
Histone Modifications and Their Targeting in Lymphoid Malignancies

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
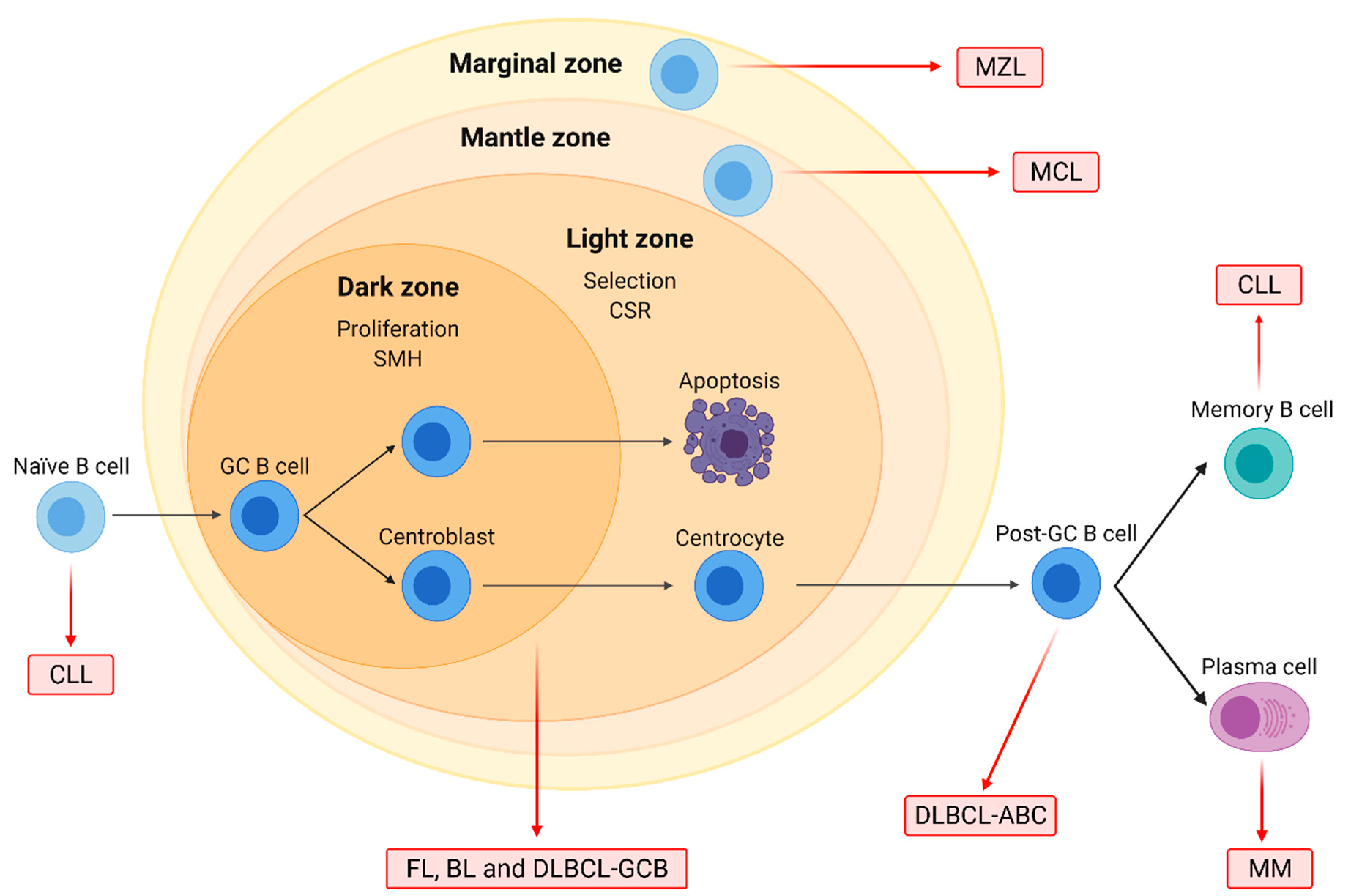
1.1. Characteristics of the Main B-NHL Subtypes
1.2. Epigenetic Modification of Histone Proteins in B-NHL
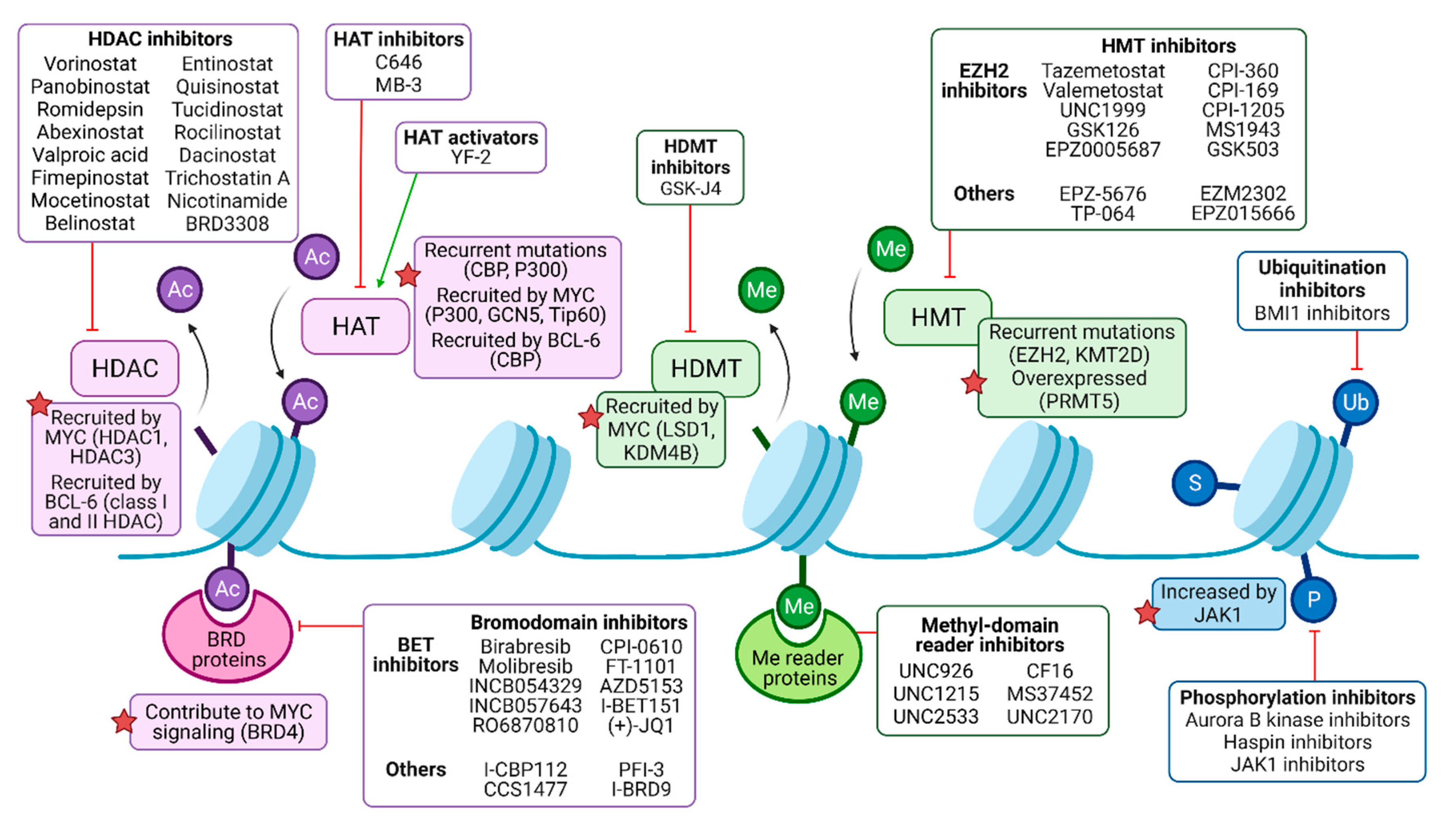
2. The Pharmacological Targeting of Histone Acetylation
2.1. Extensive Preclinical Development of HDAC Inhibitors
2.2. Preclinical Evaluation of HATs as Targets for B-NHL Therapy
2.3. Experimental Insight in the Utility BET Inhibitors
2.4. Ongoing Clinical Development of HDACi, BETi and HATi
2.4.1. HDACi
2.4.2. HATi
2.4.3. BETi
3. Targeting Histone Methylation and Some Other PTMs
3.1. Preclinical Development of Histone Methyl Transferase and Demethylase Inhibitors
3.2. EZH2 Inhibitors in Clinical Development
3.3. Targeting Other Posttranslational Modifications of Histones
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Elsen, M.B.; Davis, R.E.; Ma, C.L.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Gascoyne, R.D.; Campo, E.; Jaffe, E.S.; Chan, W.C. Diffuse Large B-Cell Lymphoma, NOS. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 291–297. [Google Scholar]
- Liu, Y.; Barta, S.K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Harris, N.L.; Swerdlow, S.H.; Ott, G.; Nathwani, B.N.; de Jong, D.; Yoshino, T.; Spagnolo, D.; Gascoyne, R.D. Follicular lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 266–277. [Google Scholar]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Prim. 2019, 5, 83. [Google Scholar] [CrossRef]
- Dreyling, M.; Ghielmini, M.; Rule, S.; Salles, G.A.; Vitolo, U.; Ladetto, M. Newly diagnosed and relapsed follicular lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v83–v90. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, L.; Campo, E.; Stein, H.; Harris, N.L.; Jaffe, E.S.; Kluin, P.M. Burkitt Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 330–334. [Google Scholar]
- Casulo, C.; Friedberg, J.W. Burkitt lymphoma- a rare but challenging lymphoma. Best Pract. Res. Clin. Haematol. 2018, 31, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Piris, M.A.; Isaacson, P.G.; Swerdlow, S.H.; Thieblemont, C.; Pittaluga, S.; Rossi, D.; Harris, N.L. Splenic Marginal Zone Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 223–225. [Google Scholar]
- Campo, E.; Pileri, S.A.; Jaffe, E.S.; Nathwani, B.N.; Stein, H.; Müller-Hermelink, H.K. Nodal Marginal Zone Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 263–265. [Google Scholar]
- Cook, J.R.; Isaacson, P.G.; Chott, A.; Nakamura, S.; Müller-Hermelink, H.K.; Harris, N.L.; Swerdlow, S.H. Extranodal Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissue (MALT Lymphoma). In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 259–262. [Google Scholar]
- Sindel, A.; Al-Juhaishi, T.; Yazbeck, V. Marginal Zone Lymphoma: State-of-the-Art Treatment. Curr. Treat. Options Oncol. 2019, 20, 90. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Seto, M.; Müller-Hermelink, H.K. Mantle Cell Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 285–290. [Google Scholar]
- Roué, G.; Sola, B. Management of drug resistance in mantle cell lymphoma. Cancers 2020, 12, 1565. [Google Scholar] [CrossRef]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef]
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, E.M.; Dekker, J. Mechanisms and Functions of Chromosome Compartmentalization. Trends Biochem. Sci. 2020, 45, 385–396. [Google Scholar] [CrossRef]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef]
- Shaban, H.A.; Barth, R.; Recoules, L.; Bystricky, K. Hi-D: Nanoscale mapping of nuclear dynamics in single living cells. Genome Biol. 2020, 21, 1–21. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Garcia-Saez, I.; Menoni, H.; Boopathi, R.; Shukla, M.S.; Soueidan, L.; Noirclerc-Savoye, M.; Le Roy, A.; Skoufias, D.A.; Bednar, J.; Hamiche, A.; et al. Structure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation. Mol. Cell 2018, 72, 902–915.e7. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Tropberger, P.; Schneider, R. Scratching the (lateral) surface of chromatin regulation by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef]
- Bosch-Presegué, L.; Vaquero, A. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015, 282, 1745–1767. [Google Scholar] [CrossRef]
- Morera, L.; Lübbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 16. [Google Scholar] [CrossRef]
- Blecua, P.; Martinez-Verbo, L.; Esteller, M. The DNA methylation landscape of hematological malignancies: An update. Mol. Oncol. 2020, 14, 1616–1639. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Karkhanis, V.; Tae, S.; Yan, F.; Smith, P.; Ayers, L.W.; Agostinelli, C.; Pileri, S.; Denis, G.V.; Baiocchi, R.A.; et al. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) Silencing. J. Biol. Chem. 2013, 288, 35534–35547. [Google Scholar] [CrossRef]
- Poole, C.J.; van Riggelen, J. MYC—master regulator of the cancer epigenome and transcriptome. Genes 2017, 8, 142. [Google Scholar] [CrossRef]
- Fujita, N.; Jaye, D.L.; Geigerman, C.; Akyildiz, A.; Mooney, M.R.; Boss, J.M.; Wade, P.A. MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell 2004, 119, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Lemercier, C.; Brocard, M.P.; Puvion-Dutilleul, F.; Kao, H.Y.; Albagli, O.; Khochbin, S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 2002, 277, 22045–22052. [Google Scholar] [CrossRef]
- Gearhart, M.D.; Corcoran, C.M.; Wamstad, J.A.; Bardwell, V.J. Polycomb Group and SCF Ubiquitin Ligases Are Found in a Novel BCOR Complex That Is Recruited to BCL6 Targets. Mol. Cell. Biol. 2006, 26, 6880–6889. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Diesch, J.; Zwick, A.; Garz, A.K.; Palau, A.; Buschbeck, M.; Götze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenetics 2016, 8, 1–11. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated Silencing of MYC-Mediated miR-29 by HDAC3 and EZH2 as a Therapeutic Target of Histone Modification in Aggressive B-Cell Lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol. 2012, 6, 637–656. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of TSA and SAHA. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- Guan, X.; Lin, P.; Knoll, E.; Chakrabarti, R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: Computational and experimental studies. PLoS ONE 2014, 9, e107729. [Google Scholar]
- Gertz, M.; Fischer, F.; Nguyen, G.T.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Horinouchi, S.; Beppu, T. Rn Trichostatin A and trapoxin: Novel. BioEssays 1995, 17, 423–430. [Google Scholar] [CrossRef]
- Murugan, K.; Sangeetha, S.; Ranjitha, S.; Vimala, A.; Al-Sohaibani, S.; Rameshkumar, G. HDACiDB: A database for histone deacetylase inhibitors. Drug Des. Devel. Ther. 2015, 9, 2257–2264. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef]
- Liu, J.; Yu, Y.; Kelly, J.; Sha, D.; Alhassan, A.B.; Yu, W.; Maletic, M.M.; Duffy, J.L.; Klein, D.J.; Holloway, M.K.; et al. Discovery of Highly Selective and Potent HDAC3 Inhibitors Based on a 2-Substituted Benzamide Zinc Binding Group. ACS Med. Chem. Lett. 2020, 11, 2476–2483. [Google Scholar] [CrossRef]
- Leonhardt, M.; Sellmer, A.; Krämer, O.H.; Dove, S.; Elz, S.; Kraus, B.; Beyer, M.; Mahboobi, S. Design and biological evaluation of tetrahydro-β-carboline derivatives as highly potent histone deacetylase 6 (HDAC6) inhibitors. Eur. J. Med. Chem. 2018, 152, 329–357. [Google Scholar] [CrossRef]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunglubulin enhancers iduces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef]
- Sidman, C.L.; Denial, T.M.; Marshall, J.D.; Roths, J.B. Multiple mechanisms of tumorigenesis in E??-myc transgenic mice. Cancer Res. 1993, 53, 1665–1669. [Google Scholar]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Schleich, K.; Kase, J.; Dörr, J.R.; Trescher, S.; Bhattacharya, A.; Yu, Y.; Wailes, E.M.; Fan, D.N.Y.; Lohneis, P.; Milanovic, M.; et al. H3K9me3-mediated epigenetic regulation of senescence in mice predicts outcome of lymphoma patients. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Stubbs, M.C.; Kim, W.; Bariteau, M.; Davis, T.; Vempati, S.; Minehart, J.; Witkin, M.; Qi, J.; Krivtsov, A.V.; Bradner, J.E.; et al. Selective inhibition of HDAC1 and HDAC2 as a potential therapeutic option for B-ALL. Clin. Cancer Res. 2015, 21, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, R.K.; Newbold, A.; Whitecross, K.F.; Cluse, L.A.; Frew, A.J.; Ellis, L.; Williams, S.; Wiegmans, A.P.; Dear, A.E.; Scott, C.L.; et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc. Natl. Acad. Sci. USA 2007, 104, 8071–8076. [Google Scholar] [CrossRef]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Mehdipour, P.; Cluse, L.A.; Falkenberg, K.J.; Wang, E.; Roth, M.; Santoro, F.; Vidacs, E.; Stanley, K.; House, C.M.; et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 2015, 126, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, D.; Zhou, J. Histone Deacetylase 6 as a Therapeutic Target in B cell-associated Hematological Malignancies. Front. Pharmacol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Amengual, J.E.; Johannet, P.; Lombardo, M.; Zullo, K.; Hoehn, D.; Bhagat, G.; Scotto, L.; Jirau-Serrano, X.; Radeski, D.; Heinen, J.; et al. Dual targeting of protein degradation pathways with the selective HDAC6 inhibitor ACY-1215 and bortezomib is synergistic in lymphoma. Clin. Cancer Res. 2015, 21, 4663–4675. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Mägdefrau, A.-S.; Kleemann, M.; Beyer, M.; Linke, K.; Hansen, L.; Schaffer, A.-M.; Hoffmann, M.E.; Poepsel, S.; Heyd, F.; et al. Targeting the MYC interaction network in B-cell lymphoma via histone deacetylase 6 inhibition. BioRxiv 2021. [Google Scholar] [CrossRef]
- Bobrowska, A.; Paganetti, P.; Matthias, P.; Bates, G.P. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2011, 6, e20696. [Google Scholar] [CrossRef] [PubMed]
- Rosenjack, J.; Hodges, C.A.; Darrah, R.J.; Kelley, T.J. HDAC6 depletion improves cystic fibrosis mouse airway responses to bacterial challenge. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC inhibitors exert anti-myeloma effects through multiple modes of action. Cancers 2019, 11, 475. [Google Scholar] [CrossRef]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. AID-Dependent Activation of a MYC Transgene Induces Multiple Myeloma in a Conditional Mouse Model of Post-Germinal Center Malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef]
- Chesi, M.; Matthews, G.M.; Garbitt, V.M.; Palmer, S.E.; Shortt, J.; Lefebure, M.; Stewart, A.K.; Johnstone, R.W.; Leif Bergsagel, P. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 2012, 120, 376–385. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.-C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Götz, R.; Doose, S.; Sauer, M.; Rodríguez-Gil, A.; Nerreter, T.; Kortüm, K.M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M.; et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2021, 35, 201–214. [Google Scholar] [CrossRef]
- Bobrowicz, M.; Dwojak, M.; Pyrzynska, B.; Stachura, J.; Muchowicz, A.; Berthel, E.; Dalla-Venezia, N.; Kozikowski, M.; Siernicka, M.; Miazek, N.; et al. HDAC6 inhibition upregulates CD20 levels and increases the efficacy of anti-CD20 monoclonal antibodies. Blood 2017, 130, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Mondello, P.; Tadros, S.; Teater, M.; Fontan, L.; Chang, A.Y.; Jain, N.; Yang, H.; Singh, S.; Ying, H.Y.; Chu, C.S.; et al. Selective inhibition of HDAC3 targets synthetic vulnerabilities and activates immune surveillance in lymphoma. Cancer Discov. 2020, 10, 440–459. [Google Scholar] [CrossRef]
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 1–17. [Google Scholar]
- Wang, X.; Waschke, B.C.; Woolaver, R.A.; Chen, Z.; Zhang, G.; Piscopio, A.D.; Liu, X.; Wang, J.H. Histone deacetylase inhibition sensitizes PD1 blockade-resistant b-cell lymphomas. Cancer Immunol. Res. 2019, 7, 1318–1331. [Google Scholar] [CrossRef]
- Amengual, J.E.; Clark-Garvey, S.; Kalac, M.; Scotto, L.; Marchi, E.; Neylon, E.; Johannet, P.; Wei, Y.; Zain, J.; O’Connor, O.A. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood 2013, 122, 2104–2113. [Google Scholar] [CrossRef]
- Varano, G.; Raffel, S.; Sormani, M.; Zanardi, F.; Lonardi, S.; Zasada, C.; Perucho, L.; Petrocelli, V.; Haake, A.; Lee, A.K.; et al. The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3ß inhibition. Nature 2017, 546, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.Y.; Hwang, S.H.; Kwon, K.S.; Kim, K.R.; Choi, H.N.; Lee, N.R.; Kwak, J.Y.; Park, B.H.; Park, H.S.; Chung, M.J.; et al. SIRT1 expression is associated with poor prognosis of diffuse large B-cell lymphoma. Am. J. Surg. Pathol. 2008, 32, 1523–1531. [Google Scholar] [CrossRef]
- He, M.; Tan, B.; Vasan, K.; Yuan, H.; Cheng, F.; Ramos da Silva, S.; Lu, C.; Gao, S.J. SIRT1 and AMPK pathways are essential for the proliferation and survival of primary effusion lymphoma cells. J. Pathol. 2017, 242, 309–321. [Google Scholar] [CrossRef]
- Yu, W.; Denu, R.A.; Krautkramer, K.A.; Grindle, K.M.; Yang, D.T.; Asimakopoulos, F.; Hematti, P.; Denu, J.M. Loss of SIRT3 provides growth advantage for B cell malignancies. J. Biol. Chem. 2016, 291, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chiang, Y.L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e9. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhu, Y.; Park, S.H.; Liu, G.; O’Brien, J.; Jiang, H.; Gius, D. SIRT3-mediated dimerization of IDH2 directs cancer cell metabolism and tumor growth. Cancer Res. 2017, 77, 3990–3999. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Zhang, Y.; Fang, X.; Chen, N.; Zhou, X.; Wang, X. Sirt6 promotes tumorigenesis and drug resistance of diffuse large B-cell lymphoma by mediating PI3K/Akt signaling. J. Exp. Clin. Cancer Res. 2020, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Sripathy, S.; Webster, A.; Park, A.; Lao, U.; Hsu, J.H.; Loe, T.; Bedalov, A.; Simon, J.A. Discovery of selective SIRT2 inhibitors as therapeutic agents in B-cell lymphoma and other malignancies. Molecules 2020, 25, 455. [Google Scholar] [CrossRef]
- Zhang, J.; Vlasevska, S.; Wells, V.A.; Nataraj, S.; Holmes, A.B.; Duval, R.; Meyer, S.N.; Mo, T.; Basso, K.; Brindle, P.K.; et al. The CREBBP acetyltransferase is a haploinsufficient tumor suppressor in B-cell lymphoma. Cancer Discov. 2017, 7, 323–337. [Google Scholar] [CrossRef] [PubMed]
- He, Z.X.; Wei, B.F.; Zhang, X.; Gong, Y.P.; Ma, L.Y.; Zhao, W. Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 2021, 209, 112861. [Google Scholar] [CrossRef]
- Ogiwara, H.; Sasaki, M.; Mitachi, T.; Oike, T.; Higuchi, S.; Tominaga, Y.; Kohno, T. Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of MYC expression. Cancer Discov. 2016, 6, 430–445. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Lee, S.C.W.; El-Saafin, F.; Vanyai, H.K.; Hu, Y.; Pang, S.H.M.; Grabow, S.; Strasser, A.; Nutt, S.L.; Alexander, W.S.; et al. MOZ regulates B-cell progenitors and, consequently, Moz haploinsufficiency dramatically retards MYC-induced lymphoma development. Blood 2015, 125, 1910–1921. [Google Scholar] [CrossRef]
- Baell, J.B.; Leaver, D.J.; Hermans, S.J.; Kelly, G.L.; Brennan, M.S.; Downer, N.L.; Nguyen, N.; Wichmann, J.; McRae, H.M.; Yang, Y.; et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 2018, 560, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Farria, A.T.; Plummer, J.B.; Salinger, A.P.; Shen, J.; Lin, K.; Lu, Y.; McBride, K.M.; Koutelou, E.; Dent, S.Y.R. Transcriptional Activation of MYC-Induced Genes by GCN5 Promotes B-cell Lymphomagenesis. Cancer Res. 2020, 80, 5543–5553. [Google Scholar] [CrossRef]
- Farria, A.T.; Mustachio, L.M.; Coban Akdemir, Z.H.; Dent, S.Y.R. GCN5 HAT inhibition reduces human Burkitt lymphoma cell survival through reduction of MYC target gene expression and impeding BCR signaling pathways. Oncotarget 2019, 10, 5847–5858. [Google Scholar] [CrossRef]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef]
- Ghizzoni, M.; Wu, J.; Gao, T.; Haisma, H.J.; Dekker, F.J.; George Zheng, Y. 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding site. Eur. J. Med. Chem. 2012, 47, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Sevinç, K.; Gürhan Sevinç, G.; Cribbs, A.P.; Philpott, M.; Uyulur, F.; Morova, T.; Dunford, J.E.; Göklemez, S.; Arı, Ş.; et al. Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nat. Chem. Biol. 2019, 15, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Bertoni, F. BET proteins as targets for anticancer treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Itzen, F.; Greifenberg, A.K.; Bösken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of pTEFb by Recruiting Multiple Proteins, Including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Trabucco, S.E.; Gerstein, R.M.; Evens, A.M.; Bradner, J.E.; Shultz, L.D.; Greiner, D.L.; Zhang, H. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin. Cancer Res. 2015, 21, 113–122. [Google Scholar] [CrossRef]
- Aird, F.; Kandela, I.; Mantis, C. Replication Study: BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Elife 2017, 6, 1–15. [Google Scholar] [CrossRef]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Cummin, T.E.C.; Cox, K.L.; Murray, T.D.; Turaj, A.H.; Dunning, L.; English, V.L.; Fell, R.; Packham, G.; Ma, Y.; Powell, B.; et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression. Blood Adv. 2020, 4, 3316–3328. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Shah, B.; Fiskus, W.; Qi, J.; Rajapakshe, K.; Coarfa, C.; Li, L.; Devaraj, S.G.T.; Sharma, S.; Zhang, L.; et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 2015, 126, 1565–1574. [Google Scholar] [CrossRef]
- Recasens-Zorzo, C.; Cardesa-Salzmann, T.; Petazzi, P.; Ros-Blanco, L.; Esteve-Arenys, A.; Clot, G.; Guerrero-Hernández, M.; Rodríguez, V.; Soldini, D.; Valera, A.; et al. Pharmacological modulation of CXCR4 cooperates with BET bromodomain inhibition in diffuse large B-cell lymphoma. Haematologica 2019, 104, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for Leukemia therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021, 44, CD-20-0751. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.S.Y.; Chooi, J.Y.; Lim, J.S.L.; Toh, S.H.M.; Tan, T.Z.; Chng, W.-J. SMARCA2 is a novel interactor of NSD2 and regulates pro-metastatic PTP4A3 through chromatin remodeling in t(4;14) multiple myeloma. Cancer Res. 2021, 81, 2332–2344. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Kougnassoukou Tchara, P.E.; Filippakopoulos, P.; Lambert, J.P. Emerging tools to investigate bromodomain functions. Methods 2020, 184, 40–52. [Google Scholar] [CrossRef]
- Ryan, K.R.; Giles, F.; Morgan, G.J. Targeting both BET and CBP/EP300 proteins with the novel dual inhibitors NEO2734 and NEO1132 leads to anti-tumor activity in multiple myeloma. Eur. J. Haematol. 2021, 106, 90–99. [Google Scholar] [CrossRef]
- Hügle, M.; Regenass, P.; Warstat, R.; Hau, M.; Schmidtkunz, K.; Lucas, X.; Wohlwend, D.; Einsle, O.; Jung, M.; Breit, B.; et al. 4-Acyl Pyrroles as Dual BET-BRD7/9 Bromodomain Inhibitors Address BETi Insensitive Human Cancer Cell Lines. J. Med. Chem. 2020, 63, 15603–15620. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Gao, W. Current status in the discovery of dual BET/HDAC inhibitors. Bioorganic Med. Chem. Lett. 2021, 31, 127671. [Google Scholar] [CrossRef]
- Watanabe, T.; Kato, H.; Kobayashi, Y.; Yamasaki, S.; Morita-Hoshi, Y.; Yokoyama, H.; Morishima, Y.; Ricker, J.L.; Otsuki, T.; Miyagi-Maesima, A.; et al. Potential efficacy of the oral histone deacetylase inhibitor vorinostat in a phase I trial in follicular and mantle cell lymphoma. Cancer Sci. 2010, 101, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Frankel, P.; Popplewell, L.; Zain, J.; Delioukina, M.; Pullarkat, V.; Matsuoka, D.; Pulone, B.; Rotter, A.J.; Espinoza-Delgado, I.; et al. Phase II Study of Vorinostat for Treatment of Relapsed or Refractory Indolent Non-Hodgkin’s Lymphoma and Mantle Cell Lymphoma. J. Clin. Oncol. 2011, 29, 1198–1203. [Google Scholar] [CrossRef]
- Crump, M.; Coiffier, B.; Jacobsen, E.D.; Sun, L.; Ricker, J.L.; Xie, H.; Frankel, S.R.; Randolph, S.S.; Cheson, B.D. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2008, 19, 964–969. [Google Scholar] [CrossRef]
- Chen, R.; Frankel, P.; Popplewell, L.; Siddiqi, T.; Ruel, N.; Rotter, A.; Thomas, S.H.; Mott, M.; Nathwani, N.; Htut, M.; et al. A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica 2015, 100, 357–362. [Google Scholar] [CrossRef]
- Spurgeon, S.E.; Sharma, K.; Claxton, D.F.; Ehmann, C.; Pu, J.; Shimko, S.; Stewart, A.; Subbiah, N.; Palmbach, G.; LeBlanc, F.; et al. Phase 1–2 study of vorinostat (SAHA), cladribine and rituximab (SCR) in relapsed B-cell non-Hodgkin lymphoma and previously untreated mantle cell lymphoma. Br. J. Haematol. 2019, 186, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, T.; Frankel, P.; Beumer, J.H.; Kiesel, B.F.; Christner, S.; Ruel, C.; Song, J.Y.; Chen, R.; Kelly, K.R.; Ailawadhi, S.; et al. Phase 1 study of the Aurora kinase A inhibitor alisertib (MLN8237) combined with the histone deacetylase inhibitor vorinostat in lymphoid malignancies. Leuk. Lymphoma 2020, 61, 309–317. [Google Scholar] [CrossRef]
- Assouline, S.E.; Nielsen, T.H.; Yu, S.; Alcaide, M.; Chong, L.; MacDonald, D.; Tosikyan, A.; Kukreti, V.; Kezouh, A.; Petrogiannis-Haliotis, T.; et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood 2016, 128, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.E.; Lichtenstein, R.; Lue, J.; Sawas, A.; Deng, C.; Lichtenstein, E.; Khan, K.; Atkins, L.; Rada, A.; Kim, H.A.; et al. A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 2018, 131, 397–407. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Falchi, L.; Lue, J.K.; Marchi, E.; Kinahan, C.; Sawas, A.; Deng, C.; Montanari, F.; Amengual, J.E.; Kim, H.A.; et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: A multicenter phase 1 study. Blood 2019, 134, 1395–1405. [Google Scholar] [CrossRef]
- Reiman, T.; Savage, K.J.; Crump, M.; Cheung, M.C.; MacDonald, D.; Buckstein, R.; Couban, S.; Piliotis, E.; Imrie, K.; Spaner, D.; et al. A phase I study of romidepsin, gemcitabine, dexamethasone and cisplatin combination therapy in the treatment of peripheral T-cell and diffuse large B-cell lymphoma; the Canadian cancer trials group LY.15 study. Leuk. Lymphoma 2019, 60, 912–919. [Google Scholar] [CrossRef]
- Ribrag, V.; Kim, W.S.; Bouabdallah, R.; Lim, S.T.; Coiffier, B.; Illes, A.; Lemieux, B.; Dyer, M.J.S.; Offner, F.; Felloussi, Z.; et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: Results of a phase II study. Haematologica 2017, 102, 903–909. [Google Scholar] [CrossRef]
- Drott, K.; Hagberg, H.; Papworth, K.; Relander, T.; Jerkeman, M. Valproate in combination with rituximab and CHOP as first-line therapy in diffuse large B-cell lymphoma (VALFRID). Blood Adv. 2018, 2, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Kelly, K.R.; Flinn, I.; Patel, M.R.; Gharavi, R.; Ma, A.; Parker, J.; Hafeez, A.; Tuck, D.; Younes, A. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: Results from an expanded phase I trial. Haematologica 2017, 102, 1923–1930. [Google Scholar] [CrossRef]
- Batlevi, C.L.; Crump, M.; Andreadis, C.; Rizzieri, D.; Assouline, S.E.; Fox, S.; Van Der Jagt, R.H.C.; Copeland, A.; Potvin, D.; Chao, R.; et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 2017, 178, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Shapiro, G.I.; LoRusso, P.; Dowlati, A.; T. Do, K.; Jacobson, C.A.; Vaishampayan, U.; Weise, A.; Caimi, P.F.; Eder, J.P.; French, C.A.; et al. A Phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with NUT carcinoma, other solid tumours, or diffuse large B-cell lymphoma. Br. J. Cancer 2020, 124, 744–753. [Google Scholar] [CrossRef]
- Falchook, G.; Rosen, S.; LoRusso, P.; Watts, J.; Gupta, S.; Coombs, C.C.; Talpaz, M.; Kurzrock, R.; Mita, M.; Cassaday, R.; et al. Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies. Clin. Cancer Res. 2020, 26, 1247–1257. [Google Scholar] [CrossRef]
- Basheer, F.; Huntly, B.J.P. BET bromodomain inhibitors in leukemia. Exp. Hematol. 2015, 43, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Donato, E.; Croci, O.; Sabò, A.; Muller, H.; Morelli, M.J.; Pelizzola, M.; Campaner, S. Compensatory RNA polymerase 2 loading determines the efficacy and transcriptional selectivity of JQ1 in Myc-driven tumors. Leukemia 2017, 31, 479–490. [Google Scholar] [CrossRef]
- ClinicalTrials.gov Database. Available online: https://clinicaltrials.gov/ (accessed on 16 November 2021).
- Puvvada, S.D.; Guillén-Rodríguez, J.M.; Rivera, X.I.; Heard, K.; Inclan, L.; Schmelz, M.; Schatz, J.H.; Persky, D.O. A Phase II Exploratory Study of PXD-101 (Belinostat) Followed by Zevalin in Patients with Relapsed Aggressive High-Risk Lymphoma. Oncology 2017, 93, 401–405. [Google Scholar] [CrossRef]
- Blum, K.A.; Abramson, J.; Maris, M.; Flinn, I.; Goy, A.; Mertz, J.; Sims, R.; Garner, F.; Senderowicz, A.; Younes, A. 41OA phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with relapsed or refractory lymphoma. Ann. Oncol. 2018, 29, iii7. [Google Scholar] [CrossRef]
- Patel, M.R.; Garcia-Manero, G.; Paquette, R.; Dinner, S.; Donnellan, W.B.; Grunwald, M.R.; Ribadeneira, M.D.; Schroeder, P.; Brevard, J.; Wilson, L.; et al. The BET Inhibitor FT-1101 As a Single Agent in Patients with Relapsed or Refractory Hematologic Malignancies. Blood 2019, 134, 3907. [Google Scholar] [CrossRef]
- Sarkozy, C.; Morschhauser, F.; Dubois, S.; Molina, T.; Michot, J.M.; Cullières-Dartigues, P.; Suttle, B.; Karlin, L.; Le Gouill, S.; Picquenot, J.M.; et al. A LYSA phase Ib study of tazemetostat (EPZ-6438) plus R-CHOP in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) with poor prognosis features. Clin. Cancer Res. 2020, 26, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.; Batlevi, C.L.; Gabrail, N.; Pagel, J.M.; Yang, J.; Whalen, J.; Adib, D. A Phase 1b/3 Randomized, Double-Blind, 3-Stage Study of Tazemetostat or Placebo Plus Lenalidomide and Rituximab in Patients with Relapsed/Refractory Follicular Lymphoma. Clin. Lymphoma Myeloma Leuk. 2020, 20, S279. [Google Scholar] [CrossRef]
- Harb, W.; Abramson, J.; Lunning, M.; Goy, A.; Maddocks, K.; Lebedinsky, C.; Senderowicz, A.; Trojer, P.; Bradley, W.D.; Flinn, I. A phase 1 study of CPI-1205, a small molecule inhibitor of EZH2, preliminary safety in patients with B-cell lymphomas. Ann. Oncol. 2018, 29, iii7. [Google Scholar] [CrossRef]
- Milosevich, N.; Hof, F. Chemical Inhibitors of Epigenetic Methyllysine Reader Proteins. Biochemistry 2016, 55, 1570–1583. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Teater, M.; Meydan, C.; Hoehn, K.B.; Phillip, J.M.; Soshnev, A.A.; Venturutti, L.; Rivas, M.A.; Calvo-Fernández, M.T.; Gutierrez, J.; et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 2020, 37, 655–673.e11. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.L.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef]
- Herviou, L.; Jourdan, M.; Martinez, A.M.; Cavalli, G.; Moreaux, J. EZH2 is overexpressed in transitional preplasmablasts and is involved in human plasma cell differentiation. Leukemia 2019, 33, 2047–2060. [Google Scholar] [CrossRef]
- Eich, M.-L.; Athar, M.; Ferguson, J.E.; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Pietra, A.D.; LaFrance, L.V.; Mellinger, M.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.S.; Chen, J.Y.; Tsai, H.J.; Chen, T.Y.; Hung, W.C. The SUV39H1 inhibitor chaetocin induces differentiation and shows synergistic cytotoxicity with other epigenetic drugs in acute myeloid leukemia cells. Blood Cancer J. 2015, 5, 1–6. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Veazey, K.J.; Cheng, D.; Lin, K.; Villarreal, O.D.; Gao, G.; Perez-Oquendo, M.; Van, H.T.; Stratton, S.A.; Green, M.; Xu, H.; et al. CARM1 inhibition reduces histone acetyltransferase activity causing synthetic lethality in CREBBP/EP300-mutated lymphomas. Leukemia 2020, 34, 3269–3285. [Google Scholar] [CrossRef]
- Wang, L.; Pal, S.; Sif, S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 2008, 28, 6262–6277. [Google Scholar] [CrossRef]
- Koh, C.M.; Bezzi, M.; Low, D.H.P.; Ang, W.X.; Teo, S.X.; Gay, F.P.H.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabò, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Fernando, T.M.; Lossos, C.; Yusufova, N.; Liu, F.; Fontán, L.; Durant, M.; Geng, H.; Melnick, J.; Luo, Y.; et al. PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood 2018, 132, 2026–2039. [Google Scholar] [CrossRef] [PubMed]
- Hatzi, K.; Geng, H.; Doane, A.S.; Meydan, C.; LaRiviere, R.; Cardenas, M.; Duy, C.; Shen, H.; Vidal, M.N.C.; Baslan, T.; et al. Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat. Immunol. 2019, 20, 86–96. [Google Scholar] [CrossRef]
- Mathur, R.; Sehgal, L.; Havranek, O.; Köhrer, S.; Khashab, T.; Jain, N.; Burger, J.A.; Neelapu, S.S.; Davis, R.E.; Samaniego, F. Inhibition of demethylase KDM6B sensitizes diffuse large B-cell lymphoma to chemotherapeutic drugs. Haematologica 2017, 102, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, J.C. Structural biology of the chromodomain: Form and function. Gene 2012, 496, 69–78. [Google Scholar] [CrossRef]
- Zhang, Y.; Lei, M.; Yang, X.; Feng, Y.; Yang, Y.; Loppnau, P.; Li, Y.; Yang, Y.; Min, J.; Liu, Y. Structural and histone binding studies of the chromo barrel domain of TIP60. FEBS Lett. 2018, 592, 1221–1232. [Google Scholar] [CrossRef]
- Huang, Y.; Fang, J.; Bedford, M.T.; Zhang, Y.; Xu, R.M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 2006, 312, 748–751. [Google Scholar] [CrossRef]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Peña, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef]
- Estève, P.O.; Terragni, J.; Deepti, K.; Chin, H.G.; Dai, N.; Espejo, A.; Corrêa, I.R.; Bedford, M.T.; Pradhan, S. Methyllysine reader plant homeodomain (PHD) finger protein 20-like 1 (PHF20L1) antagonizes DNA (cytosine-5) methyltransferase 1 (DNMT1) proteasomal degradation. J. Biol. Chem. 2014, 289, 8277–8287. [Google Scholar] [CrossRef]
- Scott, C.L.; Gil, J.; Hernando, E.; Teruya-Feldstein, J.; Narita, M.; Martínez, D.; Visakorpi, T.; Mu, D.; Cordon-Cardo, C.; Peters, G.; et al. Role of the chromobox protein CBX7 in lymphomagenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 5389–5394. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Duncan, E.M.; Masui, O.; Gil, J.; Heard, E.; Allis, C.D. Mouse Polycomb Proteins Bind Differentially to Methylated Histone H3 and RNA and Are Enriched in Facultative Heterochromatin. Mol. Cell. Biol. 2006, 26, 2560–2569. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, C.; Gignac, M.C.; Anderson, C.J.; Milosevich, N.; Dheri, A.; Prashar, N.; Flemmer, R.T.; Dev, A.; Henderson, T.G.; Douglas, S.F.; et al. Structure-Activity Relationships of Cbx7 Inhibitors, Including Selectivity Studies against Other Cbx Proteins. ACS Omega 2016, 1, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Scott, M.P.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- FDA Approves Tazemetostat for Relapsed/Refractory Follicular Lymphoma. Available online: https://www.targetedonc.com/view/fda-approves-tazemetostat-for-relapsed-refractory-follicular-lymphoma (accessed on 29 April 2021).
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef]
- Qi, W.; Chan, H.M.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef]
- Campbell, J.E.; Kuntz, K.W.; Knutson, S.K.; Warholic, N.M.; Keilhack, H.; Wigle, T.J.; Raimondi, A.; Klaus, C.R.; Rioux, N.; Yokoi, A.; et al. EPZ011989, A potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med. Chem. Lett. 2015, 6, 491–495. [Google Scholar] [CrossRef]
- Gehling, V.S.; Vaswani, R.G.; Nasveschuk, C.G.; Duplessis, M.; Iyer, P.; Balasubramanian, S.; Zhao, F.; Good, A.C.; Campbell, R.; Lee, C.; et al. Discovery, design, and synthesis of indole-based EZH2 inhibitors. Bioorganic Med. Chem. Lett. 2015, 25, 3644–3649. [Google Scholar] [CrossRef]
- Chamorro-Jorganes, A.; Ribeiro, M.L.; Profitos-Peleja, N.; Reyes-Garau, D.; Recasens-Zorzo, C.; Valero, J.G.; Armengol, M.; Perez-Galan, P.; Butler, R.; Postigo, A.; et al. Abstract 2925: Safety and efficacy of EZH2 and BRD4 dual targeting in EZH2 Y641mut germinal centre-derived lymphoma. In Proceedings of the Cancer Research, American Association for Cancer Research (AACR). Philadelphia, PA, USA, 22–24 June 2020; Volume 80, p. 2925. [Google Scholar]
- Vaswani, R.G.; Gehling, V.S.; Dakin, L.A.; Cook, A.S.; Nasveschuk, C.G.; Duplessis, M.; Iyer, P.; Balasubramanian, S.; Zhao, F.; Good, A.C.; et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase i Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 2016, 59, 9928–9941. [Google Scholar] [PubMed]
- Lakhani, N.J.; Gutierrez, M.; Duska, L.R.; Do, K.T.; Sharma, M.; Gandhi, L.; Papadopoulos, K.P.; Truong, J.; Fan, X.; Lee, J.H.; et al. Phase 1/2 first-in-human (FIH) study of CPI-0209, a novel small molecule inhibitor of enhancer of zeste homolog 2 (EZH2) in patients with advanced tumors. J. Clin. Oncol. 2021, 39, 3104. [Google Scholar] [CrossRef]
- Bisserier, M.; Wajapeyee, N. Mechanisms of resistance to ezh2 inhibitors in diffuse large b-cell lymphomas. Blood 2018, 131, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, Y.; Hsu, Y.J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.C.; Orkin, S.H. EZH1 Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; MacNevin, C.J.; Liu, F.; Gao, C.; Huang, X.P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Honma, D.; Kanno, O.; Watanabe, J.; Kinoshita, J.; Hirasawa, M.; Nosaka, E.; Shiroishi, M.; Takizawa, T.; Yasumatsu, I.; Horiuchi, T.; et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017, 108, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, D.; Tobinai, K.; Makita, S.; Ishida, T.; Kusumoto, S.; Ishitsuka, K.; Yoshimitsu, M.; Imaizumi, Y.; Sawayama, Y.; Takeuchi, S.; et al. First-in-Human Study of the EZH1/2 Dual Inhibitor DS-3201b in Patients with Relapsed or Refractory Non-Hodgkin Lymphomas—Preliminary Results. Blood 2017, 130, 4070. [Google Scholar]
- Kung, P.P.; Bingham, P.; Brooun, A.; Collins, M.; Deng, Y.L.; Dinh, D.; Fan, C.; Gajiwala, K.S.; Grantner, R.; Gukasyan, H.J.; et al. Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J. Med. Chem. 2018, 61, 650–665. [Google Scholar]
- Luger, K.; Richmond, T.J. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 1998, 8, 140–146. [Google Scholar] [CrossRef]
- Weake, V.M.; Workman, J.L. Histone Ubiquitination: Triggering Gene Activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cadahía, B.; Drobic, B.; Davie, J.R. H3 phosphorylation: Dual role in mitosis and interphaseThis paper is one of a selection of papers published in this Special Issue entitled 30th Annual International Asilomar Chromatin and Chromosomes Conference and has undergone the Journal’s usual peer r. Biochem. Cell Biol. 2009, 87, 695–709. [Google Scholar] [CrossRef]
- Hegyi, K.; Méhes, G. Mitotic Failures in Cancer: Aurora B Kinase and its Potential Role in the Development of Aneuploidy. Pathol. Oncol. Res. 2012, 18, 761–769. [Google Scholar] [CrossRef]
- Ramani, P.; Taylor, S.; Miller, E.; Sowa-Avugrah, E.; May, M.T. High Phosphohistone H3 Expression Correlates with Adverse Clinical, Biological, and Pathological Factors in Neuroblastomas. J. Histochem. Cytochem. 2015, 63, 397–407. [Google Scholar] [CrossRef]
- Hale, C.S.; Qian, M.; Ma, M.W.; Scanlon, P.; Berman, R.S.; Shapiro, R.L.; Pavlick, A.C.; Shao, Y.; Polsky, D.; Osman, I.; et al. Mitotic Rate in Melanoma. Am. J. Surg. Pathol. 2013, 37, 882–889. [Google Scholar] [CrossRef]
- Ginter, P.S.; Shin, S.J.; Liu, Y.; Chen, Z.; D’Alfonso, T.M. Phosphohistone H3 expression correlates with manual mitotic counts and aids in identification of “hot spots” in fibroepithelial tumors of the breast. Hum. Pathol. 2016, 49, 90–98. [Google Scholar] [CrossRef]
- Khieu, M.L.; Broadwater, D.R.; Aden, J.K.; Coviello, J.M.; Lynch, D.T.; Hall, J.M. The Utility of Phosphohistone H3 (PHH3) in Follicular Lymphoma Grading: A Comparative Study with Ki-67 and H&E Mitotic Count. Am. J. Clin. Pathol. 2019, 151, 542–550. [Google Scholar]
- Méhes, G.; Hegyi, K.; Jobanputra, R.; Beke, L.; Vereb, G.; Bedekovics, J. Distinct Dynamics of Mitotic Transition in B-Cell Lymphoma and Reactive B-Cell Lymphoproliferations Determined by H3S10 Phosphohistone Immunolabeling. Pathobiology 2017, 84, 243–250. [Google Scholar] [CrossRef]
- Crosio, C.; Fimia, G.M.; Loury, R.; Kimura, M.; Okano, Y.; Zhou, H.; Sen, S.; Allis, C.D.; Sassone-Corsi, P. Mitotic Phosphorylation of Histone H3: Spatio-Temporal Regulation by Mammalian Aurora Kinases. Mol. Cell. Biol. 2002, 22, 874–885. [Google Scholar] [CrossRef]
- Ikezoe, T.; Takeuchi, T.; Yang, J.; Adachi, Y.; Nishioka, C.; Furihata, M.; Koeffler, H.P.; Yokoyama, A. Analysis of Aurora B kinase in non-Hodgkin lymphoma. Lab. Investig. 2009, 89, 1364–1373. [Google Scholar] [CrossRef]
- Floc’h, N.; Ashton, S.; Ferguson, D.; Taylor, P.; Carnevalli, L.S.; Hughes, A.M.; Harris, E.; Hattersley, M.; Wen, S.; Curtis, N.J.; et al. Modeling Dose and Schedule Effects of AZD2811 Nanoparticles Targeting Aurora B Kinase for Treatment of Diffuse Large B-cell Lymphoma. Mol. Cancer Ther. 2019, 18, 909–919. [Google Scholar] [CrossRef]
- Kelly, K.R.; Friedberg, J.W.; Park, S.I.; McDonagh, K.; Hayslip, J.; Persky, D.; Ruan, J.; Puvvada, S.; Rosen, P.; Iyer, S.P.; et al. Phase I Study of the Investigational Aurora A Kinase Inhibitor Alisertib plus Rituximab or Rituximab/Vincristine in Relapsed/Refractory Aggressive B-cell Lymphoma. Clin. Cancer Res. 2018, 24, 6150–6159. [Google Scholar] [CrossRef]
- Huertas, D.; Soler, M.; Moreto, J.; Villanueva, A.; Martinez, A.; Vidal, A.; Charlton, M.; Moffat, D.; Patel, S.; McDermott, J.; et al. Antitumor activity of a small-molecule inhibitor of the histone kinase Haspin. Oncogene 2012, 31, 1408–1418. [Google Scholar] [CrossRef]
- Rzymski, T.; Zarebski, A.; Windak, R.; Sibinska, Z.; Klosowska-Wardega, A.; Trebacz, E.; Cholody, M.; Szamborska-Gbur, A.; Milik, M.; Prymula, K.; et al. Abstract 3845: Antitumor activity of SEL120: An orally available dual inhibitors of Haspin/CDK9, for standalone and combination therapy with AuroraB inhibitors in solid tumors and hematopoietic malignancies. In Proceedings of the Experimental and Molecular Therapeutics. Am. Assoc. Cancer Res. 2012, 72, 3845. [Google Scholar]
- Ayyappan, S.; Maddocks, K. Novel and emerging therapies for B cell lymphoma. J. Hematol. Oncol. 2019, 12, 82. [Google Scholar] [CrossRef]
- Chen, E.; Staudt, L.M.; Green, A.R. Janus Kinase Deregulation in Leukemia and Lymphoma. Immunity 2012, 36, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Drennan, A.C.; Ceribelli, M.; Zhu, F.; Wright, G.W.; Huang, D.W.; Xiao, W.; Li, Y.; Grindle, K.M.; Lu, L.; et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7260–E7267. [Google Scholar] [CrossRef]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Zibellini, S. JAK inhibitors and risk of B-cell lymphomas. Blood 2019, 133, 2251–2253. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Pascaud, J.; Ly, B.; Tahmasebi, F.; Mariette, X. JAK inhibitors alter NK cell functions and may impair immunosurveillance against lymphomagenesis. Cell. Mol. Immunol. 2020, 17, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Paakinaho, V.; Lempiäinen, J.K.; Sigismondo, G.; Niskanen, E.A.; Malinen, M.; Jääskeläinen, T.; Varjosalo, M.; Krijgsveld, J.; Palvimo, J.J. SUMOylation regulates the protein network and chromatin accessibility at glucocorticoid receptor-binding sites. Nucleic Acids Res. 2021, 49, 1951–1971. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Kunkalla, K.; Vaghefi, A.; Frederiksen, J.K.; Liu, Y.; Chapman, J.R.; Blonska, M.; Bernal-Mizrachi, L.; Alderuccio, J.P.; Lossos, I.S.; et al. Smoothened stabilizes and protects TRAF6 from degradation: A novel non-canonical role of smoothened with implications in lymphoma biology. Cancer Lett. 2018, 436, 149–158. [Google Scholar] [CrossRef]
- Pham, L.V.; Zhou, H.-J.; Lin-Lee, Y.-C.; Tamayo, A.T.; Yoshimura, L.C.; Fu, L.; Darnay, B.G.; Ford, R.J. Nuclear Tumor Necrosis Factor Receptor-associated Factor 6 in Lymphoid Cells Negatively Regulates c-Myb-mediated Transactivation through Small Ubiquitin-related Modifier-1 Modification. J. Biol. Chem. 2008, 283, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Guppy, B.J.; Sawchuk, L.; Davie, J.R.; McManus, K.J. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013, 32, 363–376. [Google Scholar] [CrossRef]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef]
- Nagel, S.; Ehrentraut, S.; Tomasch, J.; Quentmeier, H.; Meyer, C.; Kaufmann, M.; Drexler, H.G.; MacLeod, R.A.F. Ectopic Expression of Homeobox Gene NKX2-1 in Diffuse Large B-Cell Lymphoma Is Mediated by Aberrant Chromatin Modifications. PLoS ONE 2013, 8, e61447. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Type | Indication | Year of Approval | |
|---|---|---|---|---|
| Vorinostat | Zolinza | HDACi | CTCL | 2006 |
| Romidepsin | Istodax | HDACi | CTCL | 2009 |
| PTCL | 2011 | |||
| Belinostat | Beleodaq | HDACi | PTCL | 2014 |
| Panobinostat | Farydak | HDACi | MM | 2015 |
| Tazemetostat | Tazverik | EZH2i | R/R FL with or without mEZH2 | 2020 |
| Sarcoma | 2020 |
| Drug/Regimen | Type | Trial ID | Phase | Number of Patients | Disease | Response | Toxicity | Ref |
|---|---|---|---|---|---|---|---|---|
| Vorinostat | HDACi | NCT00253630 | 2 | 35 | FL, MCL, MZL | ORR = 29% OS (2 years) = 77% PFS (2 years) = 37% | 26% SAEs: TP (8.5%) 100% AEs: TP (86%) | [130] |
| Vorinostat | HDACi | NCT00127140 | 1 | 10 | FL, MCL, DLBCL | ORR = 75%/50%/0% | Grade 3–4 AEs: NP (30%) AEs: TP (70%), anemia (70%), leukopenia (60%) | [129] |
| Vorinostat | HDACi | NCT00875056 | 2 | 56 | FL, MCL | ORR = 49%/28% | 23%/27% SAEs: TP (5/9%) 100% AEs: TP (95/91%), diarrhea (72%/82%), NP (72%/64%) | [149] |
| Vorinostat | HDACi | NCT00097929 | 2 | 18 | DLBCL | ORR = 5% | 39% SAEs AEs: diarrhea (61%), fatigue (50%), nausea (39%) | [131] |
| Vorinostat +rituximab | HDACi | NCT00720876 | 2 | 28 | FL, MCL, MZL, LPL | ORR = 46% PFS = 29 months | 43% SAEs: thrombosis (13%) 100% AEs: fatigue (87%), diarrhea (80%), nausea (73%) | [132] |
| Vorinostat +R-ICE | HDACi | NCT00601718 | 1/2 | 29 | DLBCL, MCL, MZL | ORR = 66% | 35% SAEs: NP (10%) 86% AEs: hypophosphatemia (41%), hypokalemia (34%) | [149] |
| Vorinostat +rituximab +cladribine | HDACi | NCT00764517 | 2 | 49 | MCL (39 frontline/10 R/R) | ORR = 97%/30% OS = 25/6 months PFS = 20/15 months | 46%/50% SAEs: NP (23%/22%) 72%/78% AEs: TP (36%/28%), NP (23%/28%) | [133] |
| Vorinostat + R-CHOP | HDACi | NCT00972478 | 1/2 | 83 | DLBCL | ORR = 81% OS (2 years) = 86% PFS (2 years) = 73% | 68% SAEs: NP (35%), anemia (22%) 100% AEs: anemia (85%), TP (59%), fatigue (74%) | [149] |
| Vorinostat +azacitidine +rituximab and others (pre-ASCT) | HDACi | NCT01983969 | 1/2 | 26 | DLBCL | EFS (100 days post-transplant) = 65% | 0% SAEs 100% AEs: NP (97%), mucositis (93%), nausea (90%) | [149] |
| Vorinostat +bortezomib | HDACi | NCT00703664 | 2 | 65 | MCL. DLBCL | ORR (9 years) = 27%/8% PFS = 7.6 months/1.8 months | 38%/56% SAEs: TP (0%/15%) 100% AEs: TP (81%/67%), diarrhea (85%/62%) | [149] |
| Vorinostat +bortezomib (post-ASCT) | HDACi | NCT00992446 | 2 | 19 | DLBCL, FL, MCL, T-NHL | OS (6.6 years post ASCT) = 84% EFS (6.6 years post ASCT) = 74% | 33% SAEs: all <10% 100% AEs: NP (68%), TP (10%) | [149] |
| Vorinostat +niacinamide +etoposide | HDACi | NCT00691210 | 1 | 25 | DLBCL, FL, HL | ORR = 24% | Grade 3–4 AEs: TP (12%), infection (12%) AEs: fatigue (84%), nausea (80%), diarrhea (72%) | [82] |
| Vorinostat +alisertib | HDACi | NCT01567709 | 1 | 12 | DLBCL | ORR = 17% | Grade 3–4 AEs: NP (22%), leukopenia (18%), anemia (17%) | [134] |
| Panobinostat | HDACi | NCT01261247 | 2 | 39 | DLBCL, MZL, BL | ORR = 21% OS = 14.9 months PFS = 3.1 months | 83% SAEs: TP (80%), NP (29%) 93% AEs: fatigue (85%), diarrhea (76%), nausea (72%) | [149] |
| Panobinostat | HDACi | NCT01523834 | 2 | 35 | DLBCL (R/R) | ORR = 17% OS = 7.6 months PFS = 2.4 months | 35% SAEs: all <10% 23% AEs | [149] |
| Panobinostat +rituximab | HDACi | NCT01238692 | 2 | 40 | DLBCL (21 single/19 combo) | ORR = 29%/26% | Grade 3–4 AEs: TP (71%/68%), NP (24%/32%) AEs: TP (76%/79%), diarrhea (76%/58%), nausea (71%/58%) | [135] |
| Panobinostat +rituximab | HDACi | NCT01282476 | 2 | 18 | DLBCL | ORR = 11% PFS (6 months) = 6% | 56% SAEs: TP (33%) 100% AEs: fatigue (72%), anemia (67%), TP (67%) | [149] |
| Panobinostat +everolimus | HDACi | NCT00918333 | 1/2 | 116 | DLBCL, FL, BL, MZL, HL, T-NHL | ORR = 33% OS = 35 months PFS = 4.2 months | 24% SAEs: all <10% 99% AEs: decreased Hb (99%), TP (91%), fatigue (90%) | [149] |
| Panobinostat +everolimus | HDACi | NCT00967044 | 1/2 | 30 | NHL, HL | N/A | 64% SAEs: TP (63%), NP (47%) 100% AEs: fatigue (83%), hyperglycemia (63%), mucositis (60%) | [149] |
| Panobinostat +everolimus | HDACi | NCT00978432 | 2 | 33 | DLBCL | ORR = 15% | 25% SAEs: all <10% 100% AEs: TP (73%), diarrhea (58%), fatigue (48%) | [149] |
| Romidepsin | HDACi | NCT00383565 | 2 | 9 | DLBCL, MCL | ORR = 11% OS = 20 months PFS = 4 months | 67% SAEs: TP (22%) 100% AEs: TP (89%), anemia (79%), lymphopenia (67%) | [149] |
| Romidepsin +pralatrexate | HDACi | NCT01947140 | 1/2 | 7 | FL, DLBCL, BL | ORR = 75% (FL) OS = 34 months PFS = 1.8 months | Grade 3–4 AEs: anemia (29%), TP (28%), NP (14%) Grade 1–2 AEs: nausea (66%), fatigue (52%), anorexia (24%) | [136] |
| Romidepsin +azacitidine | HDACi | NCT01998035 | 1/2 | 20 | DLBCL, FL, HL | ORR = 10% PFS = 2.5 months | Grade 3–4 AEs: NP (42%), lymphopenia (42%), TP (27%), 100% grade 1–2 AEs: hyperglycemia (81%), nausea (54%), vomiting (46%) | [137] |
| Romidepsin +GDP | HDACi | NCT01846390 | 1 | 20 | DLBCL, PTCL | ORR = 50% OS = 5.5 months (DLBCL) PFS = 2 months (DLBCL) | 60% SAEs: 1 grade 5 sepsis Grade 2–4 AEs: infection (75%), TP (55%), NP (30%), anemia (30%) | [138] |
| Abexinostat | HDACi | NCT00724984 | 1/2 | 30 | FL, MCL | ORR = 56% FL/21% MCL | 38%/36% SAEs: all <10% 100%/93% AEs: TP (63%/29%), nausea (69%/50%), diarrhea (50%/50%) | [149] |
| Abexinostat | HDACi | EudraCT-2009-013691-47 | 2 | 100 | FL, DLBCL, MCL, MZL, T-NHL, CLL | ORR = 56% FL/31% DLBCL PFS = 10.2 months FL/2.8 months DLBCL | 73% SAEs: TP (54%), NP (11%) 82% grade 3–4 AEs: TP (80%), NP (27%), anemia (12%) 98% AEs, any grade | [139] |
| Valproic acid +R-CHOP | HDACi | NCT01622439 | 1/2 | 33 | DLBCL | ORR = 90% OS (2 years) = 97% PFS (2 years) = 85% | Grade 3–4 AEs: NP (81%), TP (33%), infection (27%) Auditory AEs | [140] |
| Fimepinostat +rituximab | HDACi | NCT01742988 | 1 | 37 | DLBCL (25 single/12 combo) | ORR = 47%/18% PFS = 5.7/1.3 months | 28% SAEs 43% grade 3–5 AEs: TP (32%), NP (16%) AEs: diarrhea (57%), TP (54%), fatigue (41%) | [141] |
| Mocetinostat | HDACi | NCT00359086 | 2 | 72 | NHL (41 DLBCL/31 FL) | ORR = 19%/12% OS = 12 months/N.R. PFS = 2 months/3.7 months | 36% SAEs: all <10% 57% grade 3–4 AEs: fatigue (24%), NP (15%), TP (12%) 99% AEs: fatigue (75%), nausea (70%), diarrhea (61%) | [142] |
| Belinostat | HDACi | NCT00303953 | 2 | 22 | DLBCL, BL, PMBCL | ORR = 0% OS = 0.9 years PFS = 0.2 years | 15% SAEs: all <10% 90% AEs: fatigue (40%), nausea (40%) | [149] |
| Belinostat +rituximab +ibritumomab tiuxetan | HDACi | NCT01686165 | 2 | 5 | DLBCL | ORR = 0% PFS (2 years) = 0% | 20% SAEs: thrombosis (20%) 60% grade 3–4 AEs: TP (40%), pain (20%), hypoglycemia (20%) 100% AEs: nausea (80%), pain (60%), TP (60%) | [150] |
| Birabresib | BETi | NCT01713582 | 1 | 33 | DLBCL, FL, MCL, BL, MZL | ORR = 10% | AEs: TP (96%), anemia (91%), neutropenia (51%) | [143] |
| Birabresib | BETi | NCT02698189 | 1 | 6 | DLBCL | ORR = 17% | 17% SAEs: infection (17%) 100% AEs: TP (50%), abdominal pain (50%), diarrhea (50%) | [149] |
| RO6870810 | BETi | NCT01987362 | 1 | 19 | DLBCL | ORR = 10.5% PFS = 29 days | 53% SAEs 74% grade 3–4 TRAEs: all <10% 95% all-grade TRAEs: fatigue (42%), nausea (31%), diarrhea (26%) | [144] |
| INCB054329 | BETi | NCT02431260 | 1/2 | 4 | Lymphoma | ORR = 0% | 23% grade 3–4 TRAEs: TP (13%) 78% all-grade TRAEs: nausea (35%), TP (33%), fatigue (29%) | [145] |
| INCB057643 | BETi | NCT02711137 | 1/2 | 16 | Lymphoma | ORR = 25% (FL) | 36% grade 3–4 TRAEs: TP (18%), anemia (10%) 86% all-grade TRAEs: TP (32%), fatigue (30%), nausea (30%) | [145] |
| CPI-0610 | BETi | NCT01949883 | 1 | 64 | DLBCL, FL | ORR = 7% | TRAEs: TP (45%), fatigue (34%), nausea (27%) | [151] |
| FT-1101 | BETi | NCT02543879 | 1 | 10 | NHL | ORR = 0% | Grade 3–4 TRAEs: pleural effusion (20%) TRAEs: diarrhea (60%), nausea (40%), pleural effusion (40%) | [152] |
| Tazemetostat +R-CHOP | EZH2i | NCT02889523 | 1b | 17 | DLBCL | mCR = 76% | Grade 3–4 AEs: NP (47%), leukopenia (29%), constipation (24%) AEs: constipation (59%), nausea (59%), vomiting (53%), NP (47%) | [153] |
| Tazemetostat +lenalidomide +rituximab | EZHi | NCT04224493 | 1b/3 | 518 (planned) | FL | N/A | N/A | [154] |
| Lirametostat | EZHi | NCT02395601 | 1 | 32 | DLBCL, FL, MZL | ORR = 3% SD = 15% | Grade 3–4 TRAEs: lymphopenia (9%), nausea (3%), anemia (3%) TRAEs: nausea, diarrhea, anemia, fatigue (all >5%) | [155] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Serrano, M.; Winkler, R.; Santos, J.C.; Le Pannérer, M.-M.; Buschbeck, M.; Roué, G. Histone Modifications and Their Targeting in Lymphoid Malignancies. Int. J. Mol. Sci. 2022, 23, 253. https://doi.org/10.3390/ijms23010253
Fernández-Serrano M, Winkler R, Santos JC, Le Pannérer M-M, Buschbeck M, Roué G. Histone Modifications and Their Targeting in Lymphoid Malignancies. International Journal of Molecular Sciences. 2022; 23(1):253. https://doi.org/10.3390/ijms23010253
Chicago/Turabian StyleFernández-Serrano, Miranda, René Winkler, Juliana C. Santos, Marguerite-Marie Le Pannérer, Marcus Buschbeck, and Gaël Roué. 2022. "Histone Modifications and Their Targeting in Lymphoid Malignancies" International Journal of Molecular Sciences 23, no. 1: 253. https://doi.org/10.3390/ijms23010253
APA StyleFernández-Serrano, M., Winkler, R., Santos, J. C., Le Pannérer, M.-M., Buschbeck, M., & Roué, G. (2022). Histone Modifications and Their Targeting in Lymphoid Malignancies. International Journal of Molecular Sciences, 23(1), 253. https://doi.org/10.3390/ijms23010253