
The Neuroprotective Effects of GPR4 Inhibition through the Attenuation of Caspase Mediated Apoptotic Cell Death in an MPTP Induced Mouse Model of Parkinson’s Disease

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
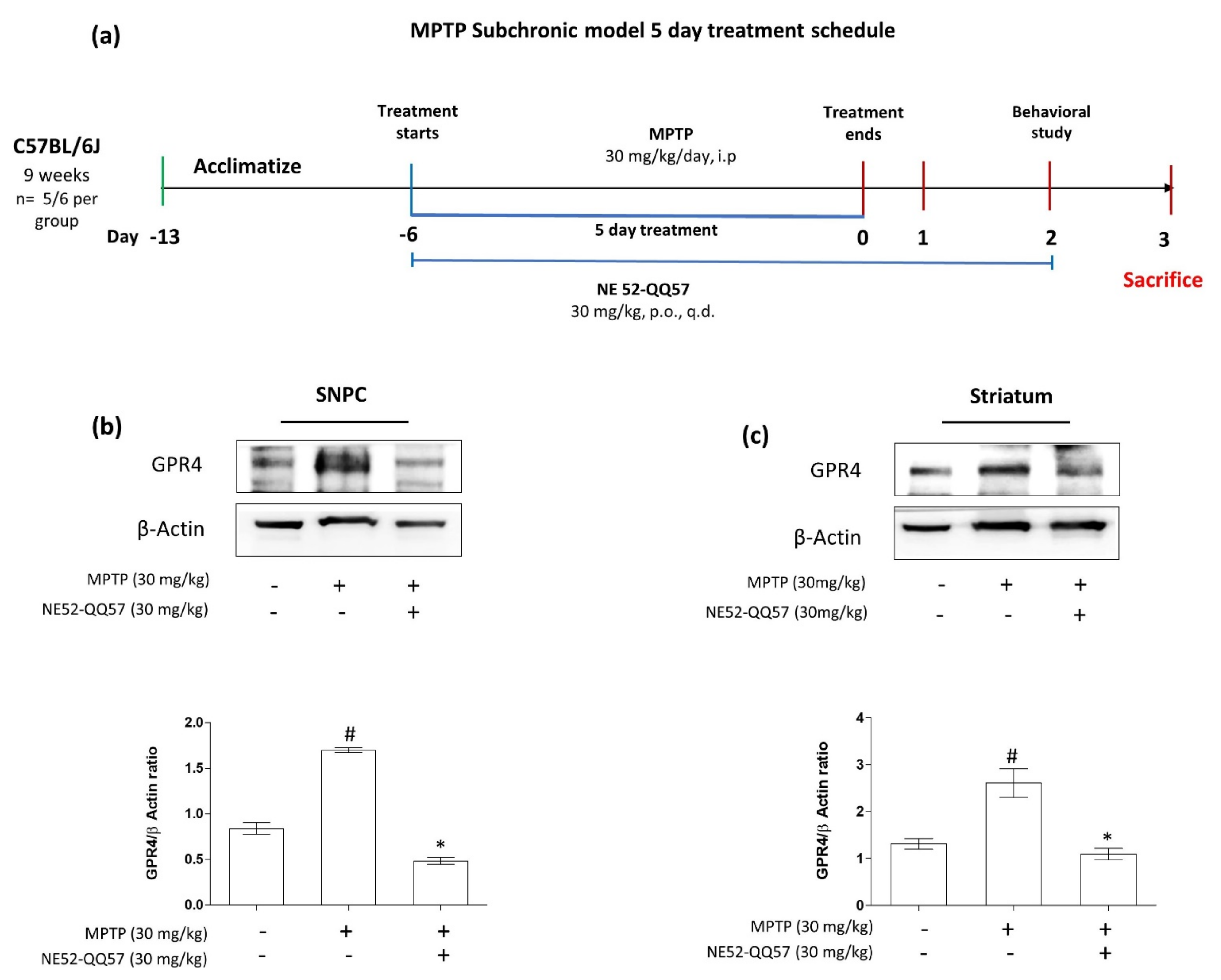
2.1. Expression of GPR4 Is Upregulated in an MPTP-Induced Mouse Model of PD
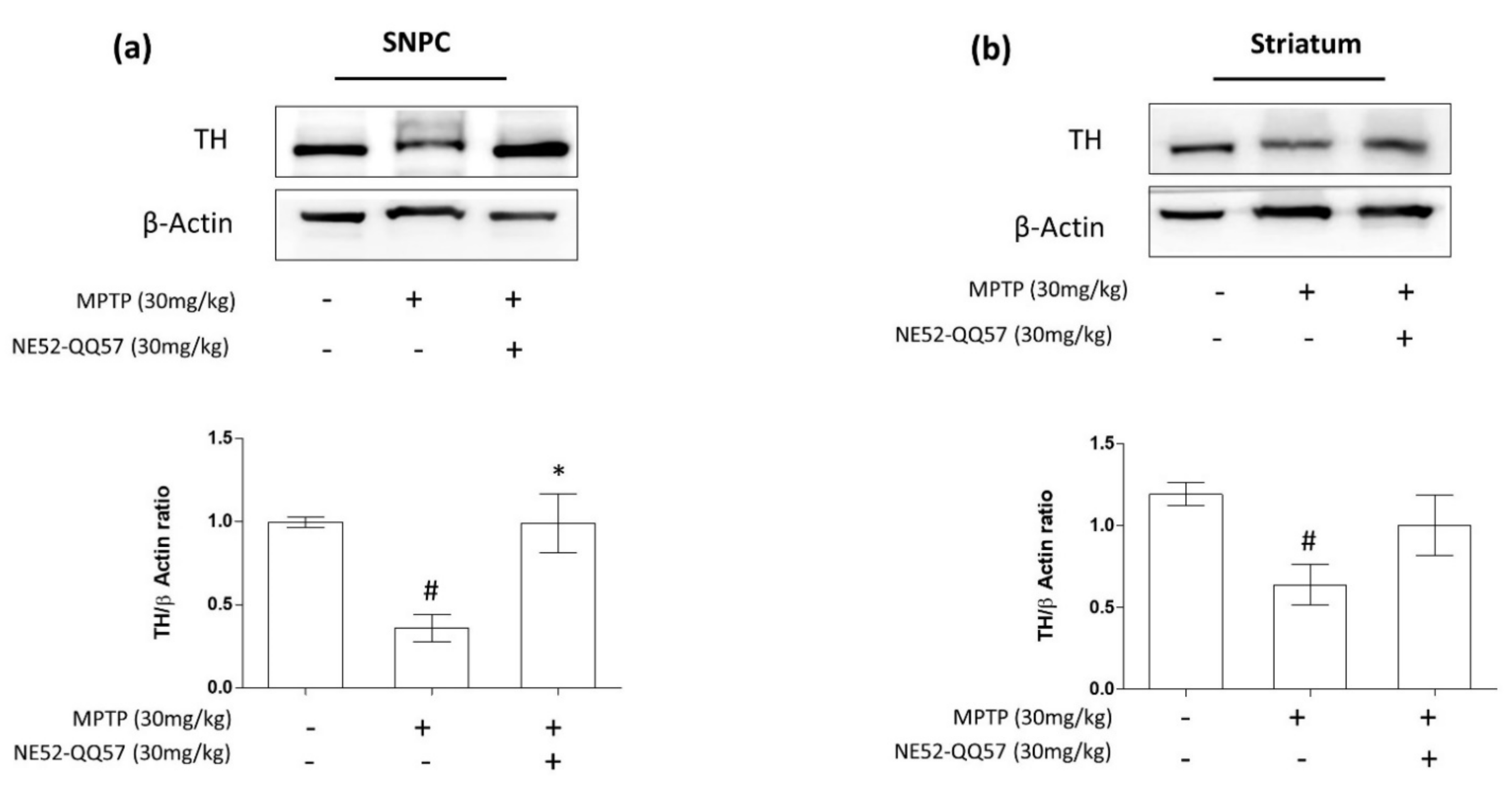
2.2. Inhibition of GPR4 Protects Against the MPTP-Induced Depletion of TH in the SNpc and Striatum
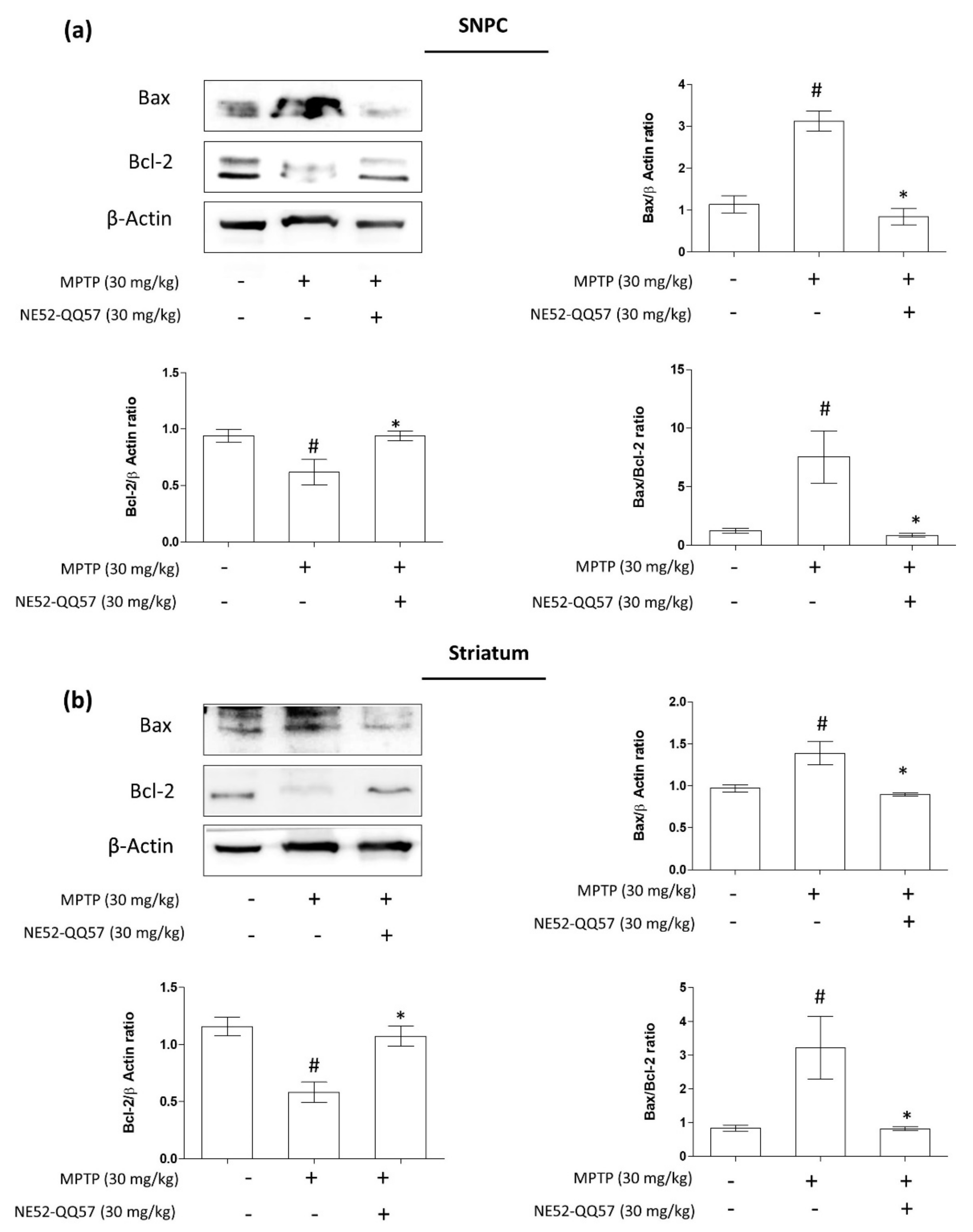
2.3. Inhibition of GPR4 Decreases MPTP-Induced Increase in the Pro-Apoptotic Bax/Bcl-2 Ratio in the SNpc and Striatum
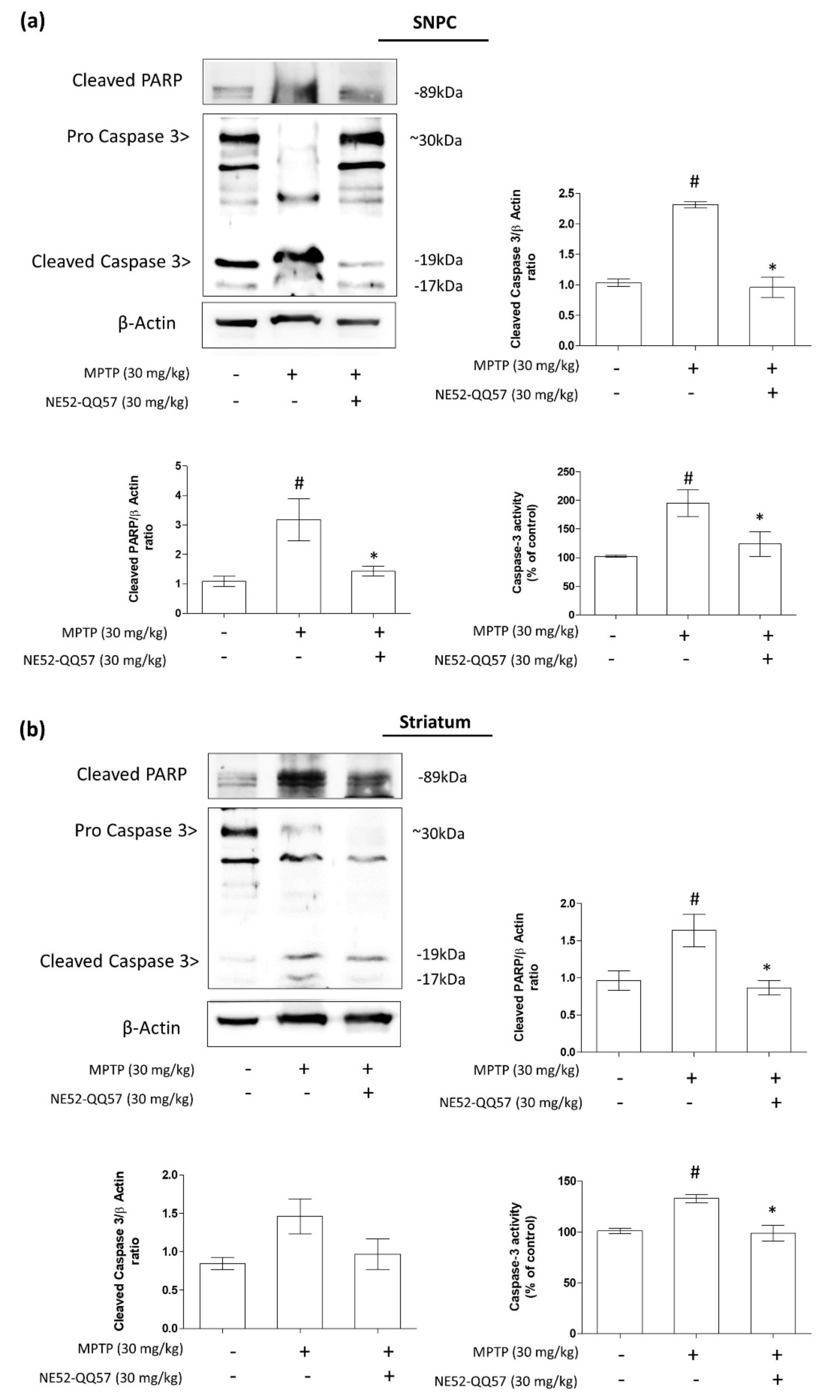
2.4. Effects of GPR4 Inhibition on PARP Cleavage and Caspase 3 Activity in the SNpc and Striatum of MPTP-Induced PD Model Mice
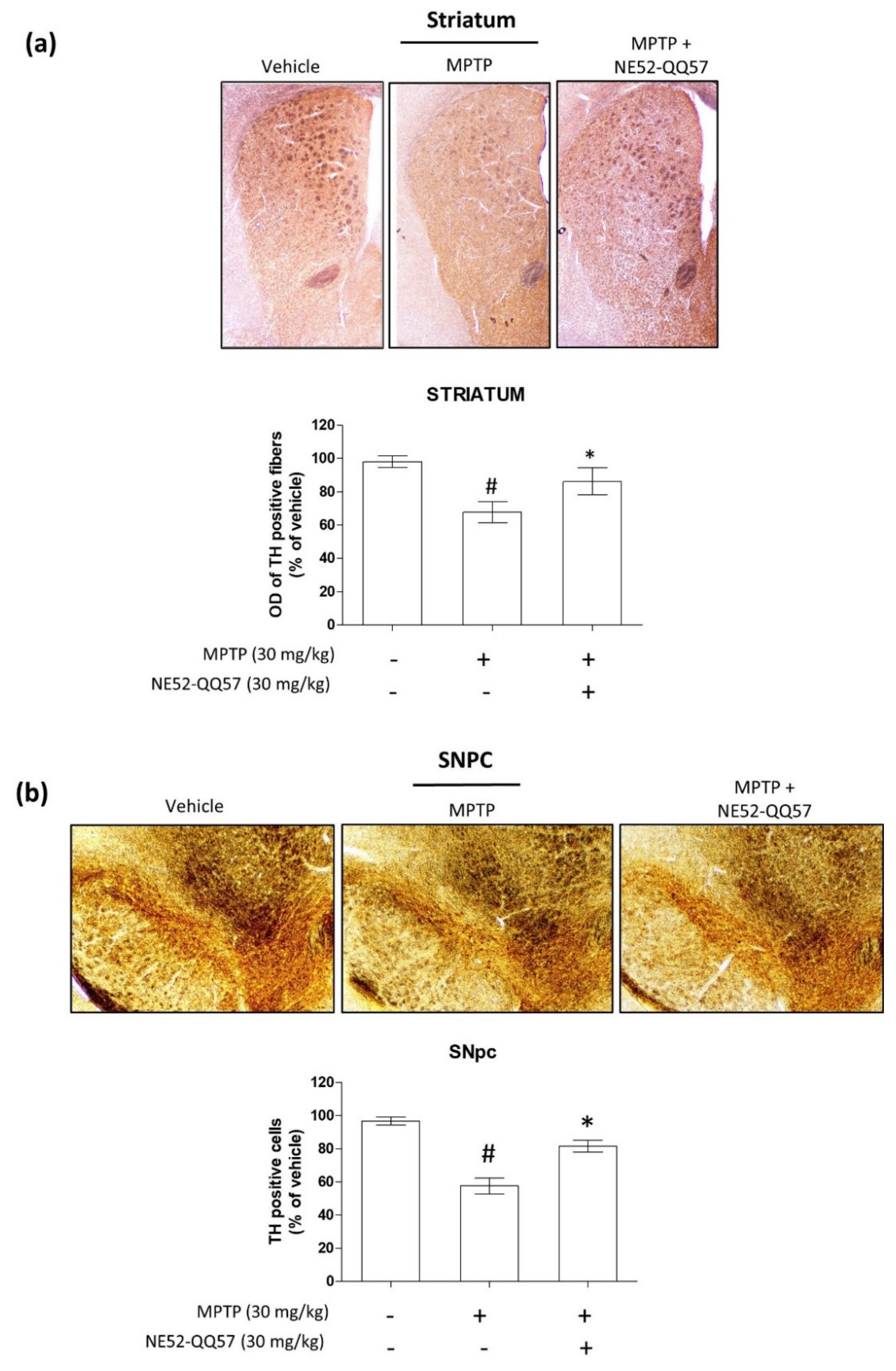
2.5. Effect of GPR4 Antagonist on the MPTP-Induced Degradation of TH-Positive Cells in SNpc and Striatum
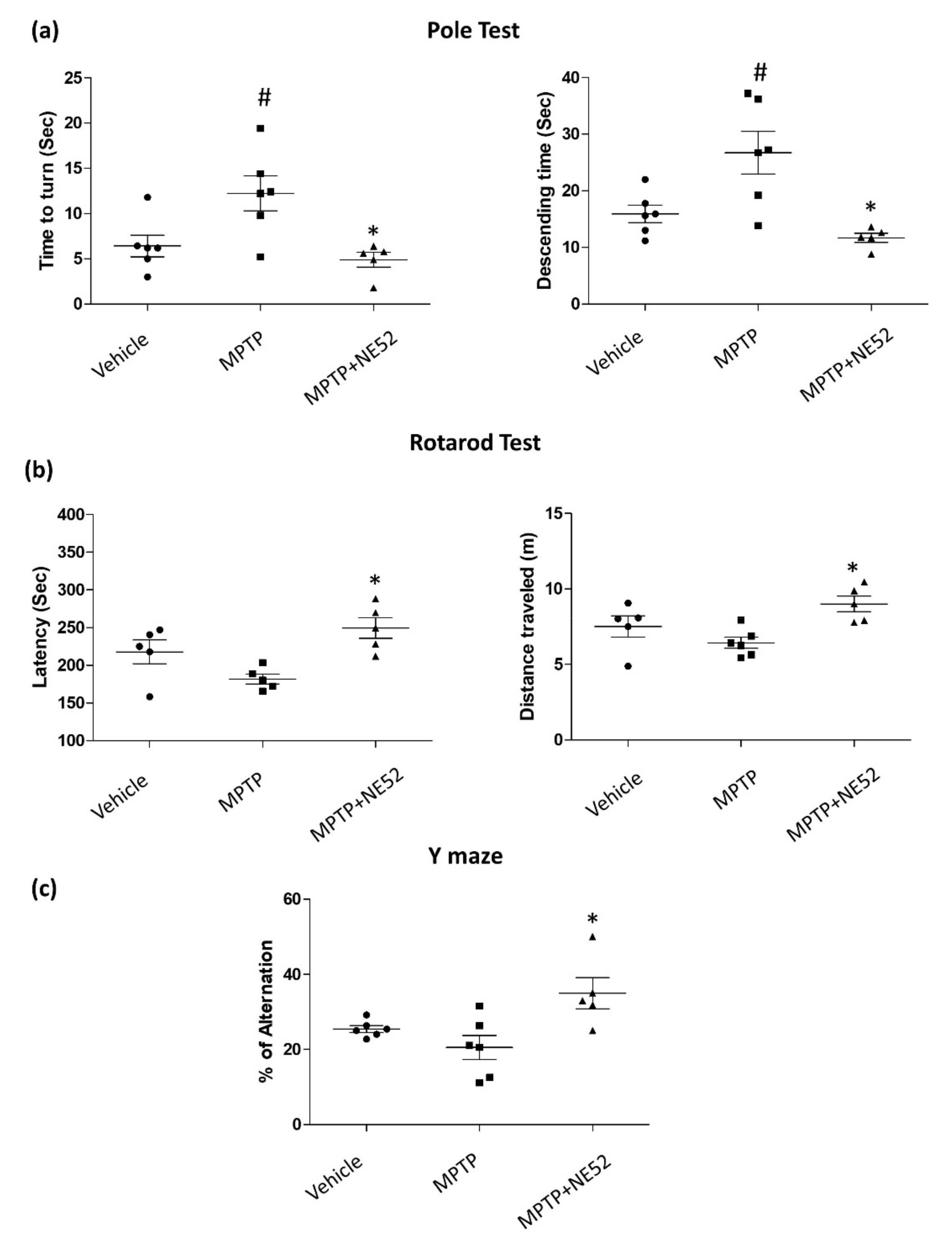
2.6. Effect of GPR4 Antagonist on the Motor Coordination Function (Rotarod), Bradykinesia (Pole Test) and Spontaneous Alternation Performance (Y-maze Test) on MPTP-Induced PD Mouse Model
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Animals and MPTP and NE 52QQ57 Administration
4.3. Immunoblot Analysis
4.4. Assessment of Caspase-3 Activity
4.5. Immunohistochemistry
4.6. Behavioural Studies
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MPTP | 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine |
| MPP+ | 1-Methyl-4-Phenylpyridinium Ion |
| MTT | 3-(3,4-Dimehylthiazol-2-Yl)-2,5-Diphenyl-Tetrazolium Bromide |
| ER | Endoplasmic Reticulum |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| L-DOPA | L-tyrosine to L-3,4-dihydroxyphenylalanine |
| MMP | Mitochondrial Membrane Potential |
| mPTP | Mitochondrial Permeability Transition Pore |
| PIP2 | Phosphatidylinositol Biphosphate |
| PARP | Poly (ADP-Ribose) Polymerase |
| SNpc | Substantia Nigra Pars Compacta |
| TH | Tyrosine hydroxylase |
| TDAG8 | T-Cell Death-Associated Gene 8 |
| OGR1 | The Ovarian Cancer G Protein-Coupled Receptor 1 |
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Velkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers. 2017, 3, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lew, M. Therapy, Overview of Parkinson’s disease. Pharmacother. J. Human Pharmacol. Drug Ther. 2007, 27, 155S–160S. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M. M The role of oxidative stress in Parkinson’s disease. J. Parkinson Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Pain, S.; Gochard, A.; Bodard, S.; Gulhan, Z.; Prunier-Aesch, C.; Chalon, S. Toxicity of MPTP on neurotransmission in three mouse models of Parkinson’s disease. Exp. Toxicol. Pathol. 2013, 65, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Brotchie, J.M. MPTP-Induced Models of Parkinson’s Disease in Mice and Non-Human Primates. Curr. Protoc. Pharmacol. 2005, 29, 5.42.1–5.42.13. [Google Scholar] [CrossRef] [PubMed]
- Perfeito, R.; Cunha-Oliveira, T.; Rego, A.C. Revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease—Resemblance to the effect of amphetamine drugs of abuse. Free. Radic. Biol. Med. 2012, 53, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Lozano, A.M. Parkinson’s disease. N. Eng. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Tobo, M.; Tomura, H.; Mogi, C.; Wang, J.-Q.; Liu, J.-P.; Komachi, M.; Damirin, A.; Kimura, T.; Murata, N.; Kurose, H. Previously postulated “ligand-independent” signaling of GPR4 is mediated through proton-sensing mechanisms. Cell. Signal. 2007, 19, 1745–1753. [Google Scholar] [CrossRef]
- Hosford, P.; Mosienko, V.; Kishi, K.; Jurisic, G.; Seuwen, K.; Kinzel, B.; Ludwig, M.; Wells, J.; Christie, I.; Koolen, L.; et al. CNS distribution, signalling properties and central effects of G-protein coupled receptor 4. Neuropharmacology 2018, 138, 381–392. [Google Scholar] [CrossRef]
- Dong, B.; Zhou, H.; Han, C.; Yao, J.; Xu, L.; Zhang, M.; Fu, Y.; Xia, Q. Ischemia/Reperfusion-Induced CHOP Expression Promotes Apoptosis and Impairs Renal Function Recovery: The Role of Acidosis and GPR4. PLoS ONE 2014, 9, e110944. [Google Scholar] [CrossRef]
- Brown, D.; Wagner, C.A. Molecular mechanisms of acid-base sensing by the kidney. J. Am. Soc. Nephrol. 2012, 23, 774–780. [Google Scholar] [CrossRef]
- Shi, Y.; Abe, C.; Holloway, B.B.; Shu, S.; Kumar, N.N.; Weaver, J.L.; Sen, J.; Perez-Reyes, E.; Stornetta, R.L.; Guyenet, P.G.; et al. Nalcn Is a “Leak” Sodium Channel That Regulates Excitability of Brainstem Chemosensory Neurons and Breathing. J. Neurosci. 2016, 36, 8174–8187. [Google Scholar] [CrossRef]
- Sanderlin, E.J.; Marie, M.; Velcicky, J.; Loetscher, P.; Yang, L.V. Pharmacological inhibition of GPR4 remediates intestinal inflammation in a mouse colitis model. Eur. J. Pharmacol. 2019, 852, 218–230. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, Y.; Cai, H.; Ma, H.; Zhao, D.; Zhang, X.; Li, Z.; Wang, S.; Wang, J.; Liu, R.; et al. Human GPR4 and the Notch signaling pathway in endothelial cell tube formation. Mol. Med. Rep. 2016, 14, 1235–1240. [Google Scholar] [CrossRef]
- Jing, Z.; Xu, H.; Chen, X.; Zhong, Q.; Huang, J.; Zhang, Y.; Guo, W.; Yang, Z.; Ding, S.; Chen, P.; et al. The Proton-Sensing G-Protein Coupled Receptor GPR4 Promotes Angiogenesis in Head and Neck Cancer. PLoS ONE 2016, 11, e0152789. [Google Scholar] [CrossRef]
- Fukuda, H.; Ito, S.; Watari, K.; Mogi, C.; Arisawa, M.; Okajima, F.; Kurose, H.; Shuto, S. Identification of a Potent and Selective GPR4 Antagonist as a Drug Lead for the Treatment of Myocardial Infarction. ACS Med. Chem. Lett. 2016, 7, 493–497. [Google Scholar] [CrossRef]
- Sun, X.; Tommasi, E.; Molina, D.; Sah, R.; Brosnihan, K.B.; Diz, D.I.; Petrovic, S. Deletion of proton-sensing receptor GPR4 associates with lower blood pressure and lower binding of angiotensin II receptor in SFO. Am. J. Physiol. Physiol. 2016, 311, F1260–F1266. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, X.; Fan, Y.; Cao, S.; Zhang, X. GPR4 knockout improves renal ischemia–reperfusion injury and inhibits apoptosis via suppressing the expression of CHOP. Biochem. J. 2017, 474, 4065–4074. [Google Scholar] [CrossRef]
- Wang, Y.; De Vallière, C.; Silva, P.H.I.; Leonardi, I.; Gruber, S.; Gerstgrasser, A.; Melhem, H.; Weber, A.; Leucht, K.; Wolfram, L.; et al. The Proton-activated Receptor GPR4 Modulates Intestinal Inflammation. J. Crohns Colitis. 2018, 12, 355–368. [Google Scholar] [CrossRef]
- Haque, M.E.; Akther, M.; Azam, S.; Choi, D.K.; Kim, I.S. GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Do-paminergic Neuronal Cell. Int. J. Mol. Sci. 2020, 21, 7517. [Google Scholar] [CrossRef]
- Kim, B.-W.; Koppula, S.; Kumar, H.; Park, J.-Y.; Kim, I.-W.; More, S.V.; Kim, I.-S.; Han, S.-D.; Kim, S.-K.; Yoon, S.-H.; et al. α-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NF kappa B activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson’s disease. Neuropharmacology 2015, 97, 46–57. [Google Scholar] [CrossRef]
- Korsmeyer, S.J. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999, 59, 1693–1700. [Google Scholar] [CrossRef]
- Gobeil, S.; Boucher, C.C.; Nadeau, D.; Poirier, G.G. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): Implication of lysosomal proteases. Cell Death Differ. 2001, 8, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Mandir, A.S.; Przedborski, S.; Lewis-Jackson, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly (ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef]
- More, S.; Choi, D.-K. Neuroprotective role of atractylenolide-I in an in vitro and in vivo model of Parkinson’s disease. Nutrients 2017, 9, 451. [Google Scholar] [CrossRef]
- Kramer, B.C.; Mytilineou, C. Alterations in the cellular distribution of bcl-2, bcl-x and bax in the adult rat substantia nigra following striatal 6-hydroxydopamine lesions. J. Neurocytology 2004, 33, 213–223. [Google Scholar] [CrossRef]
- Rekha, K.R.; Selvakumar, G.P. Gene expression regulation of Bcl2, Bax and cytochrome-C by geraniol on chronic MPTP/probenecid induced C57BL/6 mice model of Parkinson’s disease. Chem. Interact. 2014, 217, 57–66. [Google Scholar] [CrossRef]
- Vila, M.; Lewis-Jackson, V.; Vukosavic, S.; Djaldetti, R.; Liberatore, G.; Offen, D.; Korsmeyer, S.J.; Przedborski, S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 2837–2842. [Google Scholar] [CrossRef]
- Bagci, E.; Vodovotz, Y.; Billiar, T.; Ermentrout, G.; Bahar, I. Bistability in Apoptosis: Roles of Bax, Bcl-2, and Mitochondrial Permeability Transition Pores. Biophys. J. 2006, 90, 1546–1559. [Google Scholar] [CrossRef]
- Hu, M.; Li, F.; Wang, W. Vitexin protects dopaminergic neurons in MPTP-induced Parkinson’s disease through PI3K/Akt signaling pathway. Drug Des. Dev. Ther. 2018, 12, 565–573. [Google Scholar] [CrossRef]
- Pawlowski, J.; Kraft, A.S. Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef]
- Schriewer, J.M.; Peek, C.B.; Bass, J.; Schumacker, P.T. ROS-Mediated PARP Activity Undermines Mitochondrial Function After Permeability Transition Pore Opening During Myocardial Ischemia–Reperfusion. J. Am. Heart. Assoc. 2013, 2, e000159. [Google Scholar] [CrossRef]
- Cosi, C.; Marien, M. Implication of Poly (ADP-Ribose) Polymerase (PARP) in Neurodegeneration and Brain Energy Metabolism: Decreases in Mouse Brain NAD+ and ATP Caused by MPTP Are Prevented by the PARP Inhibitor Benzamide. Ann. N. Y. Acad. Sci. 1999, 890, 227–239. [Google Scholar] [CrossRef]
- Kumar, H.; Kim, I.-S.; More, S.V.; Kim, B.-W.; Bahk, Y.-Y.; Choi, D.-K. Gastrodin Protects Apoptotic Dopaminergic Neurons in a Toxin-Induced Parkinson’s Disease Model. Evid. Based Complement. Altern. Med. 2013, 2013, 1–13. [Google Scholar] [CrossRef]
- Matsuura, K.; Kabuto, H.; Makino, H.; Ogawa, N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 1997, 73, 45–48. [Google Scholar] [CrossRef]
- Rozas, G.; López-Martín, E.; Guerra, M.; Labandeira-García, J. The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J. Neurosci. Methods 1998, 83, 165–175. [Google Scholar] [CrossRef]
- Moriguchi, S.; Yabuki, Y.; Fukunaga, K. Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in MPTP-treated mice. J. Neurochem. 2012, 120, 541–551. [Google Scholar] [CrossRef]
- Han, N.-R.; Kim, Y.-K.; Ahn, S.; Hwang, T.-Y.; Lee, H.; Park, H.-J. A Comprehensive Phenotype of Non-motor Impairments and Distribution of Alpha-Synuclein Deposition in Parkinsonism-Induced Mice by a Combination Injection of MPTP and Probenecid. Front. Aging Neurosci. 2021, 12, 512. [Google Scholar] [CrossRef]
- Braga, R.; Kouzmine, I.; Canteras, N.S.; Da Cunha, C. Lesion of the substantia nigra, pars compacta impairs delayed alternation in a Y-maze in rats. Exp. Neurol. 2005, 192, 134–141. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Jiang, M.; Cai, Q.; Fang, J.; Jin, L. MANF improves the MPP+/MPTP-induced Parkinson’s disease via improvement of mitochondrial function and inhibition of oxidative stress. Am. J. Transl. Res. 2018, 10, 1284–1294. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, M.E.; Azam, S.; Akther, M.; Cho, D.-Y.; Kim, I.-S.; Choi, D.-K. The Neuroprotective Effects of GPR4 Inhibition through the Attenuation of Caspase Mediated Apoptotic Cell Death in an MPTP Induced Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4674. https://doi.org/10.3390/ijms22094674
Haque ME, Azam S, Akther M, Cho D-Y, Kim I-S, Choi D-K. The Neuroprotective Effects of GPR4 Inhibition through the Attenuation of Caspase Mediated Apoptotic Cell Death in an MPTP Induced Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(9):4674. https://doi.org/10.3390/ijms22094674
Chicago/Turabian StyleHaque, Md Ezazul, Shofiul Azam, Mahbuba Akther, Duk-Yeon Cho, In-Su Kim, and Dong-Kug Choi. 2021. "The Neuroprotective Effects of GPR4 Inhibition through the Attenuation of Caspase Mediated Apoptotic Cell Death in an MPTP Induced Mouse Model of Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 9: 4674. https://doi.org/10.3390/ijms22094674
APA StyleHaque, M. E., Azam, S., Akther, M., Cho, D.-Y., Kim, I.-S., & Choi, D.-K. (2021). The Neuroprotective Effects of GPR4 Inhibition through the Attenuation of Caspase Mediated Apoptotic Cell Death in an MPTP Induced Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences, 22(9), 4674. https://doi.org/10.3390/ijms22094674