
Endothelial ADAM17 Expression in the Progression of Kidney Injury in an Obese Mouse Model of Pre-Diabetes

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Endothelial Adam17 Deletion Modifies Blood Glucose Levels and Renal Function in Obese Mice
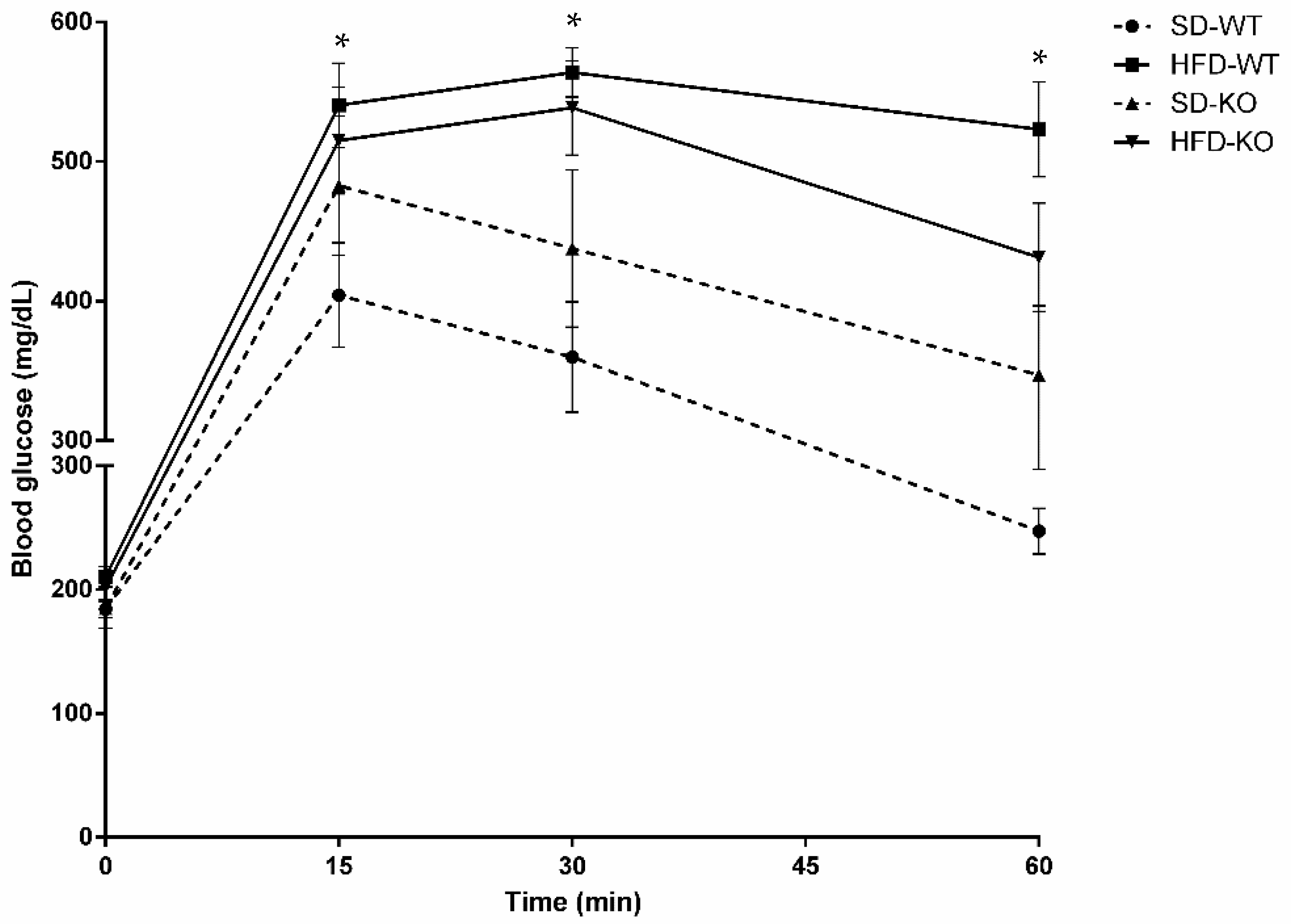
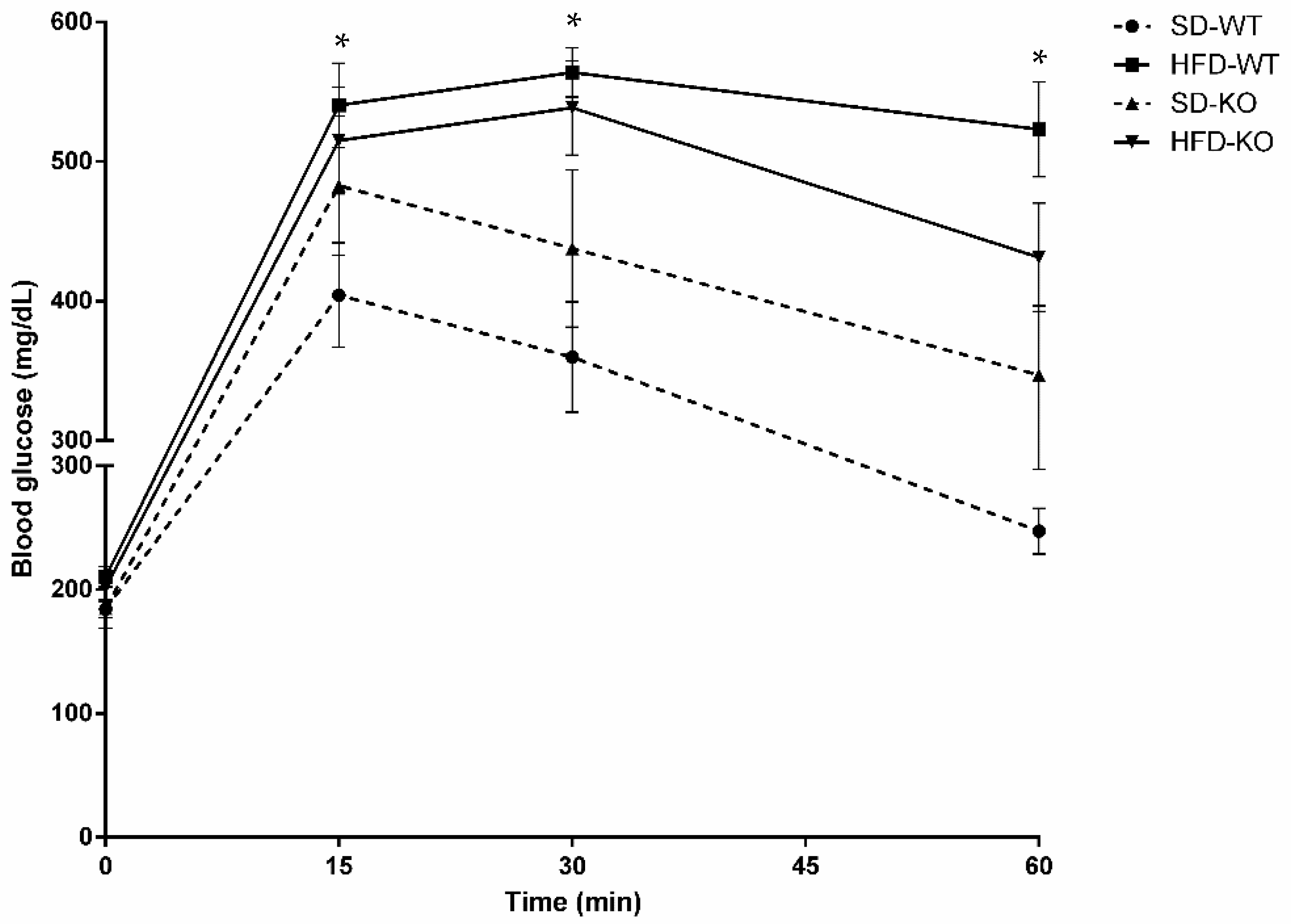
2.2. Glucose Tolerance was Modulated by Adam17 Deletion in Endothelial Cells
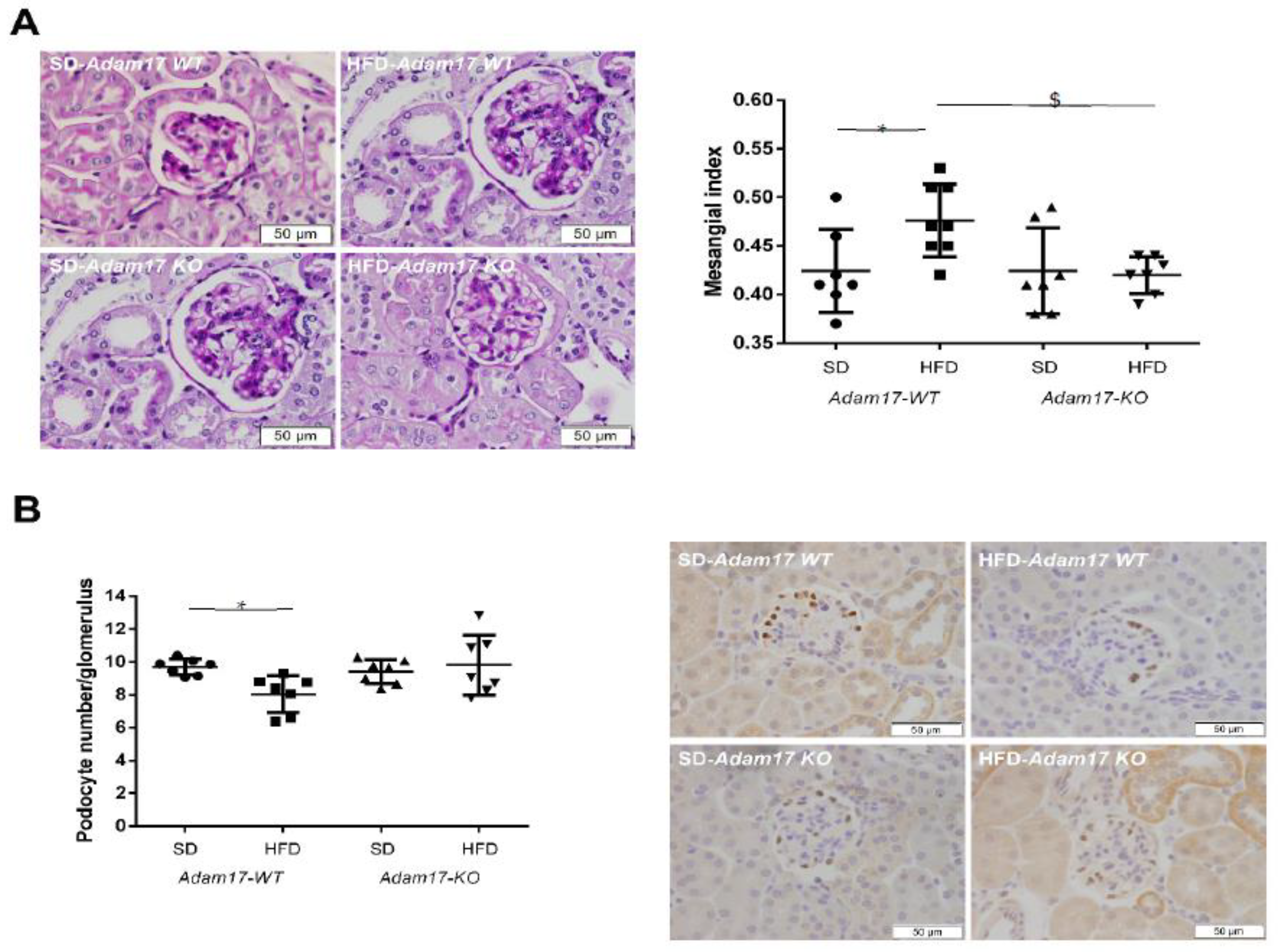
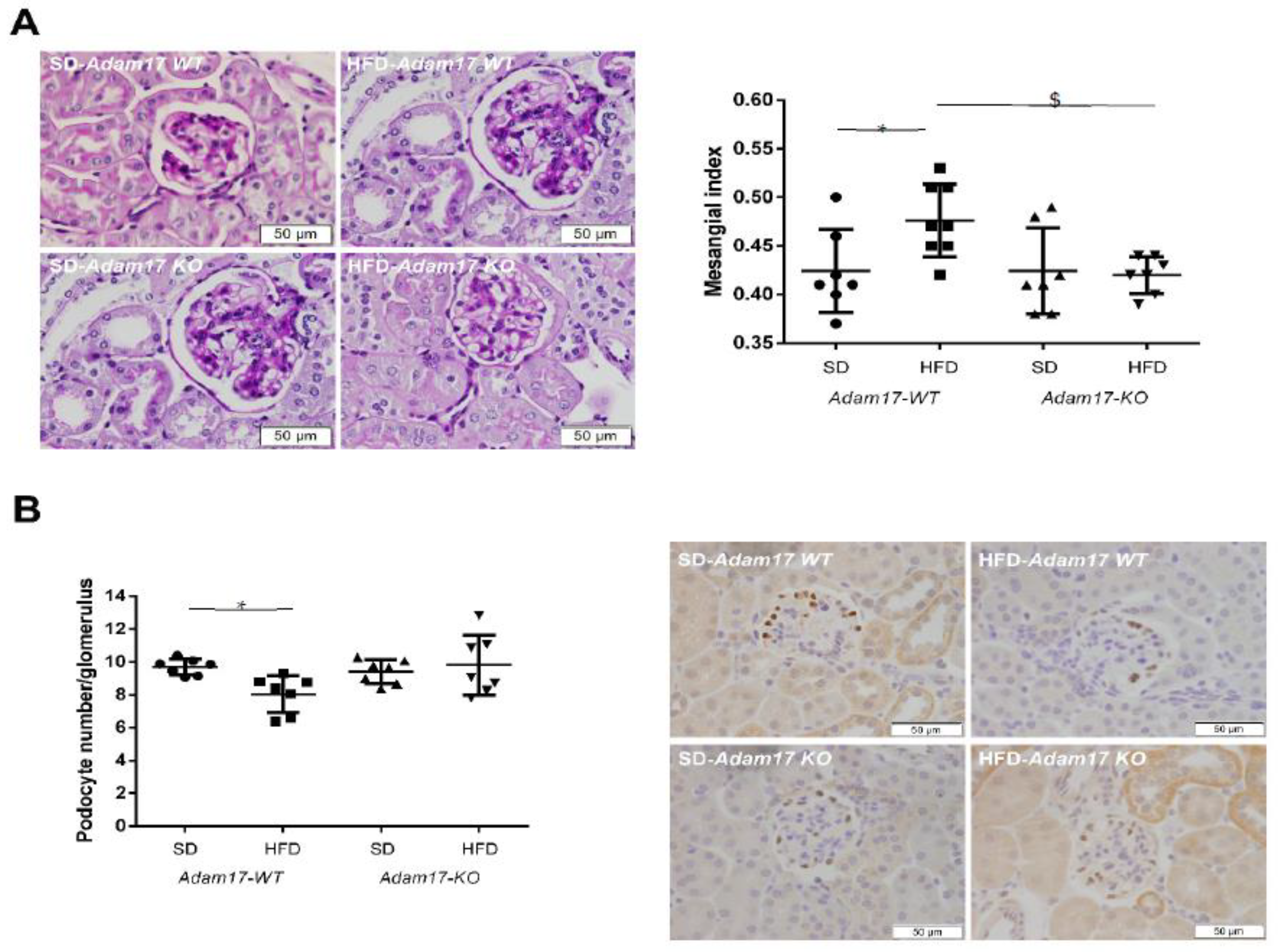
2.3. Adam17 Deletion in Endothelial Cells Protects Obese Mice from Glomerular Alterations
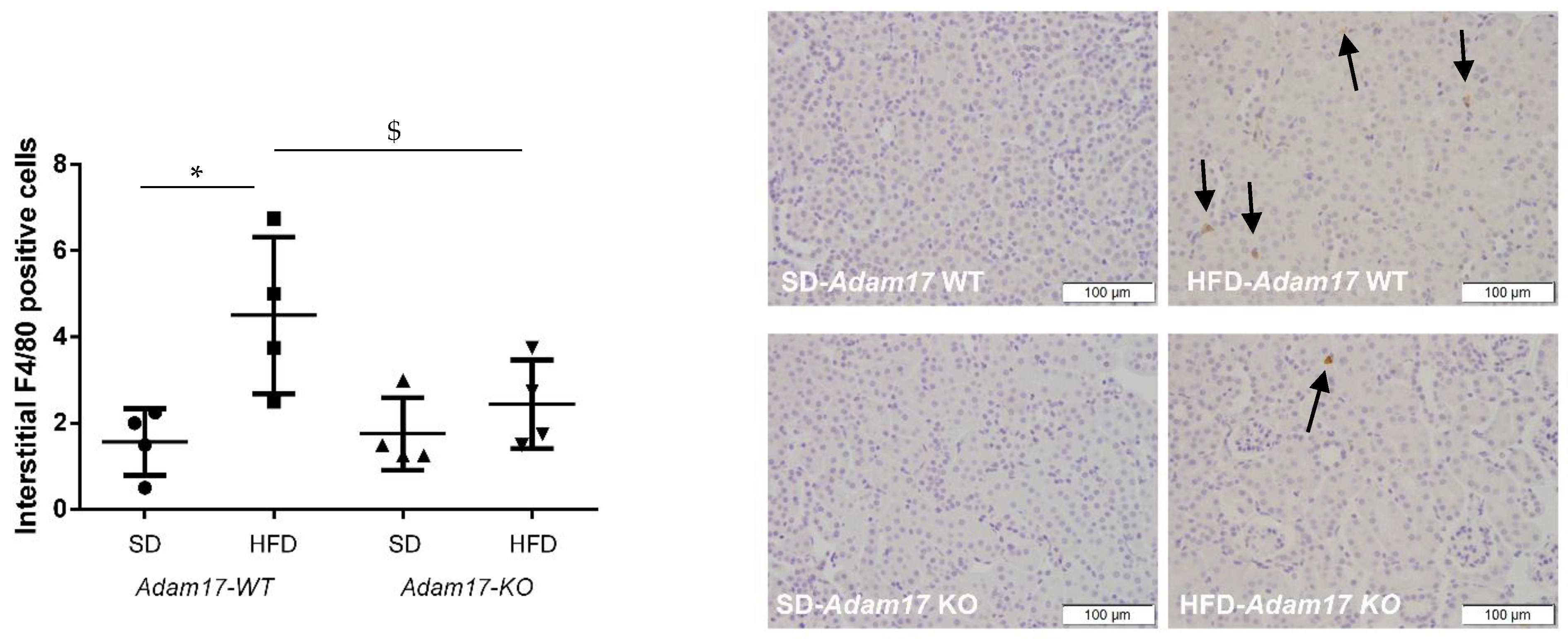
2.4. Endothelial Adam17 Promotes Renal Macrophage Infiltration in Obese Mice
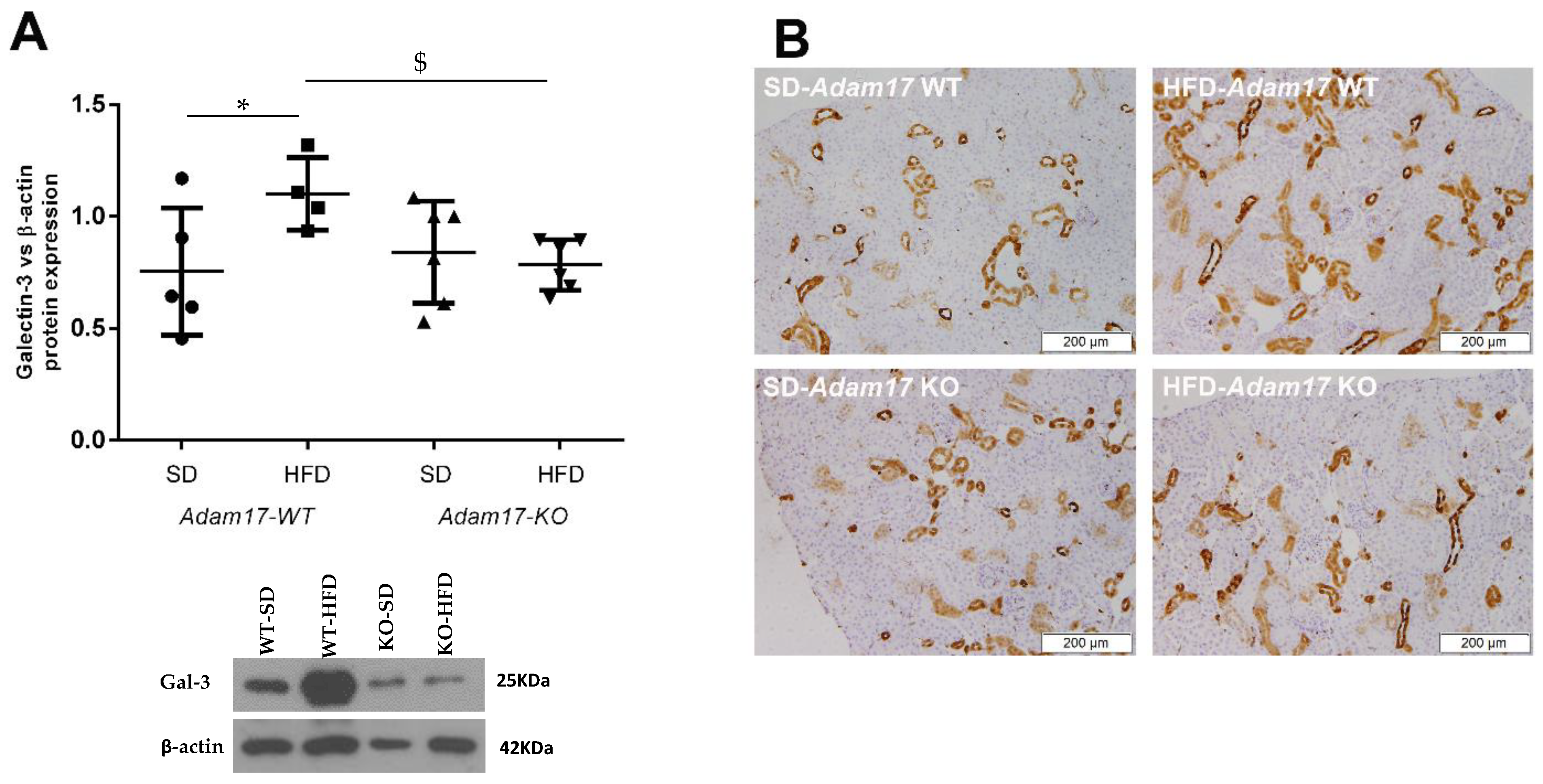
2.5. Renal Galectin-3 Expression Is Modified by Endothelial Adam17 Deletion
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Glucose Tolerance Test
4.3. Urinary Albumin-to-Creatinine Ratio
4.4. Immunohistochemistry on Paraffined-Embedded Tissue
4.5. Western Blot
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Kwaifa, I.K.; Bahari, H.; Yong, Y.K.; Md Noor, S. Endothelial dysfunction in obesity-induced inflammation: Molecular mechanisms and clinical implications. Biomolecules 2020, 10, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, G.B., Jr.; Bentes, A.C.S.N.; Daher, E.D.F.; de Matos, S.M.A. Obesity and kidney disease. J. Bras. Nefrol. 2017, 39, 65–69. [Google Scholar]
- Segovia, S.A.; Vickers, M.H.; Gray, C.; Reynolds, C.M. Maternal obesity, inflammation, and developmental programming. Biomed. Res. Int. 2014, 2014, 418975. [Google Scholar] [CrossRef]
- Segovia, S.A.; Vickers, M.H.; Reynolds, C.M. The impact of maternal obesity on inflammatory processes and consequences for later offspring health outcomes. J. Dev. Orig. Health Dis. 2017, 8, 529–540. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Furth, S.L.; Zoccali, C. Obesity and kidney disease: Hidden consequences of the epidemic. J. Bras. Nefrol. 2017, 39, 1–10. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Dempsey, P.J.; Eguchi, S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am. J. Physiol. Cell Physiol. 2006, 291, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Giricz, O.; Calvo, V.; Peterson, E.A.; Abouzeid, C.M.; Kenny, P.A. TACE-dependent TGFα shedding drives triple-negative breast cancer cell invasion. Int. J. Cancer 2013, 133, 2587–2595. [Google Scholar] [CrossRef]
- Menghini, R.; Fiorentino, L.; Casagrande, V.; Lauro, R.; Federici, M. The role of ADAM17 in metabolic inflammation. Atherosclerosis 2013, 228, 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem. J. 2000, 347, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [Green Version]
- Lownik, J.C.; Farrar, J.S.; Pearce, J.V.; Celi, F.S.; Martin, R.K. Adipocyte ADAM17 plays a limited role in metabolic inflammation. Adipocyte 2020, 9, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Menghini, R.; Menini, S.; Amoruso, R.; Fiorentino, L.; Casagrande, V.; Marzano, V.; Tornei, F.; Bertucci, P.; Iacobini, C.; Serino, M.; et al. Tissue Inhibitor of Metalloproteinase 3 Deficiency Causes Hepatic Steatosis and Adipose Tissue Inflammation in Mice. Gastroenterology 2009, 136, 663–672. [Google Scholar] [CrossRef] [PubMed]
- de Meijer, V.E.; Le, H.D.; Meisel, J.A.; Sharma, A.K.; Popov, Y.; Puder, M. Tumor necrosis factor α-converting enzyme inhibition reverses hepatic steatosis and improves insulin sensitivity markers and surgical outcome in mice. PLoS ONE 2011, 10, e0124260. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, H.; Anzai, T.; Horiuchi, K.; Morimoto, K.; Anzai, A.; Nagai, T.; Sugano, Y.; Maekawa, Y.; Itoh, H.; Yoshikawa, T.; et al. Tumor necrosis factor-α converting enzyme inactivation ameliorates high-fat diet-induced insulin resistance and altered energy homeostasis. Circ. J. 2011, 75, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, M.; Tadaki, H.; Tomimoto, D.; Okuma, C.; Sano, R.; Ishii, Y.; Katsuda, Y.; Yoshiuchi, H.; Kakefuda, R.; Ohta, T.; et al. A novel TNF-α converting enzyme (TACE) selective inhibitor JTP-96193 prevents insulin resistance in KK-Ay type 2 diabetic mice and diabetic peripheral neuropathy in type 1 diabetic mice. Biol. Pharm. Bull. 2019, 42, 1906–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melenhorst, W.B.; Visser, L.; Timmer, A.; van den Heuvel, M.C.; Stegeman, C.A.; van Goor, H. ADAM17 upregulation in human renal disease: A role in modulating TGF-α availability? Am. J. Physiol. Physiol. 2009, 297, 781–790. [Google Scholar] [CrossRef]
- Ford, B.M.; Eid, A.A.; Gooz, M.; Barnes, J.L.; Gorin, Y.C.; Abboud, H.E. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. AJP Ren. Physiol. 2013, 305, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Lattenist, L.; Ochodnický, P.; Ahdi, M.; Claessen, N.; Leemans, J.C.; Satchell, S.C.; Florquin, S.; Gerdes, V.E.; Roelofs, J.J.T.H. Renal endothelial protein C receptor expression and shedding during diabetic nephropathy. J. Thromb. Haemost. 2016, 14, 1171–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Uttarwar, L.; Gao, B.; Charbonneau, M.; Shi, Y.; Chan, J.S.D.; Dubois, C.M.; Krepinsky, J.C. High glucose up-regulates ADAM17 through HIF-1α in mesangial cells. J. Biol. Chem. 2015, 290, 21603–21614. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Li, L.; Huang, M.; Tan, Y.; Shang, Y.; Meng, X.; Pang, Y.; Xu, H.; Zhao, X.; Lei, W.; et al. Sanye Tablet Ameliorates Insulin Resistance and Dysregulated Lipid Metabolism in High-Fat Diet-Induced Obese Mice. Front. Pharmacol. 2021, 12, 713750. [Google Scholar] [CrossRef] [PubMed]
- Salem, E.S.B.; Grobe, N.; Elased, K.M. Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. AJP Ren. Physiol. 2014, 306, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera, M.; Anguiano, L.; Clotet, S.; Roca-Ho, H.; Rebull, M.; Pascual, J.; Soler, M.J. Paricalcitol modulates ACE2 shedding and renal ADAM17 in NOD mice beyond proteinuria. Am. J. Physiol. Physiol. 2016, 310, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Chodavarapu, H.; Grobe, N.; Somineni, H.K.; Salem, E.S.B.; Madhu, M.; Elased, K.M. Rosiglitazone Treatment of Type 2 Diabetic db/db Mice Attenuates Urinary Albumin and Angiotensin Converting Enzyme 2 Excretion. PLoS ONE 2013, 8, e62833. [Google Scholar] [CrossRef]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef]
- Deji, N.; Kume, S.; Araki, S.I.; Soumura, M.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Koya, D.; Haneda, M.; et al. Structural and functional changes in the kidneys of high-fat diet-induced obese mice. Am. J. Physiol. Ren. Physiol. 2009, 296, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Uzu, T.; Araki, S.I.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Kubota, N.; Terauchi, Y.; Kadowaki, T.; et al. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. J. Am. Soc. Nephrol. 2007, 18, 2715–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Lane, P.H.; Lane, J.T.; Padanilam, B.J.; Sansom, S.C. Glomerular structural and functional changes in a high-fat diet mouse model of early-stage Type 2 diabetes. Diabetologia 2004, 47, 1541–1549. [Google Scholar] [CrossRef] [Green Version]
- Palau, V.; Villanueva, S.; Jarrín, J.; Benito, D.; Márquez, E.; Rodríguez, E.; Soler, M.J.; Oliveras, A.; Gimeno, J.; Sans, L.; et al. Redefining the Role of ADAM17 in Renal Proximal Tubular Cells and Its Implications in an Obese Mouse Model of Pre-Diabetes. Int. J. Mol. Sci. 2021, 22, 13093. [Google Scholar] [CrossRef]
- Mogensen, C.E.; Christensen, C.K. Predicting Diabetic Nephropathy in Insulin-Dependent Patients. N. Engl. J. Med. 1984, 311, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Viberti, G.C.; Jarrett, R.J.; Keen, H. Microalbuminuria as predictor of nephropathy in diabetics. Lancet 1982, 1, 1430–1432. [Google Scholar] [CrossRef]
- Perkins, B.A.; Ficociello, L.H.; Silva, K.H.; Finkelstein, D.M.; Warram, J.H.; Krolewski, A.S. Regression of microalbuminuria in type 1 diabetes. N. Engl. J. Med. 2003, 348, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.J.; Wei, R.B.; Zhao, J.; Su, T.Y.; Li, Q.P.; Yang, X.; Chen, X.M. Albuminuria and endothelial dysfunction in patients with non-diabetic chronic kidney disease. Med. Sci. Monit. 2017, 23, 4447–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, Y.; Ning, G.; Deb, D.K.; Kong, J.; Yan, C.L. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proc. Natl. Acad. Sci. USA 2008, 105, 15896–15901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eren, Z.; Günal, M.Y.; Bakir, E.A.; Coban, J.; Çaʇlayan, B.; Ekimci, N.; Ethemoglu, S.; Albayrak, O.; Akdeniz, T.; Demirel, G.Y.; et al. Effects of paricalcitol and aliskiren combination therapy on experimental diabetic nephropathy model in rats. Kidney Blood Press. Res. 2014, 39, 581–590. [Google Scholar] [CrossRef]
- Arcidiacono, M.V.; Yang, J.; Fernandez, E.; Dusso, A. The induction of C/EBPβ′ contributes to vitamin D inhibition of ADAM17 expression and parathyroid hyperplasia in kidney disease. Nephrol. Dial. Transplant. 2015, 30, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Harris, R. Renal Endothelial Dysfunction in Diabetic Nephropathy. Cardiovasc. Hematol. Disord. Targets 2014, 14, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Griffin, K.A.; Kramer, H.; Bidani, A.K. Adverse renal consequences of obesity. Am. J. Physiol. Ren. Physiol. 2008, 294, F685–F696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziyadeh, F.N. Mediators of Diabetic Renal Disease: The Case for TGF-β as the Major Mediator. J. Am. Soc. Nephrol. 2004, 15, S55–S57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maric, C.; Hall, J.E. Obesity, Metabolic syndrome and diabetic nephropathy. Contrib. Nephrol. 2011, 170, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.N.; Chen, X.; Li, R.; Gao, B.; Mohammed-Ali, Z.; Lu, C.; Yum, V.; Dickhout, J.G.; Krepinsky, J.C. SREBP-1 mediates angiotensin II-induced TGF-β1 upregulation and glomerular fibrosis. J. Am. Soc. Nephrol. 2015, 26, 1839–1854. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wang, T.; Uttarwar, L.; vanKrieken, R.; Li, R.; Chen, X.; Gao, B.; Ghayur, A.; Margetts, P.; Krepinsky, J.C. SREBP-1 is a novel mediator of TGFβ1 signaling in mesangial cells. J. Mol. Cell Biol. 2014, 6, 516–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttarwar, L.; Gao, B.; Ingram, A.J.; Krepinsky, J.C. SREBP-1 activation by glucose mediates TGF-β upregulation in mesangial cells. Am. J. Physiol. Ren. Physiol. 2012, 302, F329–F341. [Google Scholar] [CrossRef] [PubMed]
- King, G.L. The Role of Inflammatory Cytokines in Diabetes and Its Complications. J. Periodontol. 2008, 79, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, R.A.; Bijzet, J.; Meijers, W.C.; Yakala, G.K.; Kleemann, R.; Nguyen, T.Q.; De Boer, R.A.; Schalkwijk, C.G.; Hazenberg, B.P.C.; Tietge, U.J.F.; et al. Obesity-induced chronic inflammation in high fat diet challenged C57BL/6J mice is associated with acceleration of age-dependent renal amyloidosis. Sci. Rep. 2015, 5, 16474. [Google Scholar] [CrossRef]
- Fernandes, R.; Garver, H.; Harkema, J.R.; Galligan, J.J.; Fink, G.D.; Xu, H. Sex differences in renal inflammation and injury in high-fat diet-fed Dahl salt-sensitive rats. Hypertension 2018, 72, e43–e52. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kang, J.S.; Kim, H.M.; Lee, E.S.; Lee, J.H.; Chung, C.H.; Lee, E.Y. CCR2 knockout ameliorates obesity-induced kidney injury through inhibiting oxidative stress and ER stress. PLoS ONE 2019, 14, e0222352. [Google Scholar] [CrossRef]
- Palau, V.; Nugraha, B.; Benito, D.; Pascual, J.; Emmert, M.Y.; Hoerstrup, S.P.; Riera, M.; Soler, M.J. Both specific endothelial and proximal tubular ADAM17 deletion protect against diabetic nephropathy. Int. J. Mol. Sci. 2021, 22, 5520. [Google Scholar] [CrossRef]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Reeves, W.B. Macrophage-derived Tumor Necrosis Factor-α mediates diabetic renal injury HHS Public Access. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.C.; Kuo, P.L. The role of galectin-3 in the kidneys. Int. J. Mol. Sci. 2016, 17, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, U.C.; Pokharel, S.; Van Brakel, T.J.; Van Berlo, J.H.; Cleutjens, J.P.M.; Schroen, B.; André, S.; Crijns, H.J.G.M.; Gabius, H.J.; Maessen, J.; et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004, 110, 3121–3128. [Google Scholar] [CrossRef]
- Djoussé, L.; Matsumoto, C.; Petrone, A.; Weir, N.L.; Tsai, M.Y.; Gaziano, J.M. Plasma galectin 3 and heart failure risk in the Physicians’ Health Study. Eur. J. Heart Fail. 2014, 16, 350–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.C.B.; Cheung, C.L.; Lee, A.C.H.; Lam, J.K.Y.; Wong, Y.; Shiu, S.W.M. Galectin-3 is independently associated with progression of nephropathy in type 2 diabetes mellitus. Diabetologia 2018, 61, 1212–1219. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.Y.; Shih, C.M.; Huang, C.Y.; Wu, A.T.H.; Cheng, T.M.; Mi, F.L.; Shih, C.C. Galectin-3 modulates macrophage activation and contributes smooth muscle cells apoptosis in abdominal aortic aneurysm pathogenesis. Int. J. Mol. Sci. 2020, 21, 8257. [Google Scholar] [CrossRef]
- Kuwabara, I.; Liu, F.T. Galectin-3 promotes adhesion of human neutrophils to laminin. J. Immunol. 1996, 156, 3939–3944. [Google Scholar] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.T.; Hughes, J.; Sethi, T. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Endre, Z.H.; Walker, R.J. Biomarkers of Cardiovascular Risk in Chronic Kidney Disease. In Biomarkers of Kidney Disease; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780128030141. [Google Scholar]
- Wilson, C.L.; Gough, P.J.; Chang, C.A.; Chan, C.K.; Frey, J.M.; Liu, Y.; Braun, K.R.; Chin, M.T.; Wight, T.N.; Raines, E.W. Endothelial deletion of ADAM17 in mice results in defective remodeling of the semilunar valves and cardiac dysfunction in adults. Mech. Dev. 2013, 130, 272–289. [Google Scholar] [CrossRef] [PubMed]
- Claxton, S.; Kostourou, V.; Jadeja, S.; Chambon, P.; Hodivala-Dilke, K.; Fruttiger, M. Efficient, inducible cre-recombinase activation in vascular endothelium. Genesis 2008, 46, 74–80. [Google Scholar] [CrossRef]
- Clotet, S.; Soler, M.J.; Rebull, M.; Gimeno, J.; Gurley, S.B.; Pascual, J.; Riera, M. Gonadectomy prevents the increase in blood pressure and glomerular injury in angiotensin-converting enzyme 2 knockout diabetic male mice. Effects on renin-angiotensin system. J. Hypertens. 2016, 34, 1752–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca-Ho, H.; Palau, V.; Gimeno, J.; Pascual, J.; Soler, M.J.; Riera, M. Angiotensin-converting enzyme 2 influences pancreatic and renal function in diabetic mice. Lab. Investig. 2020, 100, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Márquez, E.; Clotet, S.; Gimeno, J.; Roca-Ho, H.; Lloreta, J.; Juanpere, N.; Batlle, D.; Pascual, J.; Soler, M.J. Effect of insulin on ACE2 activity and kidney function in the non-obese diabetic mouse. PLoS ONE 2014, 9, e84683. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fasting Blood Glucose (mg/dL) | Body Weight (g) | Kidney Weight (g) | ACR (μg Alb/mg Crea) | |
|---|---|---|---|---|
| Adam17WT-SD | 185.71 ± 16.39 | 36.83 ± 7.79 | 0.32 ± 0.04 | 27.26 ± 11.36 |
| Adam17WT-HFD | 245.38 ± 36.29 * | 50.91 ± 4.98 * | 0.39 ± 0.06 * | 50.43 ± 13.03 * |
| Adam17KO-SD | 193.45 ± 17.83 | 40.23 ± 6.49 | 0.34 ± 0.05 | 20.16 ± 6.24 |
| Adam17KO-HFD | 235.40 ± 27.31 # | 49.69 ± 6.06 # | 0.38 ± 0.07 | 29.74 ± 9.55 $ |
| AUC (mg/dL/min) | |
|---|---|
| Adam17WT-SD | 19,059.64 ± 4509.13 |
| Adam17WT-HFD | 30,221.79 ± 3366.05 * |
| Adam17KO-SD | 23,692.5 ± 7531.78 |
| Adam17KO-HFD | 27,831 ± 3955.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palau, V.; Jarrín, J.; Villanueva, S.; Benito, D.; Márquez, E.; Rodríguez, E.; Soler, M.J.; Oliveras, A.; Gimeno, J.; Sans, L.; et al. Endothelial ADAM17 Expression in the Progression of Kidney Injury in an Obese Mouse Model of Pre-Diabetes. Int. J. Mol. Sci. 2022, 23, 221. https://doi.org/10.3390/ijms23010221
Palau V, Jarrín J, Villanueva S, Benito D, Márquez E, Rodríguez E, Soler MJ, Oliveras A, Gimeno J, Sans L, et al. Endothelial ADAM17 Expression in the Progression of Kidney Injury in an Obese Mouse Model of Pre-Diabetes. International Journal of Molecular Sciences. 2022; 23(1):221. https://doi.org/10.3390/ijms23010221
Chicago/Turabian StylePalau, Vanesa, Josué Jarrín, Sofia Villanueva, David Benito, Eva Márquez, Eva Rodríguez, María José Soler, Anna Oliveras, Javier Gimeno, Laia Sans, and et al. 2022. "Endothelial ADAM17 Expression in the Progression of Kidney Injury in an Obese Mouse Model of Pre-Diabetes" International Journal of Molecular Sciences 23, no. 1: 221. https://doi.org/10.3390/ijms23010221
APA StylePalau, V., Jarrín, J., Villanueva, S., Benito, D., Márquez, E., Rodríguez, E., Soler, M. J., Oliveras, A., Gimeno, J., Sans, L., Crespo, M., Pascual, J., Barrios, C., & Riera, M. (2022). Endothelial ADAM17 Expression in the Progression of Kidney Injury in an Obese Mouse Model of Pre-Diabetes. International Journal of Molecular Sciences, 23(1), 221. https://doi.org/10.3390/ijms23010221