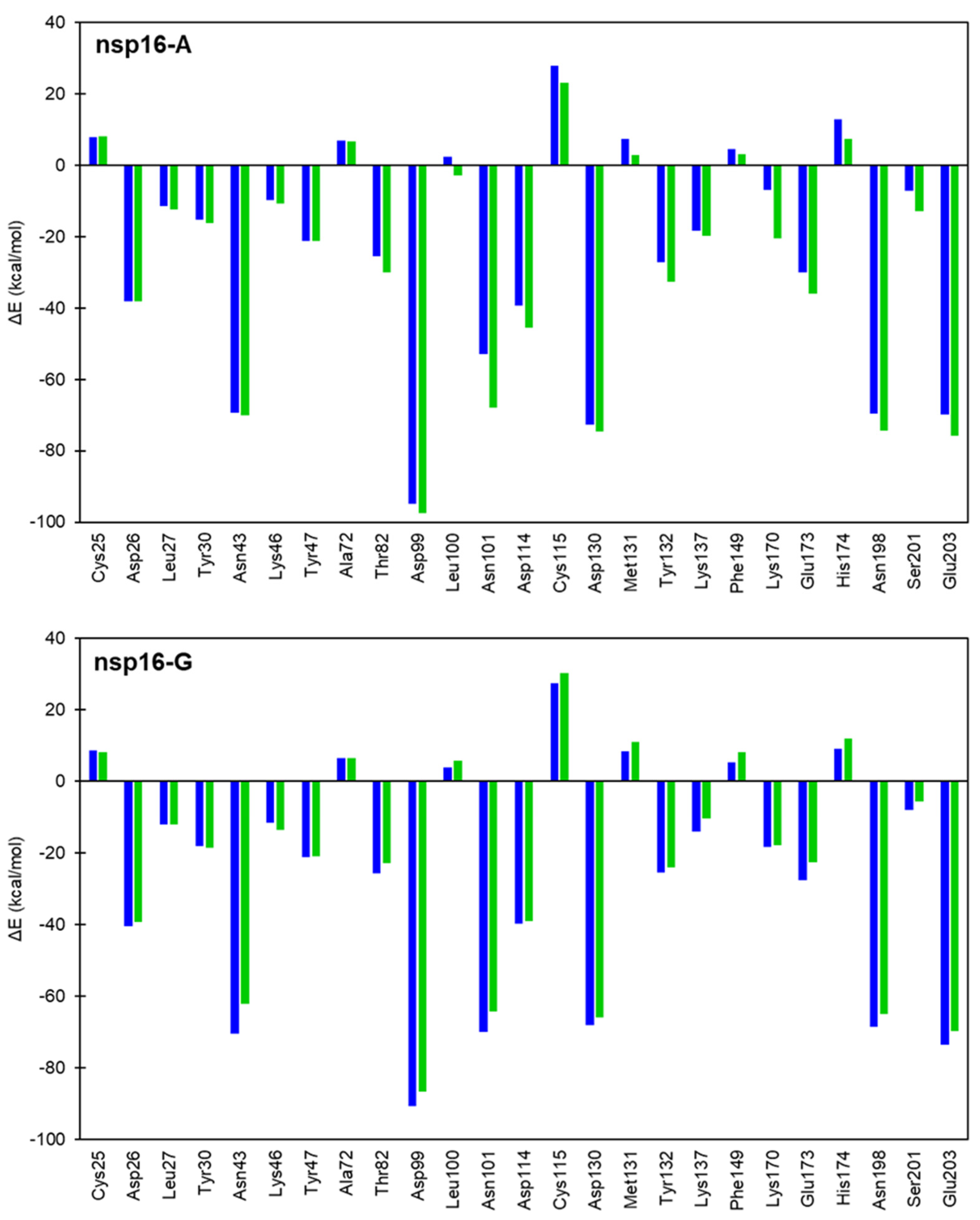
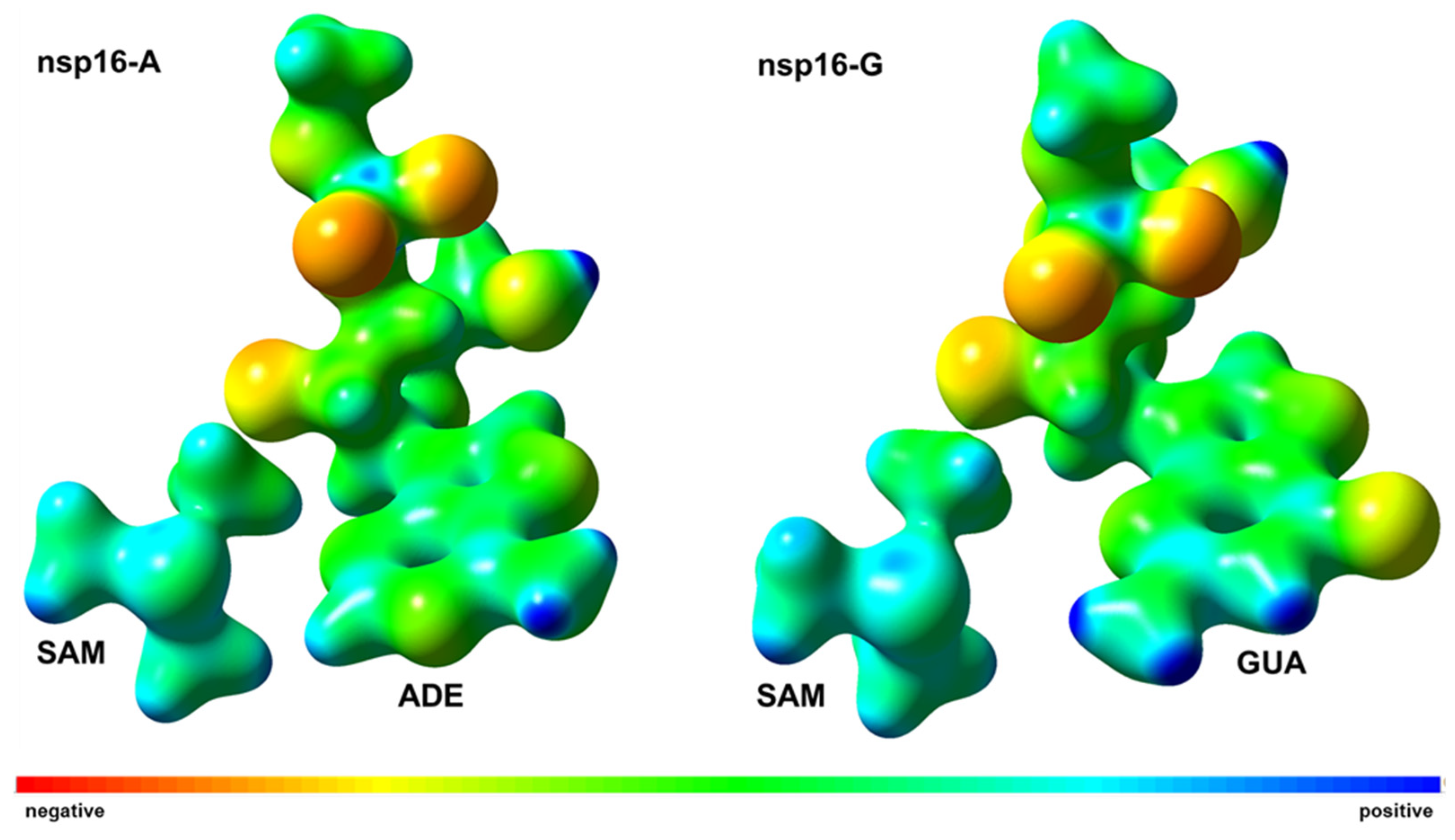
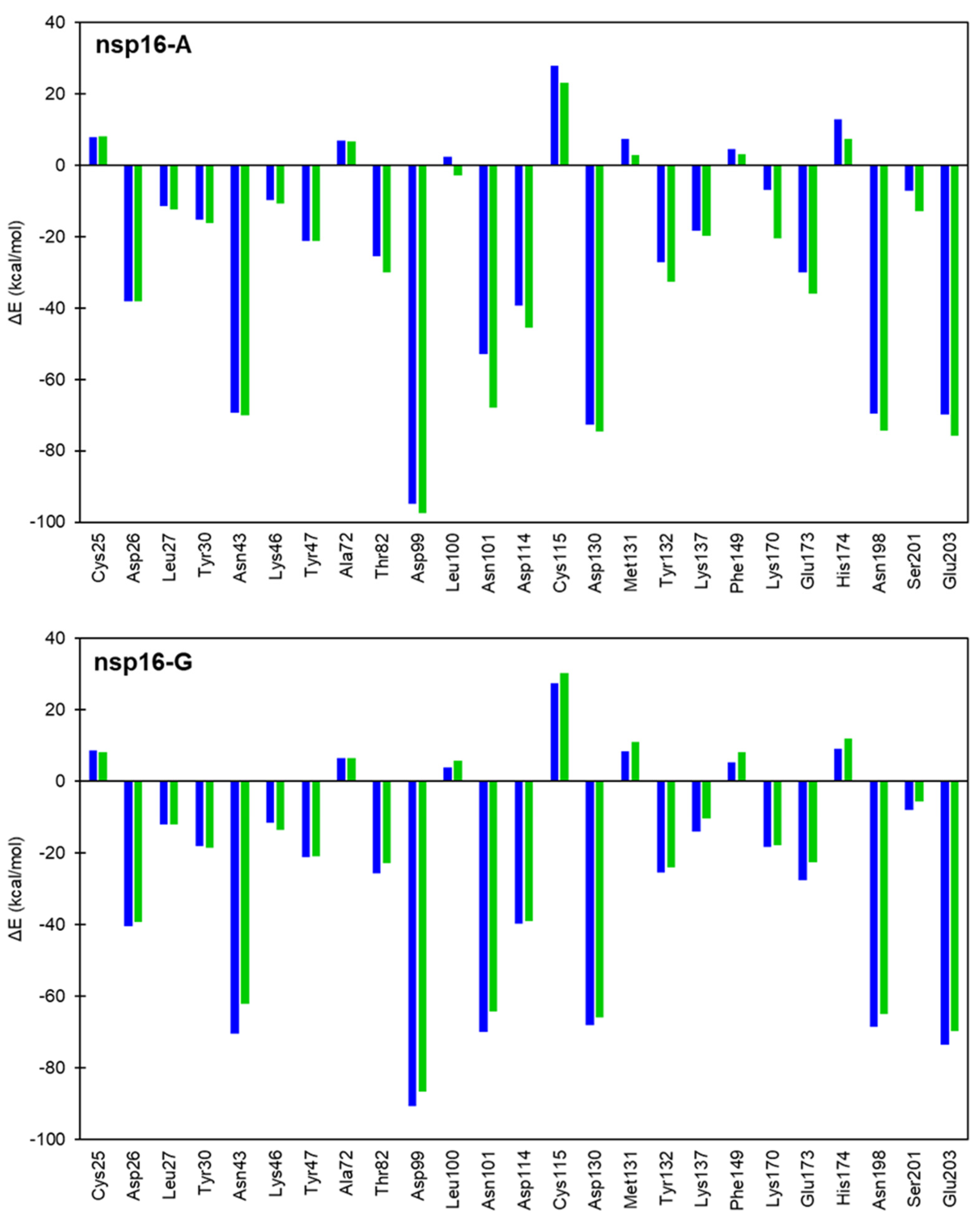
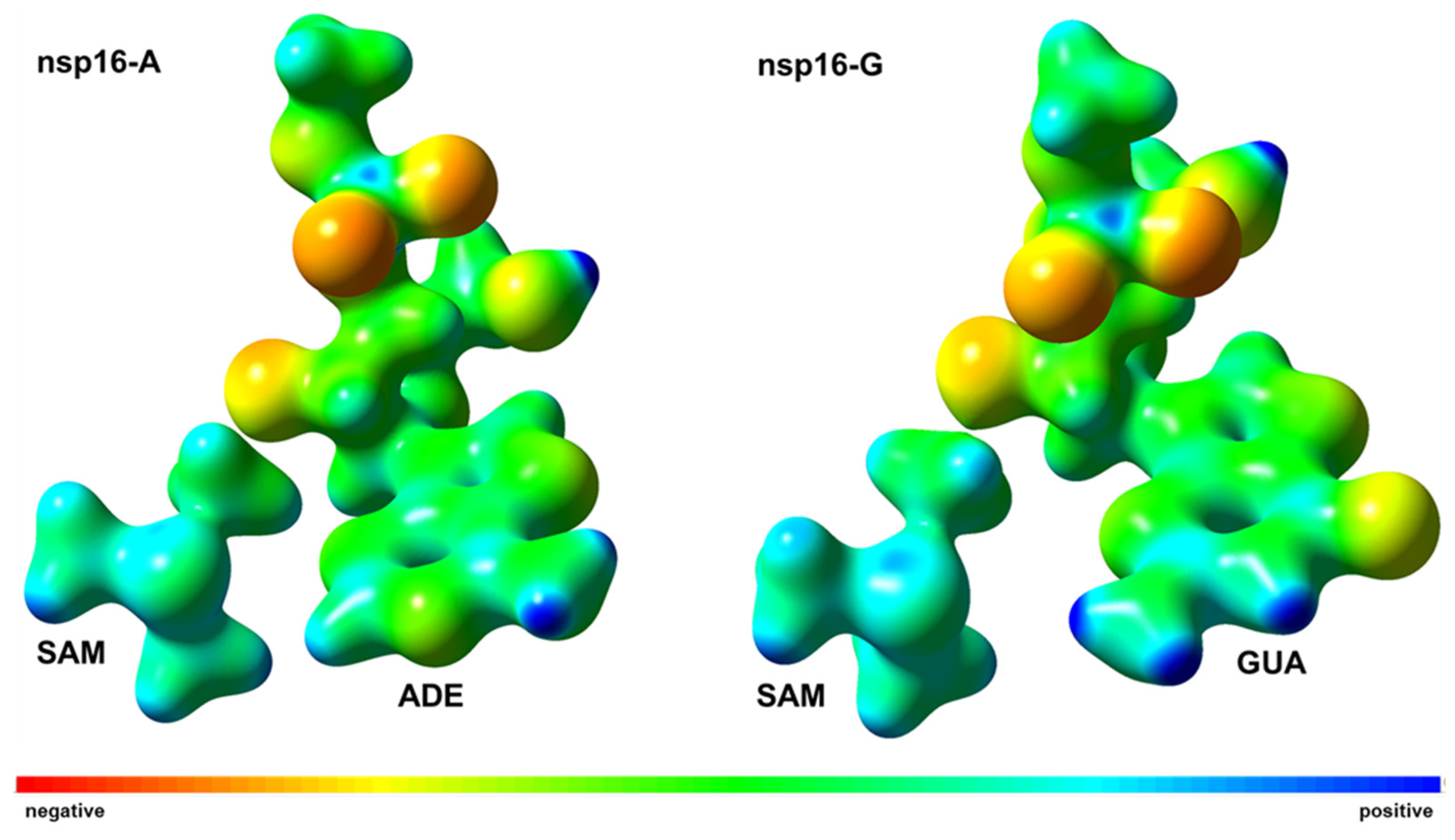
2.3. Interaction Energy Decomposition Analysis
3. Computational Methods
3.1. System Setup
3.2. QM/MM Calculations
3.3. Umbrella Sampling and Free Energy
3.4. High-Level QM/MM Corrections
3.5. Residual Decomposition Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. COVID-19 Weekly Epidemiological Update; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. Chapter three—The nonstructural proteins directing coronavirus RNA synthesis and processing. In Coronaviruses; Ziebuhr, J., Decroly, E., Snijder, E.J., Eds.; Academic Press: New York, NY, USA, 2016; Volume 96, pp. 59–126. ISBN 0065-3527. [Google Scholar]
- Yoneyama, M.; Fujita, T. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 2010, 20, 4–22. [Google Scholar] [CrossRef]
- Shuman, S. Structure, Mechanism, and Evolution of the mRNA Capping Apparatus; Academic Press: New York, NY, USA, 2000; Volume 66, pp. 1–40. ISBN 0079-6603. [Google Scholar]
- Gu, M.; Lima, C.D. Processing the message: Structural insights into capping and decapping mRNA. Curr. Opin. Struct. Biol. 2005, 15, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Diamond, M.S. Innate immune restriction and antagonism of viral RNA lacking 2′-O methylation. Virology 2015, 479, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Su, C.; Ke, M.; Jin, X.; Xu, L.; Zhang, Z.; Wu, A.; Sun, Y.; Tien, P.; Ahola, T.; et al. Biochemical and Structural Insights into the Mechanisms of SARS Coronavirus RNA Ribose 2 9 -O-Methylation by nsp16/nsp10 Protein Complex. PLoS Pathog. 2011, 7, e1002294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In Vitro Reconstitution of SARS-Coronavirus mRNA Cap Methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Decroly, E.; Imbert, I.; Coutard, B.; Bouvet, M.; Selisko, B.; Alvarez, K.; Gorbalenya, A.E.; Snijder, E.J.; Canard, B. Coronavirus Nonstructural Protein 16 Is a Cap-0 Binding Enzyme Possessing (Nucleoside-2′O)-Methyltransferase Activity. J. Virol. 2008, 82, 8071–8084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef] [PubMed]
- Aouadi, W.; Blanjoie, A.; Vasseur, J.-J.; Debart, F.; Canard, B.; Decroly, E. Binding of the Methyl Donor S-Adenosyl-l-Methionine to Middle East Respiratory Syndrome Coronavirus 2′-O-Methyltransferase nsp16 Promotes Recruitment of the Allosteric Activator nsp10. J. Virol. 2017, 91, 2217–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Brunzelle, J.; Satchell, K.J.F. High-resolution structures of the SARS-CoV-2 2′-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 2020, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.; Kiryukhina, O.; Wiersum, G.; Kim, Y.; Jedrzejczak, R.; Maltseva, N.; Endres, M.; et al. The crystal structure of nsp10-nsp16 heterodimer from SARS-CoV-2 in complex with S-adenosylmethionine. Biorxiv Prepr. Serv. Biol. 2020, 17, 1–22. [Google Scholar] [CrossRef]
- Viswanathan, T.; Arya, S.; Chan, S.-H.; Qi, S.; Dai, N.; Misra, A.; Park, J.-G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef]
- Benoni, R.; Krafcikova, P.; Baranowski, M.R.; Kowalska, J.; Boura, E.; Cahová, H. Substrate Specificity of SARS-CoV-2 Nsp10-Nsp16 Methyltransferase. Viruses 2021, 13, 1722. [Google Scholar] [CrossRef]
- Decroly, E.; Debarnot, C.; Ferron, F.; Bouvet, M.; Coutard, B.; Imbert, I.; Gluais, L.; Papageorgiou, N.; Sharff, A.; Bricogne, G.; et al. Crystal Structure and Functional Analysis of the SARS-Coronavirus RNA Cap 2′-O-Methyltransferase nsp10/nsp16 Complex. PLoS Pathog. 2011, 7, e1002059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Guo, D. Molecular mechanisms of coronavirus RNA capping and methylation. Virol. Sin. 2016, 31, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Menachery, V.D.; Yount, B.L.; Josset, L.; Gralinski, L.E.; Scobey, T.; Agnihothram, S.; Katze, M.G.; Baric, R.S. Attenuation and Restoration of Severe Acute Respiratory Syndrome Coronavirus Mutant Lacking 2′-O-Methyltransferase Activity. J. Virol. 2014, 88, 4251–4264. [Google Scholar] [CrossRef] [Green Version]
- Araújo, E.; Lima, A.H.; Lameira, J. Catalysis by solvation rather than the desolvation effect: Exploring the catalytic efficiency of SAM-dependent chlorinase. Phys. Chem. Chem. Phys. 2017, 19, 21350–21356. [Google Scholar] [CrossRef]
- Lameira, J.; Bora, R.P.; Chu, Z.T.; Warshel, A. Methyltransferases do not work by compression, cratic, or desolvation effects, but by electrostatic preorganization. Proteins Struct. Funct. Bioinform. 2015, 83, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Alves, N.; Lameira, J.; Roge, J.; Lima, A.H. Exploring Chloride Selectivity and Halogenase Regioselectivity of the SalL Enzyme through Quantum Mechanical/Molecular Mechanical Modeling. J. Chem. Inf. Modeling 2020, 60, 738–746. [Google Scholar] [CrossRef]
- Roca, M.; Andrés, J.; Moliner, V.; Tuñón, I.; Bertrán, J. On the Nature of the Transition State in Catechol O-Methyltransferase. A Complementary Study Based on Molecular Dynamics and Potential Energy Surface Explorations. J. Am. Chem. Soc. 2005, 127, 10648–10655. [Google Scholar] [CrossRef] [PubMed]
- Świderek, K.; Tuñón, I.; Williams, I.H.; Moliner, V. Insights on the Origin of Catalysis on Glycine N-Methyltransferase from Computational Modeling. J. Am. Chem. Soc. 2018, 140, 4327–4334. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef]
- O’Hagan, D.; Schmidberger, J.W. Enzymes that catalyse SN2 reaction mechanisms. Nat. Prod. Rep. 2010, 27, 900–918. [Google Scholar] [CrossRef]
- Minasov, G.; Rosas-Lemus, M.; Shuvalova, L.; Inniss, N.L.; Brunzelle, J.S.; Daczkowski, C.M.; Hoover, P.; Mesecar, A.D.; Satchell, K.J.F. Mn 2+ coordinates Cap-0-RNA to align substrates for efficient 2′- O -methyl transfer by SARS-CoV-2 nsp16. Sci. Signal. 2021, 14, 14. [Google Scholar] [CrossRef]
- Sk, M.F.; Jonniya, N.A.; Roy, R.; Poddar, S.; Kar, P. Computational Investigation of Structural Dynamics of SARS-CoV-2 Methyltransferase-Stimulatory Factor Heterodimer nsp16/nsp10 Bound to the Cofactor SAM. Front. Mol. Biosci. 2020, 7, 590165. [Google Scholar] [CrossRef]
- Lameira, J.; Alves, C.N.; Tuñón, I.; Martí, S.; Moliner, V. Enzyme molecular mechanism as a starting point to design new inhibitors: A theoretical study of O-GlcNAcase. J. Phys. Chem. B 2011, 115, 6764–6775. [Google Scholar] [CrossRef]
- Lim, S.P.; Wen, D.; Yap, T.L.; Yan, C.K.; Lescar, J.; Vasudevan, S.G. A scintillation proximity assay for dengue virus NS5 2′-O-methyltransferase—kinetic and inhibition analyses. Antivir. Res. 2008, 80, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. Nature of Forces between Large Molecules of Biological Interest. Nature 1948, 161, 707–709. [Google Scholar] [CrossRef] [Green Version]
- Amyes, T.L.; Richard, J.P. Specificity in transition state binding: The pauling model revisited. Biochemistry 2013, 52, 2021–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warshel, A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 1998, 273, 27035–27038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of p K a Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; MJG Frisch-Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Kamerlin, S.C.L.; Warshel, A. Multiscale modeling of biological functions. Phys. Chem. Chem. Phys. 2011, 13, 10401–10411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seabra, G.D.M.; Walker, R.C.; Elstner, M.; Case, D.A.; Roitberg, A.E. Implementation of the SCC-DFTB Method for Hybrid QM/MM Simulations within the Amber Molecular Dynamics Package. J. Phys. Chem. A 2007, 111, 5655–5664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warshel, A.; Weiss, R.M. An Empirical Valence Bond Approach for Comparing Reactions in Solutions and in Enzymes. J. Am. Chem. Soc. 1980, 102, 6218–6226. [Google Scholar] [CrossRef]
- Zhang, X.; Bruice, T.C. Catalytic Mechanism and Product Specificity of Rubisco Large Subunit Methyltransferase: QM/MM and MD Investigations. Biochemistry 2007, 46, 5505–5514. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Roux, B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995, 91, 275–282. [Google Scholar] [CrossRef]
- Grossfield, A. WHAM: The Weighted Histogram Analysis Method, Version 2011. Available online: http://membrane.urmc.rochester.edu/wordpress/?page_id=126 (accessed on 15 October 2021).
- Jencks, W.P. A primer for the Bema Hapothle. An empirical approach to the characterization of changing transition-state structures. Chem. Rev. 1985, 85, 511–527. [Google Scholar] [CrossRef]
- Hermann, J.C.; Hensen, C.; Ridder, L.; Mulholland, A.J.; Höltje, H.-D. Mechanisms of Antibiotic Resistance: QM/MM Modeling of the Acylation Reaction of a Class A β-Lactamase with Benzylpenicillin. J. Am. Chem. Soc. 2005, 127, 4454–4465. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, S.; Van der Kamp, M.W.; Ranaghan, K.E.; Mulholland, A.J.; Colombo, G. Understanding Complex Mechanisms of Enzyme Reactivity: The Case of Limonene-1,2-Epoxide Hydrolases. ACS Catal. 2018, 8, 5698–5707. [Google Scholar] [CrossRef] [Green Version]
- Major, D.T.; Gao, J. A Combined Quantum Mechanical and Molecular Mechanical Study of the Reaction Mechanism and α-Amino Acidity in Alanine Racemase. J. Am. Chem. Soc. 2006, 128, 16345–16357. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Viloca, M.; Truhlar, D.G.; Gao, J. Reaction-Path Energetics and Kinetics of the Hydride Transfer Reaction Catalyzed by Dihydrofolate Reductase. Biochemistry 2003, 42, 13558–13575. [Google Scholar] [CrossRef] [PubMed]
- Pierdominici-Sottile, G.; Roitberg, A.E. Proton Transfer Facilitated by Ligand Binding. An Energetic Analysis of the Catalytic Mechanism of Trypanosoma cruzi Trans-Sialidase. Biochemistry 2011, 50, 836–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierdominici-Sottile, G.; Cossio Pérez, R.; Galindo, J.F.; Palma, J. QM/MM Molecular Dynamics Study of the Galactopyranose Galactofuranose Reaction Catalysed by Trypanosoma cruzi UDP-Galactopyranose Mutase. PLoS ONE 2014, 9, e109559. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | ΔG‡ (DFTB3/MM) | ΔG° (DFTB3/MM) | ΔG‡ (M062X/MM) | ΔG° (M062X/MM) |
|---|---|---|---|---|
| nsp16-A | 13.9 | −16.7 | 28.3 | −4.7 |
| nsp16-G | 11.7 | −25.8 | 32.6 | 0.9 |
| nsp16-A | nsp16-G | |||||
|---|---|---|---|---|---|---|
| RS | TS | PS | RS | TS | PS | |
| Distances (Å) | ||||||
| d(O1–C1) | 2.76 (0.04) | 2.05 (0.04) | 1.47 (0.03) | 2.84 (0.05) | 2.10 (0.04) | 1.46 (0.03) |
| d(C1–S1) | 1.85 (0.04) | 2.36 (0.04) | 3.18 (0.04) | 1.84 (0.04) | 2.31 (0.04) | 3.27 (0.04) |
| Charges (a.u.) | ||||||
| O1 | −1.08 (0.03) | −0.80 (0.03) | −0.42 (0.02) | −1.10 (0.02) | −0.83 (0.03) | −0.42 (0.02) |
| C1 | −0.26 (0.04) | −0.11 (0.02) | 0.00 (0.02) | −0.25 (0.04) | −0.12 (0.02) | 0.02 (0.02) |
| S1 | 0.35 (0.03) | 0.03 (0.03) | −0.20 (0.03) | 0.36 (0.03) | 0.07 (0.03) | −0.18 (0.02) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, J.R.A.; Urban, J.; Araújo, E.; Lameira, J.; Moliner, V.; Alves, C.N. Exploring the Catalytic Mechanism of the RNA Cap Modification by nsp16-nsp10 Complex of SARS-CoV-2 through a QM/MM Approach. Int. J. Mol. Sci. 2022, 23, 300. https://doi.org/10.3390/ijms23010300
Silva JRA, Urban J, Araújo E, Lameira J, Moliner V, Alves CN. Exploring the Catalytic Mechanism of the RNA Cap Modification by nsp16-nsp10 Complex of SARS-CoV-2 through a QM/MM Approach. International Journal of Molecular Sciences. 2022; 23(1):300. https://doi.org/10.3390/ijms23010300
Chicago/Turabian StyleSilva, José Rogério A., Jaime Urban, Edson Araújo, Jerônimo Lameira, Vicent Moliner, and Cláudio Nahum Alves. 2022. "Exploring the Catalytic Mechanism of the RNA Cap Modification by nsp16-nsp10 Complex of SARS-CoV-2 through a QM/MM Approach" International Journal of Molecular Sciences 23, no. 1: 300. https://doi.org/10.3390/ijms23010300
APA StyleSilva, J. R. A., Urban, J., Araújo, E., Lameira, J., Moliner, V., & Alves, C. N. (2022). Exploring the Catalytic Mechanism of the RNA Cap Modification by nsp16-nsp10 Complex of SARS-CoV-2 through a QM/MM Approach. International Journal of Molecular Sciences, 23(1), 300. https://doi.org/10.3390/ijms23010300