
The Influence of Mitochondrial-DNA-Driven Inflammation Pathways on Macrophage Polarization: A New Perspective for Targeted Immunometabolic Therapy in Cerebral Ischemia-Reperfusion Injury

 ,
,
Abstract
:1. Introduction
2. The Function of Macrophages in Cerebral Ischemia-Reperfusion
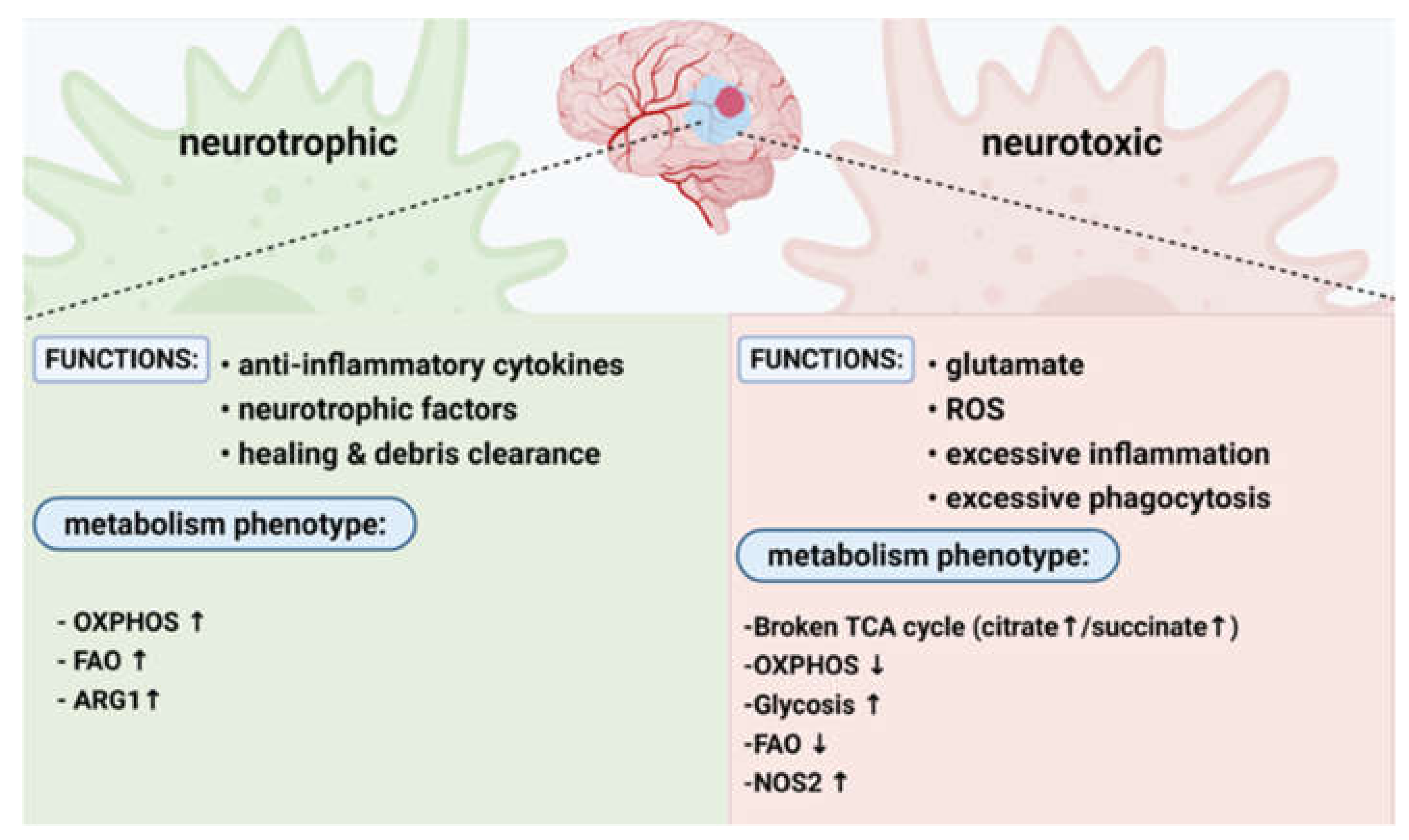
2.1. Neurotoxic Macrophages
2.2. Neuroprotective Macrophages
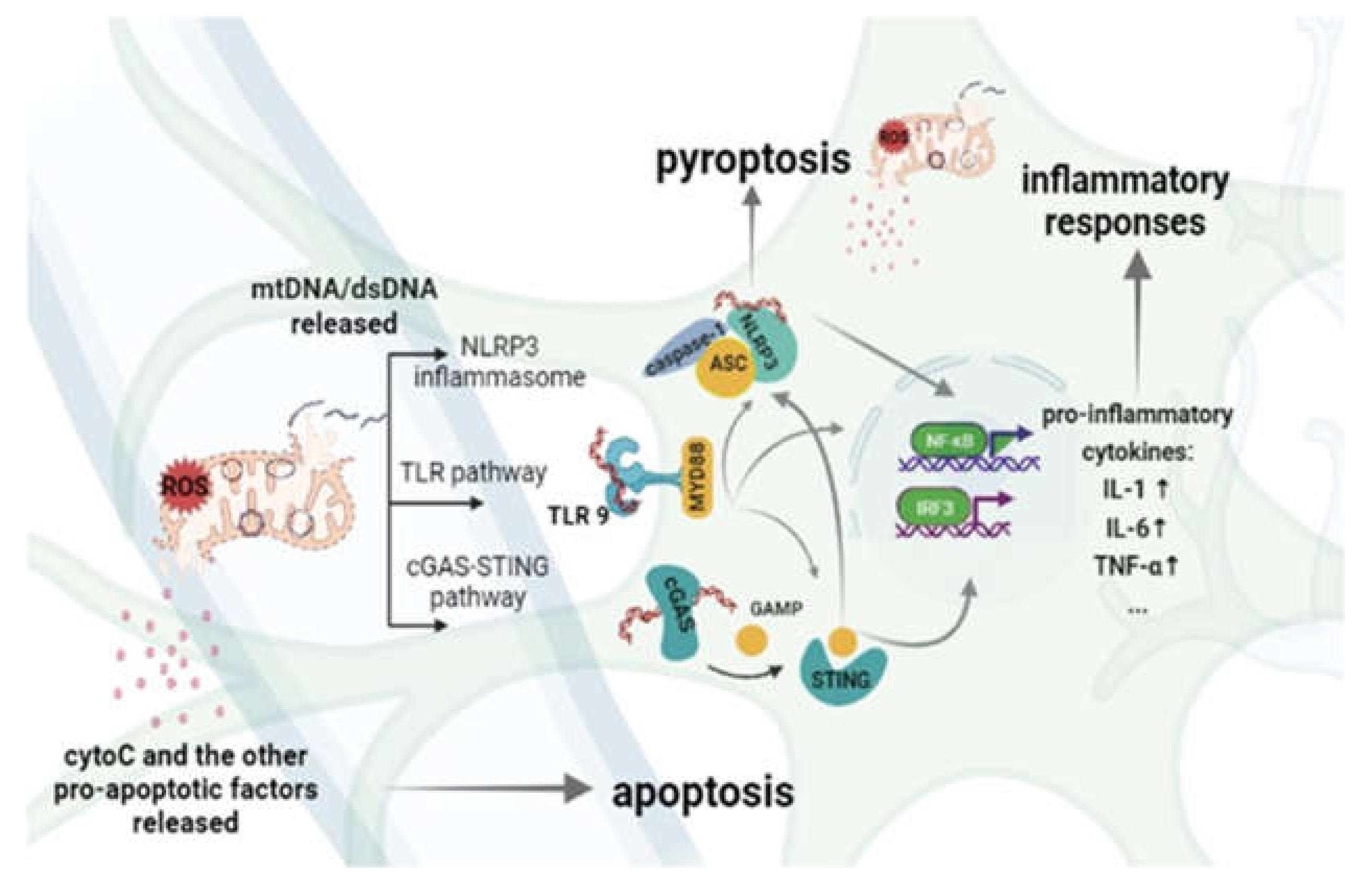
3. The Inflammatory Response Is Driven by MtDNA in Cerebral Ischemia-Reperfusion Injury
3.1. TLRs
3.2. NLRP3
3.3. STING
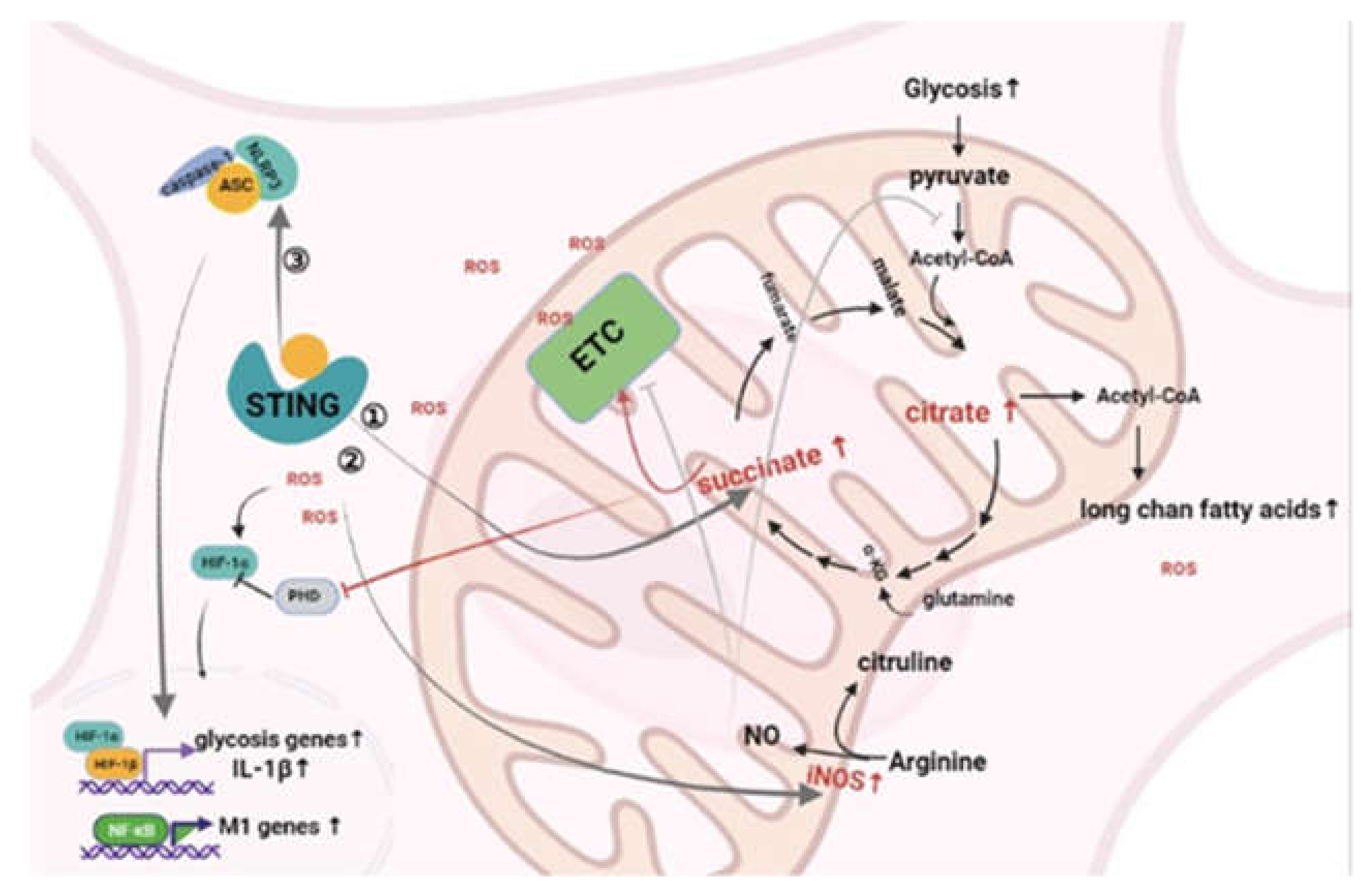
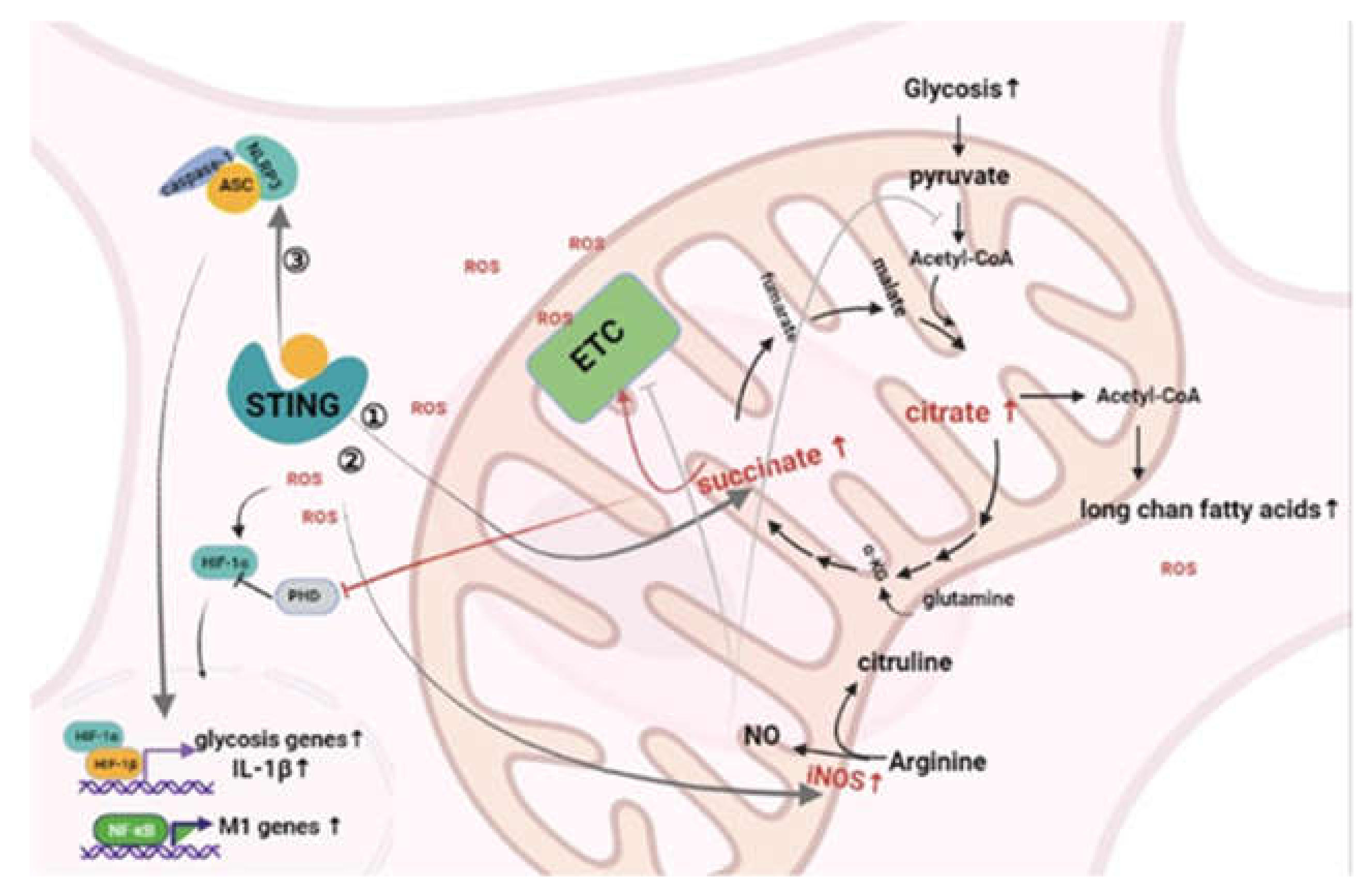
4. The Influence of MtDNA-Mediated Inflammation on the Metabolic Pattern of Macrophages
4.1. cGAS/STING
4.2. NLRP3 Inflammasome
4.3. TLRs
5. The Therapeutic Prospects of Cerebral Ischemia-Reperfusion Targeting MtDNA-Mediated Inflammation and Microglia/Macrophage Metabolism
5.1. Therapies That Target TLRs
5.2. Therapies That Target NLRP3
5.3. Therapies That Target cGAS/STING
5.4. Therapies That Target Microglia/Macrophage Metabolism
6. Conclusions and Perspectives

{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitor | Mechanism | Models | Reference |
|---|---|---|---|---|
| TLRs | Garcinol | TLR4-NFκB↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R), oxygen–glucose deprivation and reperfusion (OGD/R) | [113] |
| Kudiezi injection | TLR4-NFκB↓ | Rat models of transient middle cerebral artery occlusion (tMCAO) | [114] | |
| Pregabalin | HMGB1/TLR4-NFκB↓ | Middle cerebral artery occlusion (MCAO) model | [115] | |
| Propofol | TLR4-NFκB↓ | Retinal ischemia reperfusion injury (RIRI) | [116] | |
| Lactoferrin | TLR4-related pathways↓ | Anoxia and reoxygenation cell model, Institute for Cancer Research (ICR) mice | [117] | |
| Lixisenatide | TLR4-NFκB↓ P38/ERK↑ | Not applicable | [118] | |
| Tangeretin | Inflammatory cytokine brain injury markers↓ | Mice model of cerebral ischemia/reperfusion injury | [120] | |
| QiShenYiQi | TLR4-NFκB↓ | OGD/R | [121] | |
| Nobiletin | Akt/mTOR↑ TLR4–NFκB↓ | MCAO | [122] | |
| Argon | TLR2/TLR4/STAT3/NFκB↓ | Retinal ischemia reperfusion injury (IRI) in rats | [124] | |
| CX-10 | Nrf2/AE TLR/NFκB | Rat models of middle cerebral artery occlusion/reperfusion (MCAO/R) | [125] | |
| NLRP3 | Diazoxide | NLRP3 inflammasome activation↓ | Transient middle cerebral artery occlusion (tMCAO) rat model, oxygen–glucose deprivation/reoxygenation (OGD/R) | [98] |
| Sulforaphane | NLRP3 inflammasome activation↓ | Middle cerebral artery occlusion (MCAO) model | [126] | |
| Verapamil | NLRP3 inflammasome activation↓ | Transient middle cerebral artery occlusion (MCAO) | [127] | |
| IMM-H004 | NLRP3 inflammasome activation↓ | PMCAO model of focal ischemia | [128] | |
| Hispidulin | AMPK/GSK3β/NLRP3- pyrolysis↓ | Middle cerebral artery occlusion (MCAO), oxygen–glucose deprivation/reoxygenation (OGD/R) | [129] | |
| Astilbin | AMPK/GSK3β/NLRP3- pyrolysis↓ | middle cerebral artery occlusion (tMCAO) model with C57BL/6 J mice, oxygen–glucose deprivation and reintroduction (OGD-R) model | [131] | |
| Adiponectin peptide | AMPK/GSK3β/NLRP3- pyrolysis↓ | middle cerebral artery occlusion-reperfusion (MCAO/R) model in rats | [132] | |
| SB216763 | AMPK/GSK3β/NLRP3- pyrolysis↓ | Middle cerebral artery occlusion–reperfusion (MCAO/R) model in rats, oxygen–glucose deprivation/reoxygenation (OGD/R) | [133] | |
| Spautin-1 | Autophagy/NLRP3- pyrolysis↓ | Middle cerebral artery occlusion–reperfusion (MCAO/R) model in rats, oxygen–glucose deprivation/reoxygenation (OGD/R) | [134] | |
| l-Homocarnosine | NLRP3 expression↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R) model in rats | [135] | |
| PAP-1 | M1 polarization↓ NLRP3 inflammasome activation↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R) model in rats and oxygen–glucose deprivation/reoxygenation (OGD/R) in primary microglia | [136] | |
| TMEM59 | NLRP3- pyrolysis↓ NLRP3-inflammation↓ | middle cerebral artery occlusion (MCAO), oxygen–glucose deprivation/reperfusion (OGD/R) | [137] | |
| MCC950 | NLRP3- pyrolysis↓ | Oxygen–glucose deprivation/reoxygenation (OGD/R) | [140] | |
| Chlorpromazine and promethazine | NLRP3 inflammasome activation↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R) model in rats | [142] | |
| Idebenone | NLRP3 inflammasome activation↓ | Oxygen–glucose deprivation and reperfusion (OGD/R) | [143] | |
| Resveratrol | NLRP3 inflammasome activation↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R) model in rats | [144] | |
| CD21 | HMGB1/TLR4-NFκB↓ Cathepsin B/NLRP3 inflammasome activation↓ | Global cerebral ischemia–reperfusion model in mice | [145] | |
| Meisoindigo | TLR4-NFκB↓ Cathepsin B/NLRP3 inflammasome Activation↓ M1 polarization↓ | C57BL/6J mice middle cerebral artery occlusion (MCAO) stroke model, HT-22 and BV2 cells in vitro oxygen–glucose deprivation/reperfusion (OGD/R) model | [146] | |
| JQ1 | NLRP3- pyrolysis↓ NLRP3- inflammation↓ | Middle cerebral artery occlusion (MCAO) model | [147] | |
| Tomentosin | TLR and NLRP3 inflammation-related pathways↓ | Rats Cerebral ischemia–reperfusion model, oxygen–glucose deprivation/reperfusion (OGD-R) | [148] | |
| Anthocyanin | TLR and NLRP3 inflammation-related pathways↓ | ICR mice cerebral ischemic/reperfusion (I/R) injury | [149] | |
| Procyanidins | TLR and NLRP3 inflammation-related pathways↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R), oxygen–glucose deprivation/reoxygenation BV2 cells | [150] | |
| Salvianolic acids | M2 polarization↑ Neuronal apoptosis ↓ NLRP3- pyrolysis↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R) model in rats, oxygen–glucose deprivation/reoxygenation (OGD/R) model | [151] | |
| Buyang Huanwu decoction | NLRP3- pyrolysis↓ | Rats focal cerebral ischemia and reperfusion model | [152] | |
| Genistein (Gen) | NLRP3 inflammasome activation↓ | Not applicable | [153] | |
| cGAS/STING | 25-HC | Autophagy↑ STING degradation↑ | Liver I/R model | [157] |
| MicroRNA-24-3p | Inhibit cGAS/STING | Transient middle cerebral artery occlusion (tMCAO) | [158] | |
| Liproxstatin-1 | Inhibit cGAS/STING | Not applicable | [155] | |
| A151 | cGAS expression↓ | Not applicable | [159] | |
| Macrophage metabolism | STF31 | Glycolysis↓ | Diabetic retinopathy (DR) model | [161] |
| 2-DG | Glycolysis↓ | not applicable | [162] | |
| HK1 inhibitor | Glycolysis↓ | Middle cerebral artery occlusion/reperfusion (MCAO/R) model in rats | [163] | |
| Astragaloside IV | PPARγ- M2 polarization↑ | Rat traumatic brain injury (TBI) model | [164] | |
| CsA | Mitophagy↓- Glycolysis↓ M2 polarization↑ | Not applicable | [11,12] | |
| 3-BU | Glycolysis↓ OXPHOS↓ | Not applicable | [165] | |
| L-NMMA | Inhibit iNOS | Not applicable | [166] | |
| Midivi-1 | Mitochondrial division↓ Glycolysis↓ | Not applicable | [169] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DAMP | Damage-associated molecular pattern |
| BINCs | Brain Iba1 + /NG2 + cells |
| cGAS | cyclic GMP-AMP synthase |
| FAO | Fatty acid oxidation |
| GLP-1 | Glucagon-like peptide 1 |
| HDAC3 | Histone deacetylase 3 |
| IRF3 | Interferon regulatory factor 3 |
| JAK | Janus kinase |
| Jmjd3 | Jumonji domain containing-3 |
| MHC-Ⅱ | Major histocompatibility complex class II receptor |
| MST1 | Macrophage stimulating 1 |
| mt-DAMP | mitochondrial damage-associated molecular pattern |
| MyD88 | MYD88 innate immune signal transduction adaptor |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NOD | Nucleotide-binding and oligomerization domain |
| OGD/R | Oxygen and glucose deprivation-reperfusion |
| OXPHOS | Oxidative phosphorylation |
| P2Y6 | Pyrimidinergic receptor P2Y6 |
| PAMP | Pathogen-associated molecular pattern |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PRR | Pattern-recognition receptor |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT6 | Signal transducer and activator of transcription 6 |
| STING | Stimulator of interferon genes |
| TBK1 | TANK binding kinase 1 |
| TCA | Tricarboxylic acid |
| TLR | Toll-like receptor |
| UDP | Uridine diphosphate |
References
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; de Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Ahnstedt, H.; Sweet, J.; Cruden, P.; Bishop, N.; Cipolla, M.J. Effects of Early Post-Ischemic Reperfusion and tPA on Cere-brovascular Function and Nitrosative Stress in Female Rats. Transl. Stroke Res. 2016, 7, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.; Bastan, B. Identifying and utilizing the ischemic penumbra. Neurology 2012, 79, S79–S85. [Google Scholar] [CrossRef] [Green Version]
- Leigh, R.; Knutsson, L.; Zhou, J.; Van Zijl, P.C. Imaging the physiological evolution of the ischemic penumbra in acute ischemic stroke. Br. J. Pharmacol. 2017, 38, 1500–1516. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and Inflammation after Ischemic Stroke: What is Known and Where We Go from Here. Br. J. Pharmacol. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Zhang, Q.; Liu, Y. Analysis of knowledge bases and research focuses of cerebral ischemia-reperfusion from the perspective of mapping knowledge domain. Brain Res. Bull. 2019, 156, 15–24. [Google Scholar] [CrossRef]
- Mayevsky, A.; Kutai-Asis, H.; Tolmasov, M. Mitochondrial function and brain Metabolic Score (BMS) in ischemic Stroke: Evaluation of “neuroprotectants” safety and efficacy. Mitochondrion 2019, 50, 170–194. [Google Scholar] [CrossRef]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free. Radic. Biol. Med. 2019, 146, 45–58. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Gan, Q.; Yang, Y.; Reis, C.; Zhang, Z.; Xu, S.; Zhang, T.; Sun, C. Mitophagy in Cerebral Ischemia and Ischemia/Reperfusion Injury. Front. Aging Neurosci. 2021, 13, 687246. [Google Scholar] [CrossRef]
- Wilkins, H.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory mi-croglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Anzell, A.R.; Maizy, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial Quality Control and Disease: Insights into Ische-mia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 2547–2564. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.T.; Fukuda, D.; Nishimoto, S.; Kim-Kaneyama, J.-R.; Lei, X.-F.; Takahashi, Y.; Sato, T.; Tanaka, K.; Suto, K.; Kawabata, Y.; et al. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: A link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur. Hear. J. 2021, 42, 4336–4348. [Google Scholar] [CrossRef] [PubMed]
- Augusto-Oliveira, M.; Arrifano, G.P.; Lopes-Araujo, A.; Santos-Sacramento, L.; Takeda, P.Y.; Anthony, D.C.; Malva, J.O.; Crespo-Lopez, M.E. What Do Microglia Really Do in Healthy Adult Brain? Cells 2019, 8, 1293. [Google Scholar] [CrossRef] [Green Version]
- Surinkaew, P.; Sawaddiruk, P.; Apaijai, N.; Chattipakorn, N.; Chattipakorn, S.C. Role of microglia under cardiac and cerebral ischemia/reperfusion (I/R) injury. Metab. Brain Dis. 2018, 33, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, Y.Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.R. Microglia in Pain: Detrimental and Protective Roles in Pathogene-sis and Resolution of Pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Sahebkar, A. Macrophage plas-ticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217, e20191644. [Google Scholar] [CrossRef]
- Naito, M.G.; Xu, D.; Amin, P.; Lee, J.; Wang, H.; Li, W.; Kelliher, M.; Pasparakis, M.; Yuan, J. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. USA 2020, 117, 4959–4970. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Li, Y.; Peng, X.; Huang, D.; Gui, L.; Huang, B. Resistance of mitochondrial DNA-depleted cells against oxidized low-density lipoprotein-induced macrophage pyroptosis. Mol. Med. Rep. 2016, 13, 4393–4399. [Google Scholar] [CrossRef] [Green Version]
- McDonough, A.; Weinstein, J.R. The role of microglia in ischemic preconditioning. Glia 2019, 68, 455–471. [Google Scholar] [CrossRef]
- Abe, N.; Nishihara, T.; Yorozuya, T.; Tanaka, J. Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems. Cells 2020, 9, 2132. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.-Q.; Ma, X.-T.; Hu, Z.-W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.-J.; Tian, D.-S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delpech, J.-C.; Herron, S.; Botros, M.B.; Ikezu, T. Neuroimmune Crosstalk through Extracellular Vesicles in Health and Disease. Trends Neurosci. 2019, 42, 361–372. [Google Scholar] [CrossRef]
- Zia, S.; Rawji, K.S.; Michaels, N.J.; Burr, M.; Kerr, B.J.; Healy, L.M.; Plemel, J.R. Microglia Diversity in Health and Multiple Sclerosis. Front. Immunol. 2020, 11, 2895. [Google Scholar] [CrossRef] [PubMed]
- Stachon, P.; Peikert, A.; Michel, N.A.; Hergeth, S.; Marchini, T.; Wolf, D.; Dufner, B.; Hoppe, N.; Ayata, K.; Grimm, M.; et al. P2Y 6 Deficiency Limits Vascular Inflammation and Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2014, 34, 2237–2245. [Google Scholar] [CrossRef] [Green Version]
- Neher, J.J.; Neniskyte, U.; Hornik, T.; Brown, G.C. Inhibition of UDP/P2Y 6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 2014, 62, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Anwar, S.; Pons, V.; Rivest, S. Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases. Cells 2020, 9, 1595. [Google Scholar] [CrossRef]
- Du, X.; Zhang, Z.; Zhou, H.; Zhou, J. Differential Modulators of NG2-Glia Differentiation into Neurons and Glia and Their Crosstalk. Cell. Mol. Neurobiol. 2020, 41, 1–15. [Google Scholar] [CrossRef]
- Manich, G.; Recasens, M.; Valente, T.; Almolda, B.; González, B.; Castellano, B. Role of the CD200-CD200R Axis During Homeostasis and Neuroinflammation. Neuroscience 2018, 405, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Smirkin, A.; Matsumoto, H.; Takahashi, H.; Inoue, A.; Tagawa, M.; Ohue, S. Iba1(+)/NG2(+) macrophage-like cells ex-pressing a variety of neuroprotective factors ameliorate ischemic damage of the brain. J. Cereb. Blood Flow Metab. 2010, 30, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, S.; Tanaka, J.; Yano, H.; Takahashi, H.; Sugimoto, K.; Ohue, S. CD200+ and CD200- macrophages accumu-lated in ischemic lesions of rat brain: The two populations cannot be classified as either M1 or M2 macrophages. J. Neuroimmunol. 2015, 282, 7–20. [Google Scholar] [CrossRef]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neuro-genesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, G.; Uhlemann, R.; Richter, N.; Klempin, F.; Wegner, S.; Staerck, L.; Wolfgang, U.; Uckert, W.; Kettenmann, H.; Endres, M.; et al. Distinguishing features of microglia- and monocyte-derived macrophages after stroke. Acta Neuropathol. 2017, 135, 551–568. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kumon, Y.; Watanabe, H.; Ohnishi, T.; Shudou, M.; Chuai, M.; Imai, Y.; Takahashi, H.; Tanaka, J. Accumulation of Macrophage-Like Cells Expressing NG2 Proteoglycan and Iba1 in Ischemic Core of Rat Brain after Transient Middle Cerebral Artery Occlusion. Br. J. Pharmacol. 2007, 28, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, T.; Tanaka, J.; Sekiya, K.; Nishikawa, Y.; Abe, N.; Hamada, T.; Kitamura, S.; Ikemune, K.; Ochi, S.; Choudhury, M.E.; et al. Chronic constriction injury of the sciatic nerve in rats causes different activation modes of microglia between the anterior and posterior horns of the spinal cord. Neurochem. Int. 2020, 134, 104672. [Google Scholar] [CrossRef] [PubMed]
- Walko, T.D., 3rd; Bola, R.A.; Hong, J.D.; Au, A.K.; Bell, M.J.; Kochanek, P.M.; Aneja, R.K. Cerebrospinal fluid mitochondrial DNA: A novel DAMP in pediatric traumatic brain injury. Shock 2014, 41, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Mathew, A.; Lindsley, T.A.; Sheridan, A.; Bhoiwala, D.L.; Hushmendy, S.F.; Yager, E.J.; Crawford, D.R. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegener-ation. J. Alzheimers Dis. 2012, 30, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Chamaillard, M.; McDonald, C.; Nunez, G. NOD-LRR proteins: Role in host-microbial interactions and inflammato-ry disease. Annu. Rev. Biochem. 2005, 74, 355–383. [Google Scholar] [CrossRef]
- Wang, P.F.; Xiong, X.Y.; Chen, J.; Wang, Y.C.; Duan, W.; Yang, Q.W. Function and mechanism of toll-like receptors in cer-ebral ischemic tolerance: From preconditioning to treatment. J. Neuroinflamm. 2015, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map ki-nase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Hyakkoku, K.; Hamanaka, J.; Tsuruma, K.; Shimazawa, M.; Tanaka, H.; Uematsu, S.; Akira, S.; Inagaki, N.; Nagai, H.; Hara, H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010, 171, 258–267. [Google Scholar] [CrossRef]
- Brea, D.; Sobrino, T.; Rodríguez-Yáñez, M.; Ramos-Cabrer, P.; Agulla, J.; Rodríguez-González, R.; Campos, F.; Blanco, M.; Castillo, J. Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin. Immunol. 2011, 139, 193–198. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Shimamura, M.; Jackman, K.; Kurinami, H.; Anrather, J.; Zhou, P.; Iadecola, C. Key role of CD36 in Toll-like receptor 2 sig-naling in cerebral ischemia. Stroke 2010, 41, 898–904. [Google Scholar] [CrossRef] [Green Version]
- Lehnardt, S.; Lehmann, S.; Kaul, D.; Tschimmel, K.; Hoffmann, O.; Cho, S.; Krueger, C.; Nitsch, R.; Meisel, A.; Weber, J.R. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J. Neuroimmunol. 2007, 190, 28–33. [Google Scholar] [CrossRef]
- Ziegler, G.; Harhausen, D.; Schepers, C.; Hoffmann, O.; Rohr, C.; Prinz, V.; Trendelenburg, G. TLR2 has a detrimental role in mouse transi-ent focal cerebral ischemia. Biochem. Biophys. Res. Commun. 2007, 359, 574–579. [Google Scholar] [CrossRef]
- Cao, C.X.; Yang, Q.W.; Lv, F.L.; Cui, J.; Fu, H.B.; Wang, J.Z. Reduced cerebral ischemia-reperfusion injury in Toll-like re-ceptor 4 deficient mice. Biochem. Biophys. Res. Commun. 2007, 353, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Caso, J.R.; Pradillo, J.M.; Hurtado, O.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 is involved in brain dam-age and inflammation after experimental stroke. Circulation 2007, 115, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.-G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.-C.; Yeh, S.-J.; Li, Y.-I.; Wang, Y.-C.; Baik, S.-H.; Santro, T.; Widiapradja, A.; Manzanero, S.; Sobey, C.G.; Jo, D.-G.; et al. Evidence for a detrimental role of TLR8 in ischemic stroke. Exp. Neurol. 2013, 250, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mito-chondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Yi, F. NLRP3 Deficiency Ameliorates Neurovascular Damage in Ex-perimental Ischemic Stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Próchnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.-W.; Lee, M.-S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharm. Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Latz, E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Ureña-Peralta, J.R.; Morillo-Bargues, M.J.; la Cruz, J.O.; Guerri, C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell. Neuro-Sci. 2014, 8, 216. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Hornung, V.; Latz, E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Savage, C.D.; Lopez-Castejon, G.; Denes, A.; Brough, D. NLRP3-Inflammasome Activating DAMPs Stimulate an In-flammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Front. Immunol. 2012, 3, 288. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Cheng, J.; Kong, X.; Li, S.; Li, X.; Zhang, M.; Yuan, Z. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 2020, 10, 9644–9662. [Google Scholar] [CrossRef]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.P.; Lombardi, E.P.; Herve, S.; Manel, N. The N-Terminal Domain of cGAS Deter-mines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep. 2019, 26, 2377–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Po-sition cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Omura, H.; Ishitani, R.; Nureki, O. Cyclic GMP–AMP as an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Annu. Rev. Biochem. 2017, 86, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.; Zhang, X.; Chen, Z.J. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qin, J.-J.; Guo, S.; Zhang, P.; Gong, J.; Zhang, X.-J.; Zheng, A.; Xia, H.; Li, H. Attenuation of cerebral ischemic injury in interferon regulatory factor 3-deficient rat. J. Neurochem. 2015, 136, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Downes, C.E.; Wong, C.H.; Brody, K.M.; Guio-Agulair, P.L.; Gould, J.; Ates, R.; Hertzog, P.J.; Taylor, J.M.; Crack, P.J. Type-I interferon signalling through IFNAR1 plays a deleterious role in the outcome after stroke. Neurochem. Int. 2017, 108, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, H.; Wang, C.; Li, Y.; Tian, H.; Siraj, S.; Chen, Q. STING directly activates autophagy to tune the innate immune re-sponse. Cell Death Differ. 2019, 26, 1735–1749. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieler, M.; Hofmann, M.; Schabbauer, G. More than just protein building blocks: How amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021, 288, 3694–3714. [Google Scholar] [CrossRef]
- Gomes MT, R.; Guimarães, E.S.; Marinho, F.V.; Macedo, I.; Aguiar, E.R.; Barber, G.N.; Oliveira, S.C. STING regulates metabolic repro-gramming in macrophages via HIF-1alpha during Brucella infection. PLoS Pathog. 2021, 17, e1009597. [Google Scholar] [CrossRef] [PubMed]
- Franco MM, C.; Marim, F.; Guimarães, E.S.; Assis, N.R.; Cerqueira, D.M.; Alves-Silva, J.; Oliveira, S.C. Brucella abortus Triggers a cGAS-Independent STING Pathway To Induce Host Protection That Involves Guanylate-Binding Proteins and Inflam-masome Activation. J. Immunol. 2018, 200, 607–622. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, Q.; Zhang, X.; Wu, X.; Zhao, Y.; Ren, J. STING-dependent induction of lipid peroxidation mediates intestinal ischemia-reperfusion injury. Free. Radic. Biol. Med. 2020, 163, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Shalova, I.N.; Biswas, S.K. Metabolic regulation of macrophage phenotype and function. Immunol. Rev. 2017, 280, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflam. 2018, 15, 242. [Google Scholar] [CrossRef]
- Claycombe-Larson, K.J.; Alvine, T.; Wu, D.; Kalupahana, N.S.; Moustaid-Moussa, N.; Roemmich, J.N. Nutrients and Im-munometabolism: Role of Macrophage NLRP3. J. Nutr. 2020, 150, 1693–1704. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Bachu, M.; Park, S.H.; Kang, K.; Bae, S.; Park-Min, K.H. IFN-gamma selectively suppresses a subset of TLR4-activated genes and enhancers to potentiate macrophage activation. Nat. Commun. 2019, 10, 3320. [Google Scholar] [CrossRef]
- Das Gupta, K.; Shakespear, M.R.; Curson, J.E.; Murthy, A.M.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C.; et al. Class IIa Histone Deacetylases Drive Toll-like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep. 2020, 30, 2712–2728. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Horng, T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Nam, H.; Kim, L.E.; Jeon, Y.; Min, H.; Ha, S.; Lee, Y.; Kim, S.-Y.; Lee, S.J.; Kim, E.-K.; et al. TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy 2018, 15, 753–770. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, M.; Min, L.; Jiang, K.; Jiang, Y. Cerebral ischemia-reperfusion is modulated by macrophage-stimulating 1 through the MAPK-ERK signaling pathway. J. Cell Physiol. 2020, 235, 7067–7080. [Google Scholar] [CrossRef]
- Fan, H.; Tang, H.-B.; Chen, Z.; Wang, H.-Q.; Zhang, L.; Jiang, Y.; Li, T.; Yang, C.-F.; Wang, X.-Y.; Li, X.; et al. Inhibiting HMGB1-RAGE axis prevents pro-inflammatory macrophages/microglia polarization and affords neuroprotection after spinal cord injury. J. Neuroinflam. 2020, 17, 295. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Y.; Xu, Y.; Ruan, W.; Wang, H.; Zhang, Y.; Saavedra, J.M.; Zhang, L.; Huang, Z.; Pang, T. A Dual AMPK/Nrf2 Activator Reduces Brain Inflammation After Stroke by Enhancing Microglia M2 Polarization. Antioxid. Redox Signal. 2018, 28, 141–163. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202. [Google Scholar] [CrossRef]
- Takeda, H.; Yamaguchi, T.; Yano, H.; Tanaka, J. Microglial metabolic disturbances and neuroinflammation in cerebral infarction. J. Pharmacol. Sci. 2020, 145, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.M.; Oliveira-Junior, M.C.; Souza, R.A.; Petroni, R.C.; Soto, S.F.; Soriano, F.G.; Vieira, R.P. Creatine supplementation at-tenuates pulmonary and systemic effects of lung ischemia and reperfusion injury. J. Heart Lung Transpl. 2016, 35, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Prass, K.; Royl, G.; Lindauer, U.; Freyer, D.; Megow, D.; Dirnagl, U.; Priller, J. Improved reperfusion and neuroprotection by crea-tine in a mouse model of stroke. J. Cereb. Blood Flow Metab. 2007, 27, 452–459. [Google Scholar] [CrossRef]
- Lygate, C.A.; Bohl, S.; Hove, M.T.; Faller, K.M.; Ostrowski, P.J.; Zervou, S.; Medway, D.J.; Aksentijevic, D.; Sebag-Montefiore, L.; Wallis, J.; et al. Moderate elevation of intracellular creatine by targeting the creatine transporter protects mice from acute myocardial infarction. Cardiovasc. Res. 2012, 96, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Sun, Y.; Li, T.; Ren, Z. Garcinol protects against cerebral ischemia-reperfusion injury in vivo and in vitro by in-hibiting inflammation and oxidative stress. Mol. Cell Probes 2020, 54, 101672. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Wang, F.; Liang, X.; Zeng, Z.; Zhao, J.; Zhang, Y. Improvement in cerebral ischemia-reperfusion injury through the TLR4/NF-kappaB pathway after Kudiezi injection in rats. Life Sci. 2017, 191, 132–140. [Google Scholar] [CrossRef]
- Song, Y.; Jun, J.H.; Shin, E.-J.; Kwak, Y.-L.; Shin, J.-S.; Shim, J.-K. Effect of pregabalin administration upon reperfusion in a rat model of hyperglycemic stroke: Mechanistic insights associated with high-mobility group box 1. PLoS ONE 2017, 12, e0171147. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Eisert, L.; Buerkle, H.; Goebel, U.; Schallner, N. Propofol, but not ketamine or midazolam, exerts neuroprotec-tion after ischaemic injury by inhibition of Toll-like receptor 4 and nuclear factor kappa-light-chain-enhancer of activated B-cell signalling: A combined in vitro and animal study. Eur. J. Anaesthesiol. 2016, 33, 670–680. [Google Scholar] [CrossRef]
- Yang, H.-G.; Li, H.-Y.; Li, P.; Bao, X.-Y.; Huang, G.-X.; Xing, L.; Zheng, N.; Wang, J.-Q. Modulation activity of heat-treated and untreated lactoferrin on the TLR-4 pathway in anoxia cell model and cerebral ischemia reperfusion mouse model. J. Dairy Sci. 2020, 103, 1151–1163. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2020, 46, 101102. [Google Scholar] [CrossRef]
- Gad, S.N.; Nofal, S.; Raafat, E.M.; Ahmed, A.A.E. Lixisenatide Reduced Damage in Hippocampus CA1 Neurons in a Rat Model of Cerebral Ischemia-Reperfusion Possibly Via the ERK/P38 Signaling Pathway. J. Mol. Neurosci. 2020, 70, 1026–1037. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, G.; He, S.; Liu, X.; Zhu, L.; Yang, X.; Zhang, Y.; Orgah, J.; Feng, Y.; Wang, X.; et al. Protection against acute cerebral ischemia/reperfusion injury by QiShenYiQi via neuroinflammatory network mobilization. Biomed. Pharmacother. 2020, 125, 109945. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Wei, H.; Gu, T.; Wang, J.; Wu, Z.; Yang, Q. Genistein Attenuates Acute Cerebral Ischemic Damage by Inhibiting the NLRP3 Inflammasome in Reproductively Senescent Mice. Front. Aging Neurosci. 2020, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Bu, J.; Yu, L.; Chen, J.; Liu, H. Nobiletin improves propofol-induced neuroprotection via regulating Akt/mTOR and TLR 4/NF-kappaB signaling in ischemic brain injury in rats. Biomed Pharm. 2017, 91, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Chen, F.S.; Tan, W.F.; Fang, B.; Zhang, Z.L.; Ma, H. Elevated microRNA-129-5p level ameliorates neuroinflamma-tion and blood-spinal cord barrier damage after ischemia-reperfusion by inhibiting HMGB1 and the TLR3-cytokine pathway. J. Neuroinflam. 2017, 14, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrich, F.; Lerach, T.; Biermann, J.; Kaufmann, K.B.; Lagreze, W.A.; Buerkle, H.; Goebel, U. Argon mediates protection by interleu-kin-8 suppression via a TLR2/TLR4/STAT3/NF-kappaB pathway in a model of apoptosis in neuroblastoma cells in vitro and following ischemia-reperfusion injury in rat retina in vivo. J. Neurochem. 2016, 138, 859–873. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.Y.; Yu, Q.L.; Huang, Y.S.; Yang, G. Neuroprotective effects of andrographolide derivative CX-10 in transient focal ischemia in rat: Involvement of Nrf2/AE and TLR/NF-kappaB signaling. Pharm. Res. 2019, 144, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; He, Q.; Zheng, J.; Li, L.Y.; Hou, Y.H.; Song, F.Z. Sulforaphane improves outcomes and slows cerebral ischem-ic/reperfusion injury via inhibition of NLRP3 inflammasome activation in rats. Int. Immunopharmacol. 2017, 45, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Nasoohi, S.; Yoo, A.; Mirzahosseini, G.; Ahmed, H.A.; Ishrat, T. Verapamil as an Adjunct Therapy to Reduce tPA Toxicity in Hyperglycemic Stroke: Implication of TXNIP/NLRP3 Inflammasome. Mol. Neurobiol. 2021, 58, 3792–3804. [Google Scholar] [CrossRef]
- Ai, Q.D.; Chen, C.; Chu, S.; Zhang, Z.; Luo, Y.; Guan, F.; Chen, N. IMM-H004 therapy for permanent focal ischemic cerebral injury via CKLF1/CCR4-mediated NLRP3 inflammasome activation. Transl. Res. 2019, 212, 36–53. [Google Scholar] [CrossRef]
- An, P.; Xie, J.; Qiu, S.; Liu, Y.; Wang, J.; Xiu, X.; Li, L.; Tang, M. Hispidulin exhibits neuroprotective activities against cerebral ischemia reperfusion injury through suppressing NLRP3-mediated pyroptosis. Life Sci. 2019, 232, 116599. [Google Scholar] [CrossRef]
- Li, Y.; Wang, R.; Xue, L.; Yang, Y.; Zhi, F. Astilbin protects against cerebral ischaemia/reperfusion injury by inhibiting cel-lular apoptosis and ROS-NLRP3 inflammasome axis activation. Int. Immunopharmacol. 2020, 84, 106571. [Google Scholar] [CrossRef]
- Liu, H.; Wu, X.; Luo, J.; Zhao, L.; Li, X.; Guo, H.; Qu, Y. Adiponectin peptide alleviates oxidative stress and NLRP3 inflam-masome activation after cerebral ischemia-reperfusion injury by regulating AMPK/GSK-3beta. Exp. Neurol. 2020, 329, 113302. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Meng, C.; Zhang, J.; Wu, J.; Zhao, J. Inhibition of GSK-3beta alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int. Immunopharmacol. 2019, 68, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, Z.; Wu, T.; Zhang, Q.; Lu, F.; Gu, J.; Xue, J. Inhibition of autophagy-dependent pyroptosis attenuates cerebral is-chaemia/reperfusion injury. J. Cell Mol. Med. 2021, 25, 5060–5069. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, M.; Liu, H.; Zhu, R.; He, H.; Zhou, Y.; Huang, G. Bone marrow mesenchymal stem cell-derived exosomes attenuate cerebral ischemia-reperfusion injury-induced neuroinflammation and pyroptosis by modulating microglia M1/M2 pheno-types. Exp. Neurol. 2021, 341, 113700. [Google Scholar] [CrossRef]
- Huang, J.; Wang, T.; Yu, D.; Fang, X.; Fan, H.; Liu, Q.; Liu, Q. l-Homocarnosine attenuates inflammation in cerebral ische-mia-reperfusion injury through inhibition of nod-like receptor protein 3 inflammasome. Int. J. Biol. Macromol. 2018, 118 Pt A, 357–364. [Google Scholar] [CrossRef]
- Ma, D.C.; Zhang, N.N.; Zhang, Y.N.; Chen, H.S. Kv1.3 channel blockade alleviates cerebral ischemia/reperfusion injury by reshaping M1/M2 phenotypes and compromising the activation of NLRP3 inflammasome in microglia. Exp. Neurol. 2020, 332, 113399. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, T.; Chen, X.F.; Xu, Z.X.; Cao, J.B.; Sun, H. TMEM59 protects against cerebral ischemic stroke by sup-pressing pyroptosis and microglial activation. Biochem. Biophys. Res. Commun. 2021, 543, 72–79. [Google Scholar] [CrossRef]
- Jiang, M.; Li, R.; Lyu, J.; Li, X.; Wang, W.; Wang, Z.; Yang, W. MCC950, a selective NLPR3 inflammasome inhibitor, improves neu-rologic function and survival after cardiac arrest and resuscitation. J. Neuroinflam. 2020, 17, 256. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, H.; Jin, J.; Liu, Q.; Zhong, D.; Li, G. Inhibition of the NLRP3 inflammasome reduces brain edema and reg-ulates the distribution of aquaporin-4 after cerebral ischaemia-reperfusion. Life Sci. 2020, 251, 117638. [Google Scholar] [CrossRef]
- Diao, M.Y.; Zhu, Y.; Yang, J.; Xi, S.S.; Wen, X.; Gu, Q.; Hu, W. Hypothermia protects neurons against ische-mia/reperfusion-induced pyroptosis via m6A-mediated activation of PTEN and the PI3K/Akt/GSK-3beta signaling pathway. Brain Res. Bull. 2020, 159, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Hong, P.; Li, F.-X.; Gu, R.-N.; Fang, Y.-Y.; Lai, L.-Y.; Wang, Y.-W.; Tao, T.; Xu, S.-Y.; You, Z.-J.; Zhang, H.-F. Inhibition of NLRP3 Inflammasome Ameliorates Cerebral Ischemia-Reperfusion Injury in Diabetic Mice. Neural Plast. 2018, 2018, 9163521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Geng, X.; Lee, H.; Ding, Y. Phenothiazine Inhibits Neuroinflammation and Inflammasome Activation Independ-ent of Hypothermia After Ischemic Stroke. Mol. Neurobiol. 2021, 58, 6136–6152. [Google Scholar] [CrossRef]
- Peng, J.; Wang, H.; Gong, Z.; Li, X.; He, L.; Shen, Q.; Pan, J.; Peng, Y. Idebenone attenuates cerebral inflammatory injury in ischemia and reperfusion via dampening NLRP3 inflammasome activity. Mol. Immunol. 2020, 123, 74–87. [Google Scholar] [CrossRef]
- He, Q.I.; Li, Z.; Wang, Y.; Hou, Y.; Li, L.; Zhao, J. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhib-iting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int. Immunopharmacol. 2017, 50, 208–215. [Google Scholar] [CrossRef]
- Li, X.; Shi, M.-Q.; Chen, C.; Du, J.-R. Phthalide derivative CD21 ameliorates ischemic brain injury in a mouse model of global cerebral ischemia: Involvement of inhibition of NLRP3. Int. Immunopharmacol. 2020, 86, 106714. [Google Scholar] [CrossRef]
- Ye, Y.; Jin, T.; Zhang, X.; Zeng, Z.; Ye, B.; Wang, J.; Gu, L. Meisoindigo Protects Against Focal Cerebral Ischemia-Reperfusion Injury by Inhibiting NLRP3 Inflammasome Activation and Regulating Microglia/Macrophage Polarization via TLR4/NF-kappaB Signaling Pathway. Front. Cell Neurosci. 2019, 13, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Gu, Y.; Liu, J. BRD4 suppression alleviates cerebral ischemia-induced brain injury by blocking glial activation via the inhibition of inflammatory response and pyroptosis. Biochem. Biophys. Res. Commun. 2019, 519, 481–488. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wu, H.; Zhou, Y.; Zheng, C. Tomentosin inhibit cerebral ischemia/reperfusion induced inflammatory response via TLR4/ NLRP3 signalling pathway-in vivo and in vitro studies. Biomed Pharm. 2020, 131, 110697. [Google Scholar] [CrossRef]
- Cui, H.X.; Chen, J.H.; Li, J.W.; Cheng, F.R.; Yuan, K. Protection of Anthocyanin from Myrica rubra against Cerebral Is-chemia-Reperfusion Injury via Modulation of the TLR4/NF-kappaB and NLRP3 Pathways. Molecules 2018, 23, 1788. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Sun, Y.; Lv, C.; Zhang, W.; Chen, Y. Procyanidins exhibits neuroprotective activities against cerebral ischemia reperfusion injury by inhibiting TLR4-NLRP3 inflammasome signal pathway. Psychopharmacology 2020, 237, 3283–3293. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.C.; Zhang, N.N.; Zhang, Y.N.; Chen, H.S. Salvianolic Acids for Injection alleviates cerebral ischemia/reperfusion injury by switching M1/M2 phenotypes and inhibiting NLRP3 inflammasome/pyroptosis axis in microglia in vivo and in vitro. J. Ethnopharmacol. 2021, 270, 113776. [Google Scholar] [CrossRef] [PubMed]
- She, Y.; Shao, L.; Zhang, Y.; Hao, Y.; Cai, Y.; Cheng, Z.; Liu, X. Neuroprotective effect of glycosides in Buyang Huanwu Decoc-tion on pyroptosis following cerebral ischemia-reperfusion injury in rats. J. Ethnopharmacol. 2019, 242, 112051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, N.; Li, Z.; Xu, G.; Zhan, X.; Tang, J.; Bai, Z. The Cytosolic DNA-Sensing cGAS-STING Pathway in Liver Diseas-es. Front. Cell Dev. Biol. 2021, 9, 717610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Liu, Q.; Li, X.; Li, S.; Chen, J.; Ren, J. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis. 2020, 11, 1050. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Lin, F.; Yao, X.; Kong, C.; Liu, X.; Zhao, Z.; Rao, S.; Dai, Q. 25-Hydroxycholesterol protecting from cerebral ischemia-reperfusion injury through the inhibition of STING activity. Aging 2021, 13, 20149–20163. [Google Scholar] [CrossRef]
- Shen, A.; Zheng, D.; Luo, Y.; Mou, T.; Chen, Q.; Huang, Z.; Wu, Z. MicroRNA-24-3p alleviates hepatic ischemia and reperfusion injury in mice through the repression of STING signaling. Biochem. Biophys. Res. Commun. 2019, 522, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cao, Y.; Dang, C.; Han, B.; Han, R.; Ma, H.; Wang, L. Inhibition of double-strand DNA-sensing cGAS ameliorates brain in-jury after ischemic stroke. EMBO Mol. Med. 2020, 12, e11002. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.S.; Hisata, S.; Park, M.A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M. mTORC1-Induced HK1-Dependent Glycol-ysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Ouyang, H.; Mei, X.; Lu, B.; Yu, Z.; Chen, K.; Ji, L. Erianin alleviates diabetic retinopathy by reducing retinal in-flammation initiated by microglial cells via inhibiting hyperglycemia-mediated ERK1/2-NF-kappaB signaling pathway. FASEB J. 2019, 33, 11776–11790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Cai, J.; Xi, M.; He, Y.; Zhao, Y.; Zheng, Y.; He, Y. Neuroprotection Effect of Astragaloside IV from 2-DG-Induced Endo-plasmic Reticulum Stress. Oxid Med. Cell Longev. 2020, 2020, 9782062. [Google Scholar] [CrossRef]
- Yin, F.; Zhou, H.; Fang, Y.; Li, C.; He, Y.; Yu, L.; Wan, H.; Yang, J. Astragaloside IV alleviates ischemia reperfusion-induced apoptosis by inhibiting the activation of key factors in death receptor pathway and mitochondrial pathway. J. Ethnopharmacol. 2019, 248, 112319. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Choudhury, M.E.; Watanabe, M.; Kawasaki, S.; Nishihara, T.; Yano, H.; Matsumoto, S.; Kunieda, T.; Kumon, Y.; Yorozuya, T.; et al. Comparison of the detrimental features of microglia and infiltrated macrophages in traumatic brain injury: A study using a hypnotic bromovalerylurea. Glia 2018, 66, 2158–2173. [Google Scholar] [CrossRef] [PubMed]
- Carter, K.J.; Ward, A.T.; Kellawan, J.M.; Eldridge, M.W.; Al-Subu, A.; Walker, B.J.; Lee, J.W.; Wieben, O.; Schrage, W.G. Nitric oxide synthase inhibition in healthy adults reduces regional and total cerebral macrovascular blood flow and microvascular perfusion. J. Physiol. 2021, 599, 4973–4989. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef]
- Katoh, M.; Wu, B.; Nguyen, H.B.; Thai, T.Q.; Yamasaki, R.; Lu, H.; Rietsch, A.M.; Zorlu, M.M.; Shinozaki, Y.; Saitoh, Y.; et al. Polymorphic regulation of mitochondrial fission and fusion modifies phenotypes of microglia in neuroinflammation. Sci. Rep. 2017, 7, 4942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Xia, S.-X.; Li, Q.-Q.; Gao, Y.; Shen, X.; Ma, L.; Zhang, M.-Y.; Wang, T.; Li, Y.-S.; Wang, Z.-F.; et al. Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res. 2016, 1630, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Ochando, J.; Ordikhani, F.; Boros, P.; Jordan, S. The innate immune response to allotransplants: Mechanisms and therapeutic potentials. Cell Mol. Immunol. 2019, 16, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Tanaka, Y.; Nakagomi, N.; Matsuyama, T.; Yoshimura, S. How Long Are Reperfusion Therapies Beneficial for Patients after Stroke Onset? Lessons from Lethal Ischemia Following Early Reperfusion in a Mouse Model of Stroke. Int. J. Mol. Sci. 2020, 21, 6360. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Fu, J.; Wang, J.; Zhao, Y.; Liu, B.; Wei, J.; Yan, X.; Su, J. The Influence of Mitochondrial-DNA-Driven Inflammation Pathways on Macrophage Polarization: A New Perspective for Targeted Immunometabolic Therapy in Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2022, 23, 135. https://doi.org/10.3390/ijms23010135
Yu S, Fu J, Wang J, Zhao Y, Liu B, Wei J, Yan X, Su J. The Influence of Mitochondrial-DNA-Driven Inflammation Pathways on Macrophage Polarization: A New Perspective for Targeted Immunometabolic Therapy in Cerebral Ischemia-Reperfusion Injury. International Journal of Molecular Sciences. 2022; 23(1):135. https://doi.org/10.3390/ijms23010135
Chicago/Turabian StyleYu, Sihang, Jiaying Fu, Jian Wang, Yuanxin Zhao, Buhan Liu, Jiahang Wei, Xiaoyu Yan, and Jing Su. 2022. "The Influence of Mitochondrial-DNA-Driven Inflammation Pathways on Macrophage Polarization: A New Perspective for Targeted Immunometabolic Therapy in Cerebral Ischemia-Reperfusion Injury" International Journal of Molecular Sciences 23, no. 1: 135. https://doi.org/10.3390/ijms23010135
APA StyleYu, S., Fu, J., Wang, J., Zhao, Y., Liu, B., Wei, J., Yan, X., & Su, J. (2022). The Influence of Mitochondrial-DNA-Driven Inflammation Pathways on Macrophage Polarization: A New Perspective for Targeted Immunometabolic Therapy in Cerebral Ischemia-Reperfusion Injury. International Journal of Molecular Sciences, 23(1), 135. https://doi.org/10.3390/ijms23010135