
Visualizing Extracellular Vesicles and Their Function in 3D Tumor Microenvironment Models

 , , ,
, , ,  , and
, and
Abstract
1. Introduction: EVs in the TME
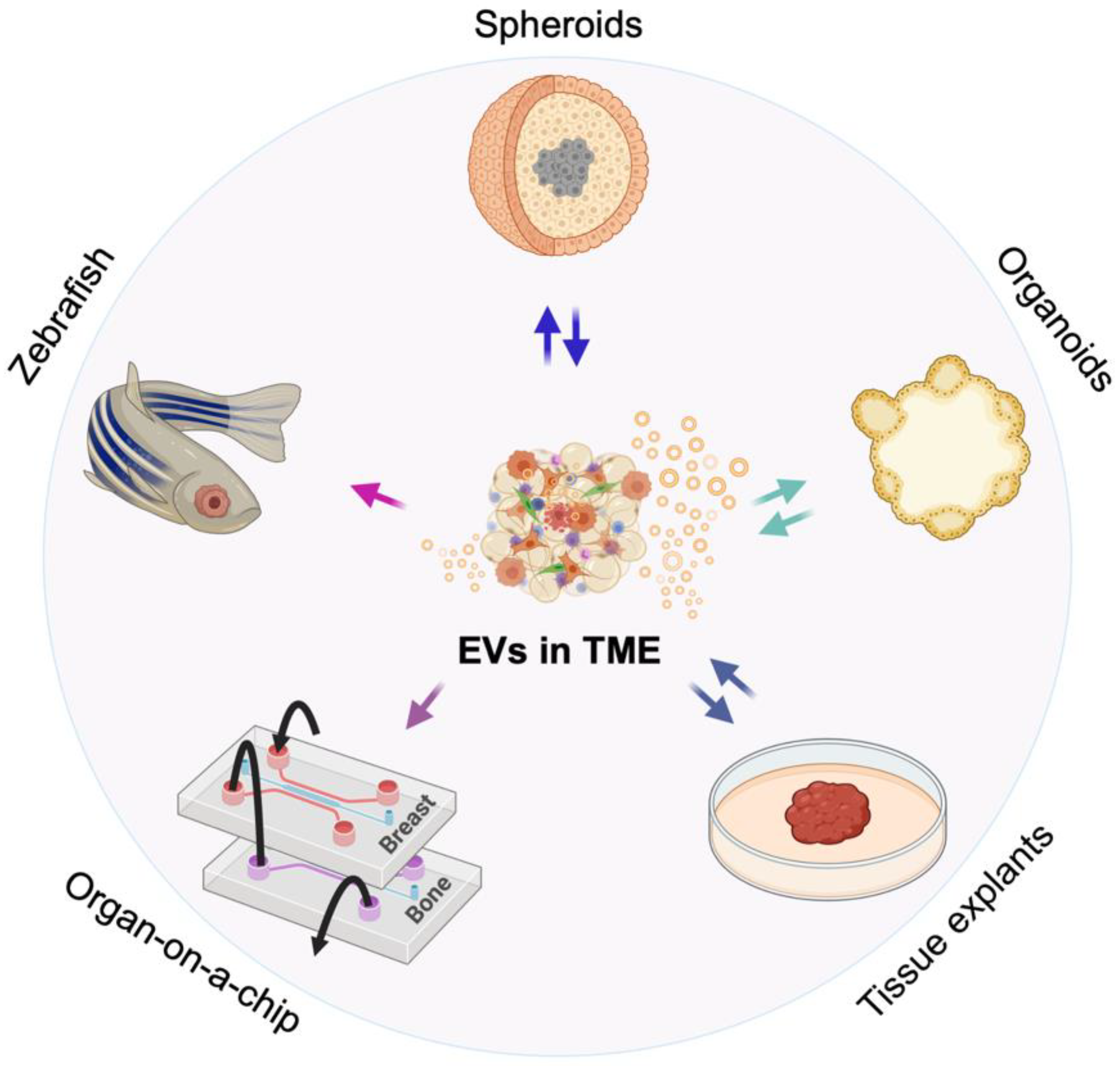
2. 3D Models of the TME
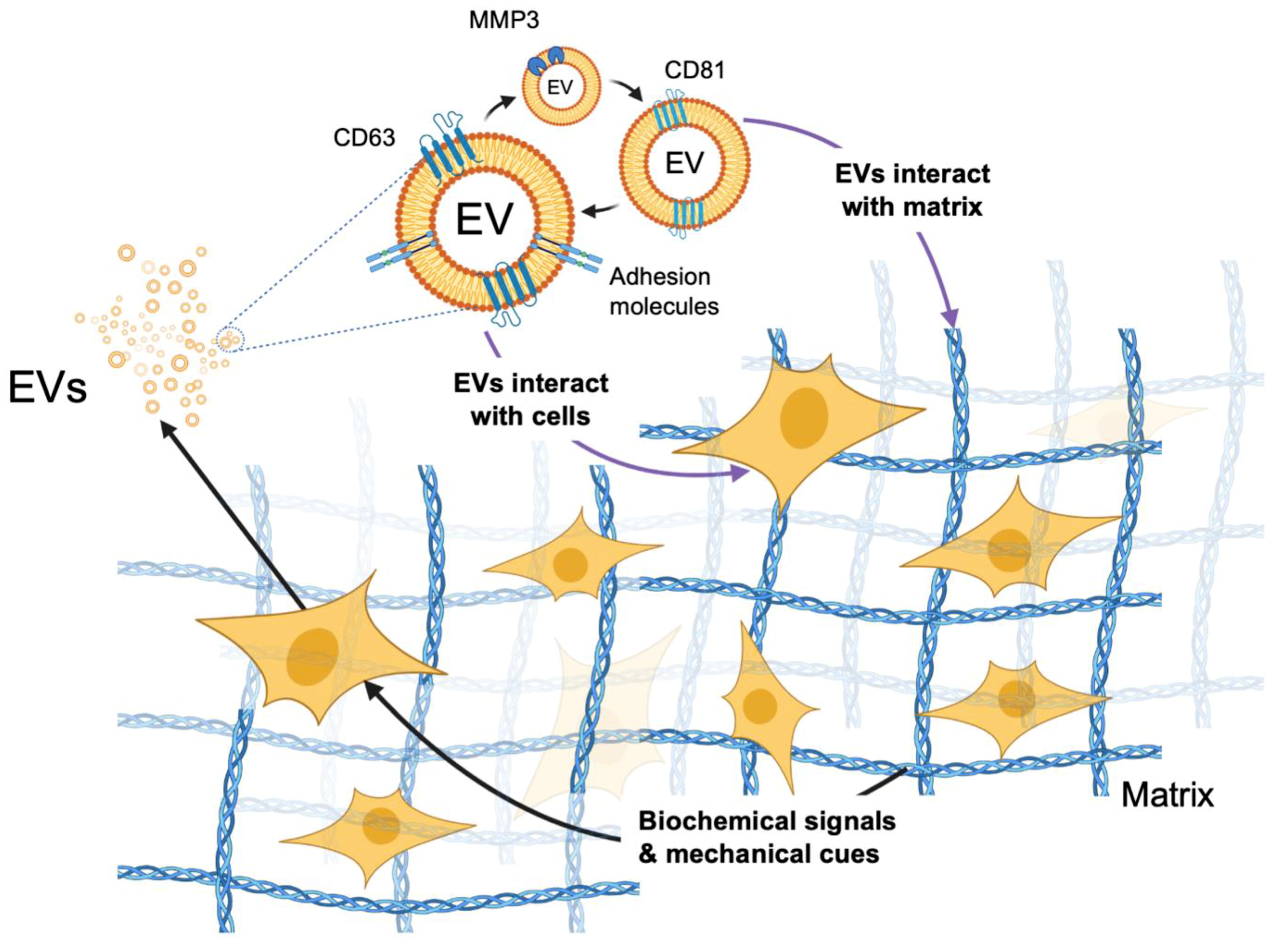
3. Advances in Visualizing EVs in 3D Models and Assessing Their Role
4. Effects of Scaffolding in 3D Cultures on EV Biogenesis
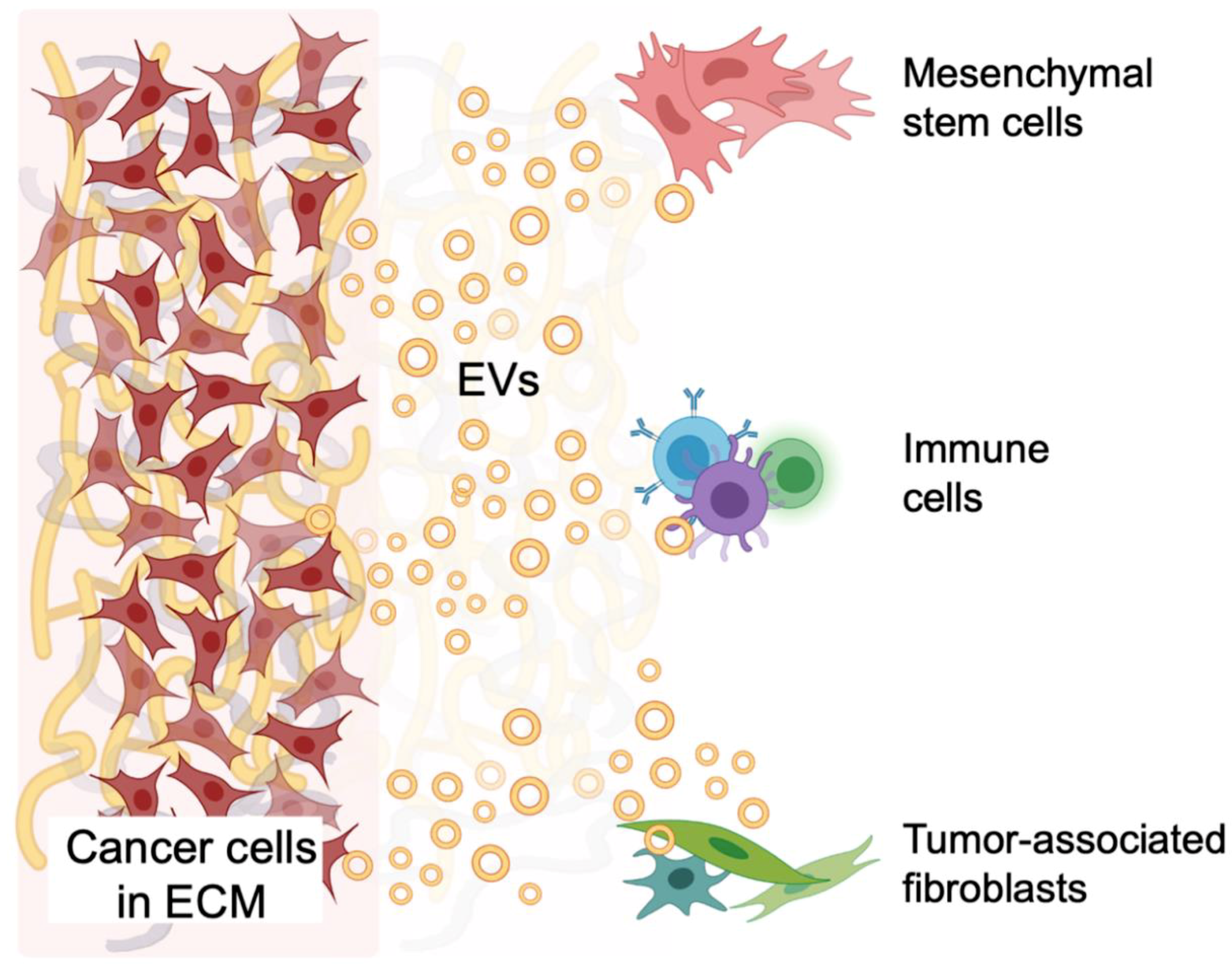
5. EV-Mediated Signaling within Models of the TME
5.1. Monitoring EVs and Their Function in Spheroids and Organoids
5.2. Cancer Stem Cells (CSCs) in 3D Cultures
5.3. Microenvironmental Stress in 3D Cultures
5.4. Tumor and Stromal Cell Crosstalk in the TME
6. Modeling How Tumors Reach Out to Adjacent and Distant Non-Malignant Tissues via EVs
6.1. Affecting the Normal Surrounding Tissue—Systemic Pathologies from Cancer EVs
6.2. Predisposition to Metastasis—EVs Cultivate the Premetastatic Niche

{kind=link}
{kind=link}
{kind=link}
| TME Model | Common Methods of EV Imaging | Advantages | Limitations |
|---|---|---|---|
| 2D cell monolayers |
|
| |
| Small mammalian animal models |
|
| |
| Cancer spheroids |
|
| |
| Stem cell-derived organoids |
|
| |
| Organ-on-a-chip |
|
| |
| Tissue explants |
| ||
| Zebrafish |
|
|
7. Future Perspectives
- Can we characterize animal models regarding their clinical relevance to human disease and use this to better inform the development of our culture correlates?
- How dramatically do EV imaging methods affect the signaling fate of EVs in monolayers, 3D organ models and in vivo?
- How do scaffold types affect signaling of tumor cell-derived EVs to stromal cells?
- How can we trace EVs undergoing transcytosis long-term and determine its relevance in human disease?
- Can we model organotropism in organ-on-a-chip cultures for various cancer types, for immune cells and in organogenesis?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TME | tumor microenvironment |
| EV | extracellular vesicle |
| ECM | extracellular matrix |
| MV | microvesicle |
| MVB | multivesicular body |
| ILV | intraluminal vesicle |
| ESCRT | endosomal complexes required for transport |
| MISEV | minimal information for studies of extracellular vesicles |
| CSC | cancer stem cell |
| iPSC | induced pluripotent stem cells |
| ESC | embryonic stem cells |
| PDE | patient-derived tissue explants |
| BBB | blood–brain barrier |
| BLI | bioluminescent imaging |
| MSC | mesenchymal stromal cell, mesenchymal stem cell |
| CRC | colorectal carcinoma |
| GBM | glioblastoma multiforme |
References
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef]
- Kanada, M.; Bachmann, M.H.; Contag, C.H. Signaling by Extracellular Vesicles Advances Cancer Hallmarks. Trends Cancer 2016, 2, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef]
- Cheng, K.W.; Lahad, J.P.; Gray, J.W.; Mills, G.B. Emerging Role of RAB GTPases in Cancer and Human Disease. Cancer Res. 2005, 65, 2516–2519. [Google Scholar] [CrossRef]
- Gingras, M.-C.; Kazan, J.M.; Pause, A. Role of ESCRT component HD-PTP/PTPN23 in cancer. Biochem. Soc. Trans. 2017, 45, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.B.A.; Wenzel, D.M.; Williams, L.K.; Guindo-Martínez, M.; Alam, S.L.; Mercader, J.M.; Torrents, D.; Ullman, K.S.; Sundquist, W.I.; Martin-Serrano, J. A cancer-associated polymorphism in ESCRT-III disrupts the abscission checkpoint and promotes genome instability. Proc. Natl. Acad. Sci. USA 2018, 115, E8900–E8908. [Google Scholar] [CrossRef]
- Morad, S.A.F.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, S.; Sandner, J.; Birod, K.; Wobst, I.; Angioni, C.; Ruckhäberle, E.; Kaufmann, M.; Ackermann, H.; Lötsch, J.; Schmidt, H.; et al. Ceramide synthases and ceramide levels are increased in breast cancer tissue. Carcinogenesis 2009, 30, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nsmase2)-dependent exosomal transfer of angiogenic micrornas regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Jin, F.; Bian, Z.; Li, L.; Liang, H.; Li, M.; Shi, L.; Pan, C.; Zhu, D.; et al. Pyruvate kinase type M2 promotes tumour cell exosome release via phosphorylating synaptosome-associated protein 23. Nat. Commun. 2017, 8, 14041. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; Bebelman, M.P.; Jimenez, C.R.; Garcia-Vallejo, J.J.; Janssen, H.; Neefjes, J.; Knol, J.C.; Haas, R.D.G.-D.; Piersma, S.R.; Baglio, S.R.; et al. Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J. Cell Biol. 2018, 217, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2009, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-Regulated Shedding of Tumor Cell-Derived Plasma Membrane Microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef]
- Latifkar, A.; Cerione, R.A.; Antonyak, M.A. Probing the mechanisms of extracellular vesicle biogenesis and function in cancer. Biochem. Soc. Trans. 2018, 46, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Di Luca, A.; Mleczko, J.; Meleady, P.; Henry, M.; Pesic, M.; Cabrera, D.; van Liempd, S.; Lima, R.T.; O’Connor, R.; et al. Identification of the metabolic alterations associated with the multidrug resistant phenotype in cancer and their intercellular transfer mediated by extracellular vesicles. Sci. Rep. 2017, 7, 44541. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Hao, Y.; He, C.; Li, L.; Zhu, G. Exosome-orchestrated hypoxic tumor microenvironment. Mol. Cancer 2019, 18, 57. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH Is a Key Factor for Exosome Traffic in Tumor Cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Logozzi, M.; Mizzoni, D.; Angelini, D.F.; Di Raimo, R.; Falchi, M.; Battistini, L.; Fais, S. Microenvironmental pH and Exosome Levels Interplay in Human Cancer Cell Lines of Different Histotypes. Cancers 2018, 10, 370. [Google Scholar] [CrossRef]
- Yamada, K.M.; Cukierman, E. Modeling Tissue Morphogenesis and Cancer in 3D. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef]
- Hoarau-Véchot, J.; Rafii, A.; Touboul, C.; Pasquier, J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? Int. J. Mol. Sci. 2018, 19, 181. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension—How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef]
- Sung, B.H.; Von Lersner, A.; Guerrero, J.; Krystofiak, E.S.; Inman, D.; Pelletier, R.; Zijlstra, A.; Ponik, S.M.; Weaver, A.M. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Mathivanan, S.; Ji, H.; Simpson, R.J. Two Distinct Populations of Exosomes Are Released from LIM1863 Colon Carcinoma Cell-derived Organoids. Mol. Cell. Proteomics 2013, 12, 587–598. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G., Jr. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef] [PubMed]
- Zanetti-Domingues, L.C.; Bonner, S.E.; Iyer, R.S.; Martin-Fernandez, M.L.; Huber, V. Cooperation and interplay between egfr signalling and extracellular vesicle biogenesis in cancer. Cells 2020, 9, 2639. [Google Scholar] [CrossRef]
- Däster, S.; Amatruda, N.; Calabrese, D.; Ivanek, R.; Turrini, E.; Droeser, R.A.; Zajac, P.; Fimognari, C.; Spagnoli, G.C.; Iezzi, G.; et al. Induction of hypoxia and necrosis in multicellular tumor spheroids is associated with resistance to chemotherapy treatment. Oncotarget 2017, 8, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Ingle, A.D. Alternatives and Refinement for Animal Experimentation in Cancer Research. In Alternatives to Animal Testing; Kojima, H., Seidle, T., Spielmann, H., Eds.; Springer: Singapore, 2019; pp. 69–75. [Google Scholar]
- Chuo, S.T.-Y.; Chien, J.C.-Y.; Lai, C.P.-K. Imaging extracellular vesicles: Current and emerging methods. J. Biomed. Sci. 2018, 25, 1–10. [Google Scholar] [CrossRef]
- Jung, K.O.; Jo, H.; Yu, J.H.; Gambhir, S.S.; Pratx, G. Development and MPI tracking of novel hypoxia-targeted theranostic exosomes. Biomaterials 2018, 177, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Contag, C.H.; Bachmann, M.H. Advances in in vivo bioluminescence imaging of gene expression. Annu. Rev. Biomed. Eng. 2002, 4, 235–260. [Google Scholar] [CrossRef]
- Wagner, K.T.; Nash, T.R.; Liu, B.; Vunjak-Novakovic, G.; Radisic, M. Extracellular vesicles in cardiac regeneration: potential applications for tissues-on-a-chip. Trends Biotechnol. 2020, S0167-7799, 30227-4. [Google Scholar] [CrossRef]
- Kanada, M.; Ashammakhi, N. Extracellular vesicles: Emerging opportunities for tissue engineering and regenerative medicine. J. Craniofac. Surg. 2021. Publish Ahead of Print. [Google Scholar] [CrossRef]
- Cekanova, M.; Rathore, K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Des. Dev. Ther. 2014, 8, 1911–1921. [Google Scholar] [CrossRef]
- Voskoglou-Nomikos, T.; Pater, J.L.; Seymour, L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 2003, 9, 4227–4239. [Google Scholar] [PubMed]
- Greek, R.; Menache, A. Systematic Reviews of Animal Models: Methodology versus Epistemology. Int. J. Med. Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Mahajan, R.; Gupta, K. Food and drug administration’s critical path initiative and innovations in drug development paradigm: Challenges, progress, and controversies. J. Pharm. Bioallied Sci. 2010, 2, 307. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Calar, K.; De La Puente, P. Mimicking tumor hypoxia and tumor-immune interactions employing three-dimensional in vitro models. J. Exp. Clin. Cancer Res. 2020, 39, 1–16. [Google Scholar] [CrossRef]
- Apu, E.H.; Akram, S.U.; Rissanen, J.; Wan, H.; Salo, T. Desmoglein 3—Influence on oral carcinoma cell migration and invasion. Exp. Cell Res. 2018, 370, 353–364. [Google Scholar] [CrossRef]
- Salo, T.; Dourado, M.R.; Sundquist, E.; Apu, E.H.; Alahuhta, I.; Tuomainen, K.; Vasara, J.; Al-Samadi, A. Organotypic three-dimensional assays based on human leiomyoma–derived matrices. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160482. [Google Scholar] [CrossRef] [PubMed]
- Fetah, K.L.; DiPardo, B.J.; Kongadzem, E.-M.; Tomlinson, J.S.; Elzagheid, A.; Elmusrati, M.; Khademhosseini, A.; Ashammakhi, N. Cancer Modeling-on-a-Chip with Future Artificial Intelligence Integration. Small 2019, 15, 1901985. [Google Scholar] [CrossRef]
- Tung, K.H.; Ernstoff, M.S.; Allen, C.; La Shu, S. A Review of Exosomes and their Role in The Tumor Microenvironment and Host-Tumor “Macroenvironment”. J. Immunol. Sci. 2019, 3, 4–8. [Google Scholar] [CrossRef]
- Mantovani, A.; Germano, G.; Marchesi, F.; Locatelli, M.; Biswas, S.K. Cancer-promoting tumor-associated macrophages: New vistas and open questions. Eur. J. Immunol. 2011, 41, 2522–2525. [Google Scholar] [CrossRef]
- Al-Zhoughbi, W.; Huang, J.; Paramasivan, G.S.; Till, H.; Pichler, M.; Guertl-Lackner, B.; Hoefler, G. Tumor Macroenvironment and Metabolism. Semin. Oncol. 2014, 41, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Dauth, S.; Hassell, B.; Herland, A.; Jain, A.; Jang, K.-J.; Karalis, K.; Kim, H.J.; MacQueen, L.; Mahmoodian, R.; et al. Engineered In Vitro Disease Models. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 195–262. [Google Scholar] [CrossRef] [PubMed]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Controlled Release 2012, 164, 192–204. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Salo, T.; Sutinen, M.; Apu, E.H.; Sundquist, E.; Cervigne, N.K.; De Oliveira, C.E.; Akram, S.U.; Ohlmeier, S.; Suomi, F.; Eklund, L.; et al. A novel human leiomyoma tissue derived matrix for cell culture studies. BMC Cancer 2015, 15, 981. [Google Scholar] [CrossRef] [PubMed]
- Almahmoudi, R.; Salem, A.; Murshid, S.; Dourado, M.R.; Apu, E.H.; Salo, T.; Al-Samadi, A. Interleukin-17F Has Anti-Tumor Effects in Oral Tongue Cancer. Cancers 2019, 11, 650. [Google Scholar] [CrossRef]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Fiorini, E.; Veghini, L.; Corbo, V. Modeling Cell Communication in Cancer with Organoids: Making the Complex Simple. Front. Cell Dev. Biol. 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, A.R.; Aldrian, C.; Bronsert, P.; Thomann, Y.; Nanko, N.; Melin, N.; Rücker, G.; Follo, M.; Grosu, A.L.; Niedermann, G.; et al. A deep conical agarose microwell array for adhesion independent three-dimensional cell culture and dynamic volume measurement. Lab Chip 2018, 18, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Carvalho, J.; Oliveira, P.; Voglstaetter, M.; Schvartz, D.; Thomsen, A.R.; Walter, N.; Khanduri, R.; Sanchez, J.-C.; Keller, A.; et al. 3D cellular architecture affects microrna and protein cargo of extracellular vesicles. Adv. Sci. 2019, 6, 1800948. [Google Scholar] [CrossRef] [PubMed]
- Villasante, A.; Marturano-Kruik, A.; Ambati, S.R.; Liu, Z.; Godier-Furnemont, A.; Parsa, H.; Lee, B.W.; Moore, M.A.S.; Vunjak-Novakovic, G. Recapitulating the Size and Cargo of Tumor Exosomes in a Tissue-Engineered Model. Theranostics 2016, 6, 1119–1130. [Google Scholar] [CrossRef]
- Hwang, W.-L.; Lan, H.-Y.; Cheng, W.-C.; Huang, S.-C.; Yang, M.-H. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon cancer. J. Hematol. Oncol. 2019, 12, 1–17. [Google Scholar] [CrossRef]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef]
- Papi, A.; De Carolis, S.; Bertoni, S.; Storci, G.; Sceberras, V.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Orlandi, M.; Bonafé, M. PPARγ and RXR Ligands Disrupt the Inflammatory Cross-talk in the Hypoxic Breast Cancer Stem Cells Niche. J. Cell. Physiol. 2014, 229, 1595–1606. [Google Scholar] [CrossRef]
- Wang, L.; Mezencev, R.; Bowen, N.J.; Matyunina, L.V.; McDonald, J.F. Isolation and characterization of stem-like cells from a human ovarian cancer cell line. Mol. Cell. Biochem. 2012, 363, 257–268. [Google Scholar] [CrossRef]
- Lopatina, T.; Gai, C.; Deregibus, M.C.; Kholia, S.; Camussi, G. Cross Talk between Cancer and Mesenchymal Stem Cells through Extracellular Vesicles Carrying Nucleic Acids. Front. Oncol. 2016, 6, 125. [Google Scholar] [CrossRef]
- Shoval, H.; Karsch-Bluman, A.; Brill-Karniely, Y.; Stern, T.; Zamir, G.; Hubert, A.; Benny, O. Tumor cells and their crosstalk with endothelial cells in 3D spheroids. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Sadovska, L.; Zandberga, E.; Sagini, K.; Jēkabsons, K.; Riekstiņa, U.; Kalniņa, Z.; Llorente, A.; Linē, A. A novel 3D heterotypic spheroid model for studying extracellular vesicle-mediated tumour and immune cell communication. Biochem. Biophys. Res. Commun. 2018, 495, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Liu, J.; Tan, Y.; Zhang, H.; Zhang, Y.; Xu, P.; Chen, J.; Poh, Y.-C.; Tang, K.; Wang, N.; Huang, B. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat. Mater. 2012, 11, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Bie, N.; Yong, T.; Tang, K.; Shi, X.; Wei, Z.; Jia, H.; Zhang, X.; Zhao, H.; Huang, W.; et al. The softness of tumour-cell-derived microparticles regulates their drug-delivery efficiency. Nat. Biomed. Eng. 2019, 3, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Ke, X.; Yan, R.; Sun, Z.; Cheng, Y.; Meltzer, A.; Lu, N.; Shu, X.; Wang, Z.; Huang, B.; Liu, X.; et al. Esophageal Adenocarcinoma-Derived Extracellular Vesicle MicroRNAs Induce a Neoplastic Phenotype in Gastric Organoids. Neoplasia 2017, 19, 941–949. [Google Scholar] [CrossRef]
- Artegiani, B.; Clevers, H. Use and application of 3D-organoid technology. Hum. Mol. Genet. 2018, 27, R99–R107. [Google Scholar] [CrossRef]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; Macfarlane, M.; et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef]
- Centenera, M.M.; Hickey, T.E.; Jindal, S.; Ryan, N.K.; Ravindranathan, P.; Mohammed, H.; Robinson, J.L.; Schiewer, M.J.; Ma, S.; Kapur, P.; et al. A patient-derived explant (PDE) model of hormone-dependent cancer. Mol. Oncol. 2018, 12, 1608–1622. [Google Scholar] [CrossRef]
- Mincheva-Nilsson, L.; Baranov, V.; Nagaeva, O.; Dehlin, E. Isolation and Characterization of Exosomes from Cultures of Tissue Explants and Cell Lines. Curr. Protoc. Immunol. 2016, 115, 14–42. [Google Scholar] [CrossRef]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Liang, X.; Pavlova, S.; Wiklander, O.P.; Corso, G.; Zhao, Y.; Saher, O.; Bost, J.; Zickler, A.M.; Piffko, A.; et al. Quantification of extracellular vesicles in vitro and in vivo using sensitive bioluminescence imaging. J. Extracell. Vesicles 2020, 9, 1800222. [Google Scholar] [CrossRef]
- Wu, A.Y.-T.; Sung, Y.-C.; Chen, Y.-J.; Chou, S.T.-Y.; Guo, V.; Chien, J.C.-Y.; Ko, J.J.-S.; Yang, A.L.; Huang, H.-C.; Chuang, J.-C.; et al. Multiresolution Imaging Using Bioluminescence Resonance Energy Transfer Identifies Distinct Biodistribution Profiles of Extracellular Vesicles and Exomeres with Redirected Tropism. Adv. Sci. 2020, 7, 2001467. [Google Scholar] [CrossRef] [PubMed]
- Lázaro-Ibáñez, E.; Faruqu, F.N.; Saleh, A.F.; Silva, A.M.; Wang, J.T.-W.; Rak, J.; Al-Jamal, K.T.; Dekker, N. Selection of Fluorescent, Bioluminescent, and Radioactive Tracers to Accurately Reflect Extracellular Vesicle Biodistribution in Vivo. ACS Nano 2021, 15, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- Cashikar, A.G.; Hanson, P.I. A cell-based assay for CD63-containing extracellular vesicles. PLoS ONE 2019, 14, e0220007. [Google Scholar] [CrossRef]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Ségaliny, A.; et al. Elucidation of Exosome Migration Across the Blood–Brain Barrier Model In Vitro. Cell. Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.P.; Tandalam, S.; Ostrager, S.; Lever, A.S.; Fung, A.R.; Hurley, D.D.; Alegre, G.B.; Espinal, J.E.; Remmel, H.L.; Mukherjee, S.; et al. Non-reversible tissue fixation retains extracellular vesicles for in situ imaging. Nat. Methods 2019, 16, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Horgan, C.C.; Nagelkerke, A.; Whittaker, T.E.; Nele, V.; Massi, L.; Kauscher, U.; Penders, J.; Bergholt, M.S.; Hood, S.R.; Stevens, M.M. Molecular imaging of extracellular vesicles in vitro via Raman metabolic labelling. J. Mater. Chem. B 2020, 8, 4447–4459. [Google Scholar]
- Wang, J.; Wuethrich, A.; Sina, A.A.I.; Lane, R.E.; Lin, L.L.; Wang, Y.; Cebon, J.; Behren, A.; Trau, M. Tracking extracellular vesicle phenotypic changes enables treatment monitoring in melanoma. Sci. Adv. 2020, 6, eaax3223. [Google Scholar] [CrossRef]
- Sun, Y.; You, S.; Tu, H.; Spillman, D.R.; Chaney, E.J.; Marjanovic, M.; Li, J.; Barkalifa, R.; Wang, J.; Higham, A.M.; et al. Intraoperative visualization of the tumor microenvironment and quantification of extracellular vesicles by label-free nonlinear imaging. Sci. Adv. 2018, 4, 12–eaau5603. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Barkalifa, R.; Chaney, E.J.; Tu, H.; Park, J.; Sorrells, J.E.; Sun, Y.; Liu, Y.-Z.; Yang, L.; Chen, D.Z.; et al. Label-free visualization and characterization of extracellular vesicles in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 24012–24018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- EL Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Haycock, J.W. 3D Cell Culture: A Review of Current Approaches and Techniques. Methods Mol. Biol. 2011, 695, 1–15. [Google Scholar] [CrossRef]
- Lee, J.; Cuddihy, M.J.; Kotov, N.A. Three-Dimensional Cell Culture Matrices: State of the Art. Tissue Eng. Part B Rev. 2008, 14, 61–86. [Google Scholar] [CrossRef]
- Richter, G.H.S.; Plehm, S.; Fasan, A.; Rössler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Löwel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentia-tion. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef]
- Huang, H.; Ding, Y.; Sun, X.S.; Nguyen, T.A. Peptide Hydrogelation and Cell Encapsulation for 3D Culture of MCF-7 Breast Cancer Cells. PLoS ONE 2013, 8, e59482. [Google Scholar] [CrossRef]
- Thippabhotla, S.; Zhong, C.; He, M. 3D cell culture stimulates the secretion of in vivo like extracellular vesicles. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Szvicsek, Z.; Oszvald, Á.; Szabó, L.; Sándor, G.O.; Kelemen, A.; Soós, A.Á.; Pálóczi, K.; Harsányi, L.; Tölgyes, T.; Dede, K.; et al. Extracellular vesicle release from intestinal organoids is modulated by Apc mutation and other colorectal cancer progression factors. Cell. Mol. Life Sci. 2019, 76, 2463–2476. [Google Scholar] [CrossRef] [PubMed]
- Rilla, K.; Mustonen, A.-M.; Arasu, U.T.; Härkönen, K.; Matilainen, J.; Nieminen, P. Extracellular vesicles are integral and functional components of the extracellular matrix. Matrix Biol. 2019, 75–76, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Lenzini, S.; Bargi, R.; Chung, G.; Shin, J.-W. Matrix mechanics and water permeation regulate extracellular vesicle transport. Nat. Nanotechnol. 2020, 15, 217–223. [Google Scholar] [CrossRef]
- Fuhrmann, G. Diffusion and transport of extracellular vesicles. Nat. Nanotechnol. 2020, 15, 168–169. [Google Scholar] [CrossRef]
- Luker, K.E.; Pata, P.; Shemiakina, I.I.; Pereverzeva, A.; Stacer, A.C.; Shcherbo, D.S.; Pletnev, V.Z.; Skolnaja, M.; Lukyanov, K.A.; Luker, G.D.; et al. Comparative study reveals better far-red fluorescent protein for whole body imaging. Sci. Rep. 2015, 5, 10332. [Google Scholar] [CrossRef] [PubMed]
- Erkan, E.P.; Senfter, D.; Madlener, S.; Jungwirth, G.; Ströbel, T.; Saydam, N.; Saydam, O. Extracellular vesicle-mediated suicide mRNA/protein delivery inhibits glioblastoma tumor growth in vivo. Cancer Gene Ther. 2016, 24, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Taha, E.A.; Sogawa, C.; Okusha, Y.; Kawai, H.; Oo, M.W.; Elseoudi, A.; Lu, Y.; Nagatsuka, H.; Kubota, S.; Satoh, A.; et al. Knockout of MMP3 Weakens Solid Tumor Organoids and Cancer Extracellular Vesicles. Cancers 2020, 12, 1260. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, T.; He, W.; Jin, H.; Liu, C.; Yang, Z.; Ren, J. Methotrexate-Loaded Extracellular Vesicles Functionalized with Therapeutic and Targeted Peptides for the Treatment of Glioblastoma Multiforme. ACS Appl. Mater. Interfaces 2018, 10, 12341–12350. [Google Scholar] [CrossRef]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.-F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef]
- Slack, J. Regeneration, Wound Healing and Cancer. In The Science of Stem Cells; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2018; pp. 217–238. ISBN 978-1-119-23529-3. [Google Scholar]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Yan, T.; Mizutani, A.; Chen, L.; Takaki, M.; Hiramoto, Y.; Matsuda, S.; Shigehiro, T.; Kasai, T.; Kudoh, T.; Murakami, H.; et al. Characterization of Cancer Stem-Like Cells Derived from Mouse Induced Pluripotent Stem Cells Transformed by Tumor-Derived Extracellular Vesicles. J. Cancer 2014, 5, 572–584. [Google Scholar] [CrossRef]
- Marzano, M.; Bejoy, J.; Cheerathodi, M.R.; Sun, L.; York, S.B.; Zhao, J.; Kanekiyo, T.; Bu, G.; Meckes, J.D.G.; Li, Y. Differential Effects of Extracellular Vesicles of Lineage-Specific Human Pluripotent Stem Cells on the Cellular Behaviors of Isogenic Cortical Spheroids. Cells 2019, 8, 993. [Google Scholar] [CrossRef]
- Oszvald, Á.; Szvicsek, Z.; Sándor, G.O.; Kelemen, A.; Soós, A.Á.; Pálóczi, K.; Bursics, A.; Dede, K.; Tölgyes, T.; Buzás, E.I.; et al. Extracellular vesicles transmit epithelial growth factor activity in the intestinal stem cell niche. Stem Cells 2020, 38, 291–300. [Google Scholar] [CrossRef]
- Eguchi, T.; Sogawa, C.; Okusha, Y.; Uchibe, K.; Iinuma, R.; Ono, K.; Nakano, K.; Murakami, J.; Itoh, M.; Arai, K.; et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. PLoS ONE 2018, 13, e0191109. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.I.; Ogawa, D.; Balaj, L.; Rienzo, G.D.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Lindoso, R.S.; Collino, F.; Camussi, G. Extracellular vesicles derived from renal cancer stem cells induce a pro-tumorigenic phenotype in mesenchymal stromal cells. Oncotarget 2015, 6, 7959–7969. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-oncogenic Signals That Maintain Intratumoral Heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Namba, Y.; Sogawa, C.; Okusha, Y.; Kawai, H.; Itagaki, M.; Ono, K.; Murakami, J.; Aoyama, E.; Ohyama, K.; Asaumi, J.; et al. Depletion of Lipid Efflux Pump ABCG1 Triggers the Intracellular Accumulation of Extracellular Vesicles and Reduces Aggregation and Tumorigenesis of Metastatic Cancer Cells. Front. Oncol. 2018, 8, 376. [Google Scholar] [CrossRef]
- Cheng, W.; Liao, T.; Lin, C.; Yuan, L.E.; Lan, H.; Lin, H.; Teng, H.; Chang, H.; Lin, C.; Yang, C.; et al. RAB27B-activated secretion of stem-like tumor exosomes delivers the biomarker microRNA-146a-5p, which promotes tumorigenesis and associates with an immunosuppressive tumor microenvironment in colorectal cancer. Int. J. Cancer 2019, 145, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Vera, N.; Acuña-Gallardo, S.; Grünenwald, F.; Caceres-Verschae, A.; Realini, O.; Acuña, R.; Lladser, A.; Illanes, S.E.; Varas-Godoy, M. Small Extracellular Vesicles Released from Ovarian Cancer Spheroids in Response to Cisplatin Promote the Pro-Tumorigenic Activity of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 4972. [Google Scholar] [CrossRef] [PubMed]
- Harmati, M.; Gyukity-Sebestyen, E.; Dobra, G.; Janovak, L.; Dekany, I.; Saydam, O.; Hunyadi-Gulyas, E.; Nagy, I.; Farkas, A.; Pankotai, T.; et al. Small extracellular vesicles convey the stress-induced adaptive responses of melanoma cells. Sci. Rep. 2019, 9, 15329. [Google Scholar] [CrossRef]
- Clayton, H.; Titley, I.; Vivanco, M. dM Growth and differentiation of progenitor/stem cells derived from the human mammary gland. Exp. Cell Res. 2004, 297, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Piva, M.; Rodriguez-Suarez, E.; Gil, D.; Royo, F.; Elortza, F.; Falcon-Perez, J.M.; Vivanco, M. Human Mammospheres Secrete Hormone-Regulated Active Extracellular Vesicles. PLoS ONE 2014, 9, e83955. [Google Scholar] [CrossRef]
- Lugini, L.; Valtieri, M.; Federici, C.; Cecchetti, S.; Meschini, S.; Condello, M.; Signore, M.; Fais, S. Exosomes from human colorectal cancer induce a tumor-like behavior in colonic mesenchymal stromal cells. Oncotarget 2016, 7, 50086–50098. [Google Scholar] [CrossRef]
- Chowdhury, R.; Webber, J.P.; Gurney, M.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015, 6, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Casson, J.; Davies, O.G.; Smith, C.-A.; Dalby, M.J.; Berry, C.C. Mesenchymal stem cell-derived extracellular vesicles may promote breast cancer cell dormancy. J. Tissue Eng. 2018, 9, 2041731418810093. [Google Scholar] [CrossRef]
- Leonora, B.; Hamar, P.; Guo, C.; Basar, E.; Perdigão-Henriques, R.; Balaj, L.; Lieberman, J. miR-200–containing extracellular vesicles promote breast cancer cell metastasis. J. Clin. Investig. 2014, 124, 5109–5128. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Israelsson, P.; Ottander, U.; Lundin, E.; Nagaev, I.; Nagaeva, O.; Dehlin, E.; Baranov, V.; Mincheva-Nilsson, L. Differential expression of ligands for NKG2D and DNAM-1 receptors by epithelial ovarian cancer-derived exosomes and its influence on NK cell cytotoxicity. Tumor Biol. 2015, 37, 5455–5466. [Google Scholar] [CrossRef]
- Thorne, S.H.; Negrin, R.S.; Contag, C.H. Synergistic Antitumor Effects of Immune Cell-Viral Biotherapy. Science 2006, 311, 1780–1784. [Google Scholar] [CrossRef]
- Javeed, N.; Mukhopadhyay, D. Exosomes and their role in the micro-/macro-environment: A comprehensive review. J. Biomed. Res. 2017, 31, 386–394. [Google Scholar]
- Rowe, J.H.; Ghouri, Y.A.; Mian, I. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar] [CrossRef]
- Kuznetsov, H.S.; Marsh, T.; Markens, B.A.; Castaño, Z.; Greene-Colozzi, A.; Hay, S.A.; Brown, V.E.; Richardson, A.L.; Signoretti, S.; Battinelli, E.M.; et al. Identification of Luminal Breast Cancers That Establish a Tumor-Supportive Macroenvironment Defined by Proangiogenic Platelets and Bone Marrow–Derived Cells. Cancer Discov. 2012, 2, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019, 156, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Honma, K.; Saji, S.; Moriwaki, H.; Ochiya, T.; Hoffman, R.M. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 2013, 65, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.W.; Choi, H.; Jang, S.C.; Yoo, M.Y.; Park, J.Y.; Choi, N.E.; Oh, H.J.; Ha, S.; Lee, Y.-S.; Jeong, J.M.; et al. Noninvasive imaging of radiolabeled exosome-mimetic nanovesicle using 99m Tc-HMPAO. Sci. Rep. 2015, 5, 15636. [Google Scholar] [CrossRef]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Bio-distribution and delivery efficiency of unmodified tumor-derived exosomes. J. Controlled Release 2015, 199, 145–155. [Google Scholar] [CrossRef]
- Busato, A.; Bonafede, R.; Bontempi, P.; Scambi, I.; Schiaffino, L.; Benati, D.; Malatesta, M.; Sbarbati, A.; Marzola, P.; Mariotti, R. Magnetic resonance imaging of ultrasmall superparamagnetic iron oxide-labeled exosomes from stem cells: A new method to obtain labeled exosomes. Int. J. Nanomed. 2016, 11, 2481–2490. [Google Scholar] [PubMed]
- Hood, J.L.; Scott, M.J.; Wickline, S.A. Maximizing exosome colloidal stability following electroporation. Anal. Biochem. 2014, 448, 41–49. [Google Scholar] [CrossRef]
- Morad, G.; Carman, C.V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.-E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-Derived Extracellular Vesicles Breach the Intact Blood–Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef]
- Whitesides, G.M. The origins and the future of microfluidics. Nat. Cell Biol. 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Tong, Z.; Tong, W.-Y.; Peng, B.; Wei, Y.; Oddo, A.; Voelcker, N.H. Using Integrated Cancer-on-Chip Platforms to Emulate and Probe Various Cancer Models. In Nanotechnology Characterization Tools for Tissue Engineering and Medical Therapy; Kumar, C.S.S.R., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 151–204. [Google Scholar]
- Ashammakhi, N.; Nasiri, R.; de Barros, N.R.; Tebon, P.; Thakor, J.; Goudie, M.; Shamloo, A.; Martin, M.G.; Khademhosseini, A. Gut-on-a-chip: Current progress and future opportunities. Biomaterials 2020, 255, 120196. [Google Scholar] [CrossRef]
- Huh, D.; Torisawa, Y.; Hamilton, G.A.; Kim, H.J.; Ingber, D.E. Microengineered physiological biomimicry: Organs-on-Chips. Lab. Chip 2012, 12, 2156–2164. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Z.; Rahim, N.A.A.; van Noort, D.; Yu, H. Towards a human-on-chip: Culturing multiple cell types on a chip with compartmentalized microenvironments. Lab. Chip 2009, 9, 3185–3192. [Google Scholar] [CrossRef]
- Tehranirokh, M.; Kouzani, A.Z.; Francis, P.S.; Kanwar, J.R. Microfluidic devices for cell cultivation and proliferation. Biomicrofluidics 2013, 7, 051502. [Google Scholar] [CrossRef] [PubMed]
- Rubiano, A.; Indapurkar, A.; Yokosawa, R.; Miedzik, A.; Rosenzweig, B.; Arefin, A.; Moulin, C.M.; Dame, K.; Hartman, N.; Volpe, D.A.; et al. Characterizing the Reproducibility in Using a Liver Microphysiological System for Assaying Drug Toxicity, Metabolism and Accumulation. Clin. Transl. Sci. 2021, 1–13. [Google Scholar] [CrossRef]
- Dioufa, N.; Clark, A.M.; Ma, B.; Beckwitt, C.H.; Wells, A. Bi-directional exosome-driven intercommunication between the hepatic niche and cancer cells. Mol. Cancer 2017, 16, 172. [Google Scholar] [CrossRef]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Caballero, D.; Kaushik, S.; Correlo, V.M.; Oliveira, J.M.; Reis, R.L.; Kundu, S.C. Organ-on-chip models of cancer metastasis for future personalized medicine: From chip to the patient. Biomaterials 2017, 149, 98–115. [Google Scholar] [CrossRef]
- Kim, J.; Lee, C.; Kim, I.; Ro, J.; Kim, J.; Min, Y.; Park, J.; Sunkara, V.; Park, Y.-S.; Michael, I.; et al. Three-Dimensional Human Liver-Chip Emulating Premetastatic Niche Formation by Breast Cancer-Derived Extracellular Vesicles. ACS Nano 2020, 14, 14971–14988. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Pang, J.; Qin, K.; Yuan, W.; Kong, J.; Ma, H.; He, J.; Yang, X.; Luo, Y.; Lu, Y.; et al. A Novel Tissue-Based Liver–Kidney-on-a-Chip Can Mimic Liver Tropism of Extracellular Vesicles Derived from Breast Cancer Cells. Biotechnol. J. 2020, 15, 1900107. [Google Scholar] [CrossRef]
- Park, T.-E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; Fitzgerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef] [PubMed]
- Hyenne, V.; Ghoroghi, S.; Collot, M.; Bons, J.; Follain, G.; Harlepp, S.; Mary, B.; Bauer, J.; Mercier, L.; Busnelli, I.; et al. Studying the Fate of Tumor Extracellular Vesicles at High Spatiotemporal Resolution Using the Zebrafish Embryo. Dev. Cell 2019, 48, 554–572. [Google Scholar] [CrossRef]
- Quiñonez-Silvero, C.; Hübner, K.; Herzog, W. Development of the brain vasculature and the blood-brain barrier in zebrafish. Dev. Biol. 2020, 457, 181–190. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Stoletov, K.; Montel, V.; Lester, R.D.; Gonias, S.L.; Klemke, R. High-resolution imaging of the dynamic tumor cell vascular interface in transparent zebrafish. Proc. Natl. Acad. Sci. USA 2007, 104, 17406–17411. [Google Scholar] [CrossRef]
- Kanada, M.; Zhang, J.; Yan, L.; Sakurai, T.; Terakawa, S. Endothelial cell-initiated extravasation of cancer cells visualized in zebrafish. PeerJ 2014, 2, e688. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; You, B.; Shi, S.; Shi, W.; Zhang, Z.; Zhang, Q.; Gu, M.; Chen, J.; Bao, L.; Liu, D.; et al. Hypoxia-Induced Matrix Metalloproteinase-13 Expression in Exosomes from Nasopharyngeal Carcinoma Enhances Metastases. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.K.; Schiavone, K.; Tazzyman, S.; Heymann, D.; Chico, T.J. Zebrafish xenograft models of cancer and metastasis for drug discovery. Expert Opin. Drug Discov. 2017, 12, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.M.; et al. BRAF Mutations Are Sufficient to Promote Nevi Formation and Cooperate with p53 in the Genesis of Melanoma. Curr. Biol. 2005, 15, 249–254. [Google Scholar] [CrossRef]
- Heilmann, S.; Ratnakumar, K.; Langdon, E.M.; Kansler, E.R.; Kim, I.S.; Campbell, N.R.; Perry, E.B.; McMahon, A.J.; Kaufman, C.K.; Van Rooijen, E.; et al. A Quantitative System for Studying Metastasis Using Transparent Zebrafish. Cancer Res. 2015, 75, 4272–4282. [Google Scholar] [CrossRef]
- Collot, M.; AshokKumar, P.; Anton, H.; Boutant, E.; Faklaris, O.; Galli, T.; Mély, Y.; Danglot, L.; Klymchenko, A.S. MemBright: A Family of Fluorescent Membrane Probes for Advanced Cellular Imaging and Neuroscience. Cell Chem. Biol. 2019, 26, 600–614. [Google Scholar] [CrossRef] [PubMed]
- van Duinen, V.; Zhu, D.; Ramakers, C.; van Zonneveld, A.J.; Vulto, P.; Hankemeier, T. Perfused 3D angiogenic sprouting in a high-throughput in vitro platform. Angiogenesis 2019, 22, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Eckford, A.W.; Haraguchi, T. Molecular Communication; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Bi, D.; Almpanis, A.; Noel, A.; Deng, Y.; Schober, R. A Survey of Molecular Communication in Cell Biology: Establishing a New Hierarchy for Interdisciplinary Applications. IEEE Commun. Surv. Tutor. 2021, PP, 1. [Google Scholar] [CrossRef]
- Veletic, M.; Barros, M.T.; Arjmandi, H.; Balasubramaniam, S.; Balasingham, I. Modeling of Modulated Exosome Release From Differentiated Induced Neural Stem Cells for Targeted Drug Delivery. IEEE Trans. NanoBiosci. 2020, 19, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Veletić, M.; Barros, M.T.; Balasingham, I.; Balasubramaniam, S. A Molecular Communication Model of Exosome-mediated Brain Drug Delivery. In Proceedings of the Proceedings of the Sixth Annual ACM International Conference on Nanoscale Computing and Communication; Association for Computing Machinery: New York, NY, USA, 2019; pp. 1–7. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ural, E.E.; Toomajian, V.; Hoque Apu, E.; Veletic, M.; Balasingham, I.; Ashammakhi, N.; Kanada, M.; Contag, C.H. Visualizing Extracellular Vesicles and Their Function in 3D Tumor Microenvironment Models. Int. J. Mol. Sci. 2021, 22, 4784. https://doi.org/10.3390/ijms22094784
Ural EE, Toomajian V, Hoque Apu E, Veletic M, Balasingham I, Ashammakhi N, Kanada M, Contag CH. Visualizing Extracellular Vesicles and Their Function in 3D Tumor Microenvironment Models. International Journal of Molecular Sciences. 2021; 22(9):4784. https://doi.org/10.3390/ijms22094784
Chicago/Turabian StyleUral, Evran E., Victoria Toomajian, Ehsanul Hoque Apu, Mladen Veletic, Ilangko Balasingham, Nureddin Ashammakhi, Masamitsu Kanada, and Christopher H. Contag. 2021. "Visualizing Extracellular Vesicles and Their Function in 3D Tumor Microenvironment Models" International Journal of Molecular Sciences 22, no. 9: 4784. https://doi.org/10.3390/ijms22094784
APA StyleUral, E. E., Toomajian, V., Hoque Apu, E., Veletic, M., Balasingham, I., Ashammakhi, N., Kanada, M., & Contag, C. H. (2021). Visualizing Extracellular Vesicles and Their Function in 3D Tumor Microenvironment Models. International Journal of Molecular Sciences, 22(9), 4784. https://doi.org/10.3390/ijms22094784