
IL-25 Induced ROS-Mediated M2 Macrophage Polarization via AMPK-Associated Mitophagy

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
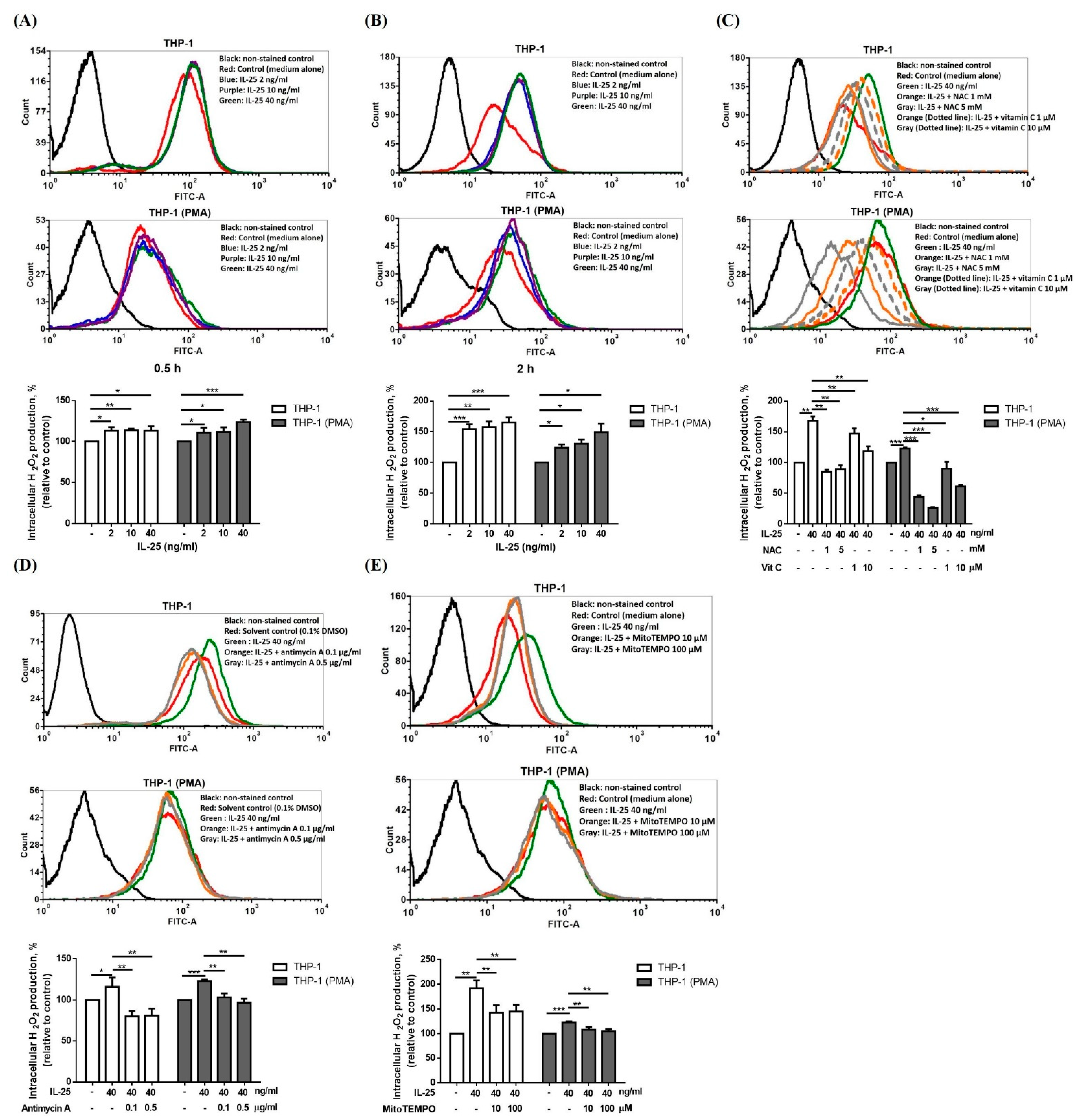
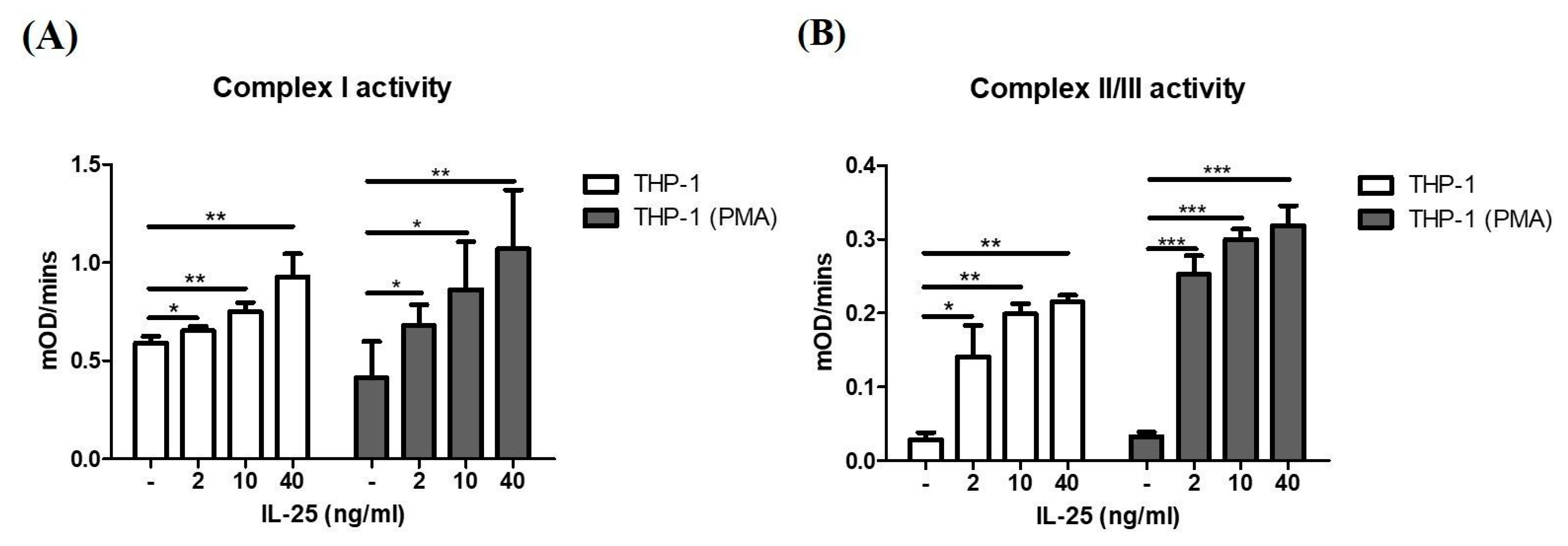
2.1. IL-25 Increased Mitochondrial Complex I and II/III Activity
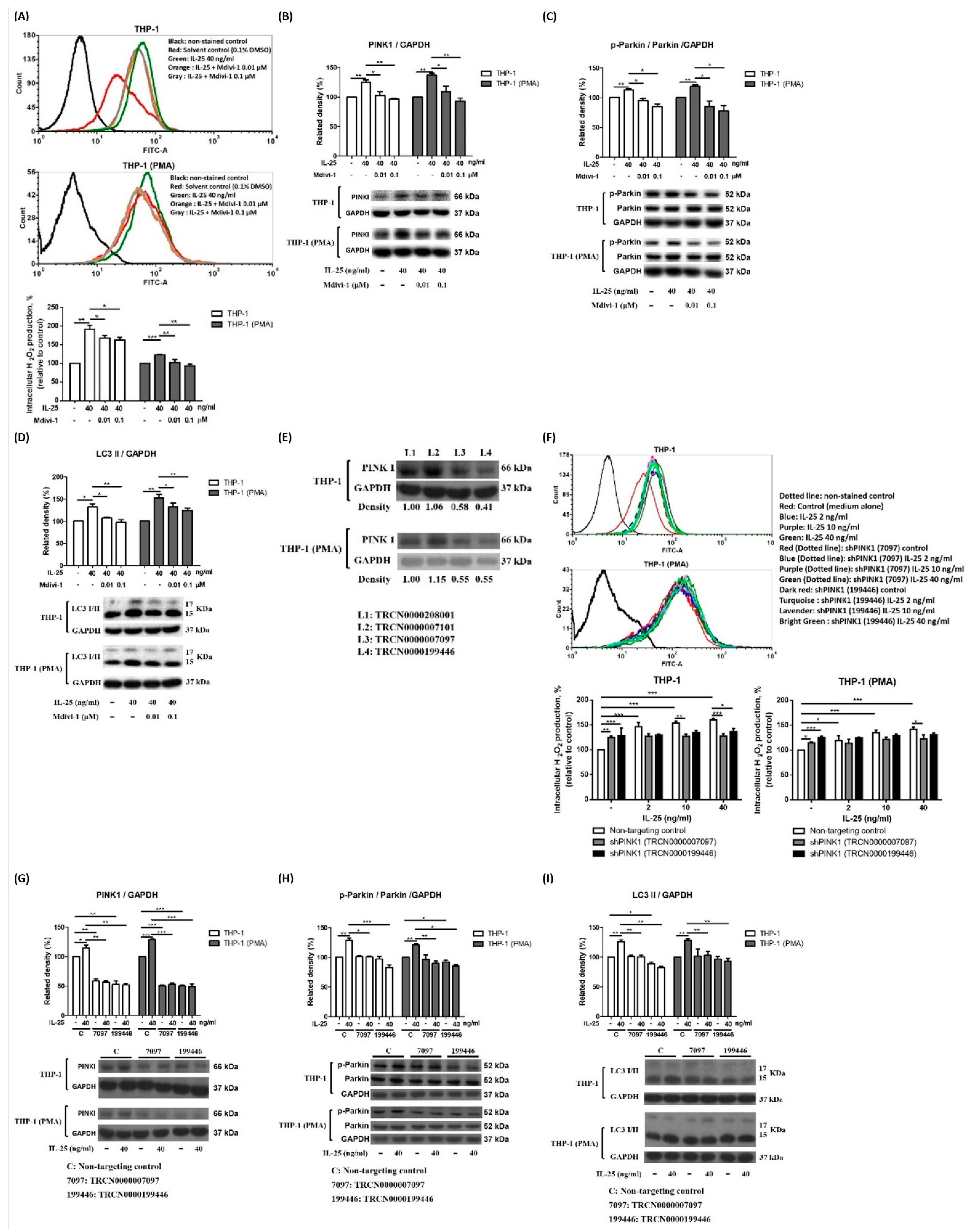
2.2. IL-25 Induced AMPK Activation and Mitophagy-Related Proteins Expression
2.3. IL-25 Induced Mitophagy through the ROS-AMPK Pathway
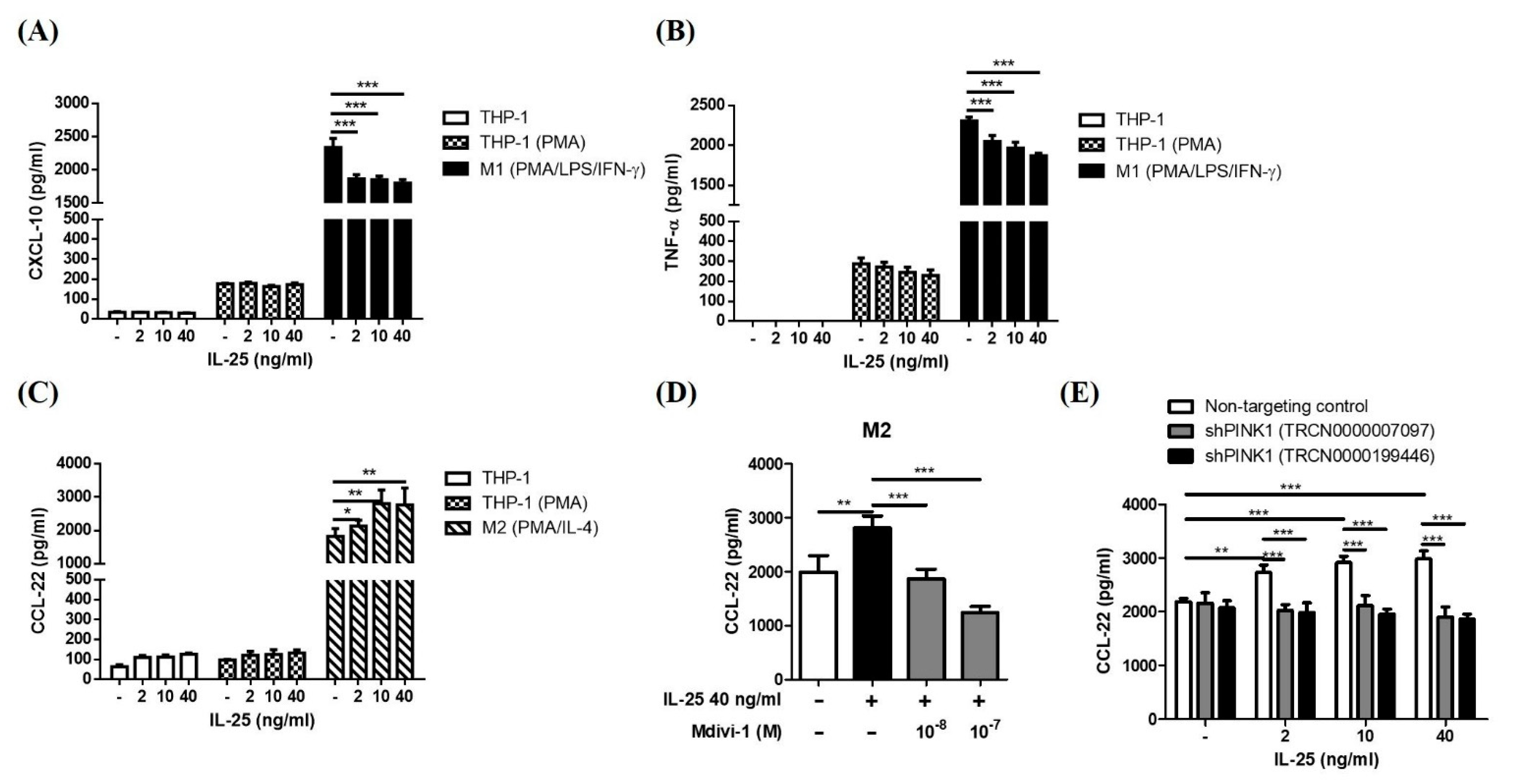
2.4. IL-25-Induced Mitophagy Was Associated with an M2 Macrophage Polarization Shift
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Measurement of ROS Production
4.3. Measurement of Mitochondrial Complex Activity
4.4. Western Blotting
4.5. Confocal Immunofluorescence Microscopy
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. PINK1 Knockdown
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saenz, S.A.; Taylor, B.C.; Artis, D. Welcome to the neighborhood: Epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 2008, 226, 172–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fux, M.; Pecaric-Petkovic, T.; Odermatt, A.; Hausmann, O.V.; Lorentz, A.; Bischoff, S.C.; Virchow, J.C.; Dahinden, C.A. IL-33 is a mediator rather than a trigger of the acute allergic response in humans. Allergy 2014, 69, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Kabata, H.; Moro, K.; Koyasu, S.; Asano, K. Group 2 innate lymphoid cells and asthma. Allergol. Int. 2015, 64, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Divekar, R.; Kita, H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Hams, E.; Armstrong, M.E.; Barlow, J.L.; Saunders, S.P.; Schwartz, C.; Cooke, G.; Fahy, R.J.; Crotty, T.B.; Hirani, N.; Flynn, R.J.; et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.; Otenbaker, N.P.; Rose, B.A.; Salisbury, K.S. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 2013, 56, 57–63. [Google Scholar] [CrossRef]
- Lee, I.T.; Yang, C.M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 2012, 84, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Ding, W.X. Targeting Pink1-Parkin-mediated mitophagy for treating liver injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Osborn-Heaford, H.L.; Ryan, A.J.; Murthy, S.; Racila, A.M.; He, C.; Sieren, J.C.; Spitz, D.R.; Carter, A.B. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J. Biol. Chem. 2012, 287, 3301–3312. [Google Scholar] [CrossRef] [Green Version]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Sun, Y.; Wang, W.; Sun, Y. Interleukin (IL)-25: Pleiotropic roles in asthma. Respirology 2016, 21, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays 2015, 37, 867–876. [Google Scholar] [CrossRef]
- Bujak, A.L.; Crane, J.D.; Lally, J.S.; Ford, R.J.; Kang, S.J.; Rebalka, I.A.; Green, A.E.; Kemp, B.E.; Hawke, T.J.; Schertzer, J.D.; et al. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 2015, 21, 883–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowczyk, J.; Shutova, M.; Brembilla, N.C.; Boehncke, W.H. IL-25 (IL-17E) in epithelial immunology and pathophysiology. J. Allergy Clin. Immunol. 2021, 148, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Angkasekwinai, P.; Lu, N.; Voo, K.S.; Arima, K.; Hanabuchi, S.; Hippe, A.; Corrigan, C.J.; Dong, C.; Homey, B.; et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J. Exp. Med. 2007, 204, 1837–1847. [Google Scholar] [CrossRef] [Green Version]
- Tworek, D.; Smith, S.G.; Salter, B.M.; Baatjes, A.J.; Scime, T.; Watson, R.; Obminski, C.; Gauvreau, G.M.; O’Byrne, P.M. IL-25 receptor expression on airway dendritic cells after allergen challenge in subjects with asthma. Am. J. Respir. Crit. Care Med. 2016, 193, 957–964. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.; Rahman, I. Oxidative stress in asthma and COPD: Antioxidants as a therapeutic strategy. Pharmacol. Ther. 2006, 111, 476–494. [Google Scholar] [CrossRef]
- Sugiura, H.; Ichinose, M. Oxidative and nitrative stress in bronchial asthma. Antioxid. Redox Signal. 2008, 10, 785–797. [Google Scholar] [CrossRef]
- Wood, L.G.; Wark, P.A.; Garg, M.L. Antioxidant and anti-inflammatory effects of resveratrol in airway disease. Antioxid. Redox Signal. 2010, 13, 1535–1548. [Google Scholar] [CrossRef]
- Reinmuth-Selzle, K.; Kampf, C.J.; Lucas, K.; Lang-Yona, N.; Frohlich-Nowoisky, J.; Shiraiwa, M.; Lakey, P.S.J.; Lai, S.; Liu, F.; Kunert, A.T.; et al. Air pollution and climate change effects on allergies in the anthropocene: Abundance, interaction, and modification of allergens and adjuvants. Environ. Sci. Technol. 2017, 51, 4119–4141. [Google Scholar] [CrossRef]
- Jiang, L.; Diaz, P.T.; Best, T.M.; Stimpfl, J.N.; He, F.; Zuo, L. Molecular characterization of redox mechanisms in allergic asthma. Ann. Allergy Asthma Immunol. 2014, 113, 137–142. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The reactive oxygen species in macrophage polarization: Reflecting its dual role in progression and treatment of human diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, J.; Jayaraman, A.; Jackson, D.J.; Macintyre, J.D.R.; Edwards, M.R.; Walton, R.P.; Zhu, J.; Man Ching, Y.; Shamji, B.; Edwards, M.; et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci. Transl. Med. 2014, 6, 256ra134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.Y.; Bentley, J.K.; Chung, Y.; Lei, J.; Steenrod, J.M.; Chen, Q.; Sajjan, U.S.; Hershenson, M.B. Neonatal rhinovirus induces mucous metaplasia and airways hyperresponsiveness through IL-25 and type 2 innate lymphoid cells. J. Allergy Clin. Immunol. 2014, 134, 429–439. [Google Scholar] [CrossRef]
- Petersen, B.C.; Dolgachev, V.; Rasky, A.; Lukacs, N.W. IL-17E (IL-25) and IL-17RB promote respiratory syncytial virus-induced pulmonary disease. J. Leukoc. Biol. 2014, 95, 809–815. [Google Scholar] [CrossRef]
- Aguilera-Aguirre, L.; Bacsi, A.; Saavedra-Molina, A.; Kurosky, A.; Sur, S.; Boldogh, I. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 2009, 183, 5379–5387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: Involvement of reactive oxygen species and cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Redout, E.M.; Wagner, M.J.; Zuidwijk, M.J.; Boer, C.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 2007, 75, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duarte, E.; Cortes-Rojo, C.; Sanchez-Briones, L.A.; Campos-Garcia, J.; Saavedra-Molina, A.; Delgado-Enciso, I.; Lopez-Lemus, U.A.; Montoya-Perez, R. Nicorandil affects mitochondrial respiratory chain function by increasing complex III activity and ROS production in skeletal muscle mitochondria. J. Membr. Biol. 2020, 253, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Green, D.R. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar] [PubMed] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, N.; Li, L.; Chen, S.; Wang, T. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016, 7, e2501. [Google Scholar] [CrossRef]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial function, and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Kirkpatrick, S.S.; Davis, B.J.; Nelson, J.S.; Wiles, W.G.T.; Schlattner, U.; Neumann, D.; Brownlee, M.; Freeman, M.B.; Goldman, M.H. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J. Biol. Chem. 2004, 279, 43940–43951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Wang, Q.; Song, P.; Zhu, Y.; Zou, M.H. Redox regulation of the AMP-activated protein kinase. PLoS ONE 2010, 5, e15420. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Irrcher, I.; Adhihetty, P.J.; Sheehan, T.; Joseph, A.M.; Hood, D.A. PPARgamma coactivator-1alpha expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Cell Physiol. 2003, 284, C1669–C1677. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Feng, J.; Li, L.; Ou, Z.; Li, Q.; Gong, B.; Zhao, Z.; Qi, W.; Zhou, T.; Zhong, J.; Cai, W.; et al. IL-25 stimulates M2 macrophage polarization and thereby promotes mitochondrial respiratory capacity and lipolysis in adipose tissues against obesity. Cell. Immunol. 2018, 15, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.P. Point: Hypoxic pulmonary vasoconstriction is mediated by increased production of reactive oxygen species. J. Appl. Physiol. 2006, 101, 993–995, discussion 999. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Liu, R.; Zhang, Z.; Chen, Z.; He, J.; Liu, Y. Mitochondrial division inhibitor 1 attenuates mitophagy in a rat model of acute lung injury. Biomed. Res. Int. 2019, 2019, 2193706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.H.; Qiu, B.Q.; Ma, M.; Zhang, R.; Hsu, S.J.; Liu, H.H.; Chen, J.; Gao, D.M.; Cui, J.F.; Ren, Z.G.; et al. Suppressing DRP1-mediated mitochondrial fission and mitophagy increases mitochondrial apoptosis of hepatocellular carcinoma cells in the setting of hypoxia. Oncogenesis 2020, 9, 67. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, M.-L.; Tsai, Y.-G.; Lin, Y.-C.; Hsu, Y.-L.; Chen, Y.-T.; Tsai, M.-K.; Liao, W.-T.; Lin, Y.-C.; Hung, C.-H. IL-25 Induced ROS-Mediated M2 Macrophage Polarization via AMPK-Associated Mitophagy. Int. J. Mol. Sci. 2022, 23, 3. https://doi.org/10.3390/ijms23010003
Tsai M-L, Tsai Y-G, Lin Y-C, Hsu Y-L, Chen Y-T, Tsai M-K, Liao W-T, Lin Y-C, Hung C-H. IL-25 Induced ROS-Mediated M2 Macrophage Polarization via AMPK-Associated Mitophagy. International Journal of Molecular Sciences. 2022; 23(1):3. https://doi.org/10.3390/ijms23010003
Chicago/Turabian StyleTsai, Mei-Lan, Yi-Giien Tsai, Yu-Chih Lin, Ya-Ling Hsu, Yi-Ting Chen, Ming-Kai Tsai, Wei-Ting Liao, Yi-Ching Lin, and Chih-Hsing Hung. 2022. "IL-25 Induced ROS-Mediated M2 Macrophage Polarization via AMPK-Associated Mitophagy" International Journal of Molecular Sciences 23, no. 1: 3. https://doi.org/10.3390/ijms23010003
APA StyleTsai, M.-L., Tsai, Y.-G., Lin, Y.-C., Hsu, Y.-L., Chen, Y.-T., Tsai, M.-K., Liao, W.-T., Lin, Y.-C., & Hung, C.-H. (2022). IL-25 Induced ROS-Mediated M2 Macrophage Polarization via AMPK-Associated Mitophagy. International Journal of Molecular Sciences, 23(1), 3. https://doi.org/10.3390/ijms23010003