
Artificial Intelligence in Bulk and Single-Cell RNA-Sequencing Data to Foster Precision Oncology

 , ,
, , 
Abstract
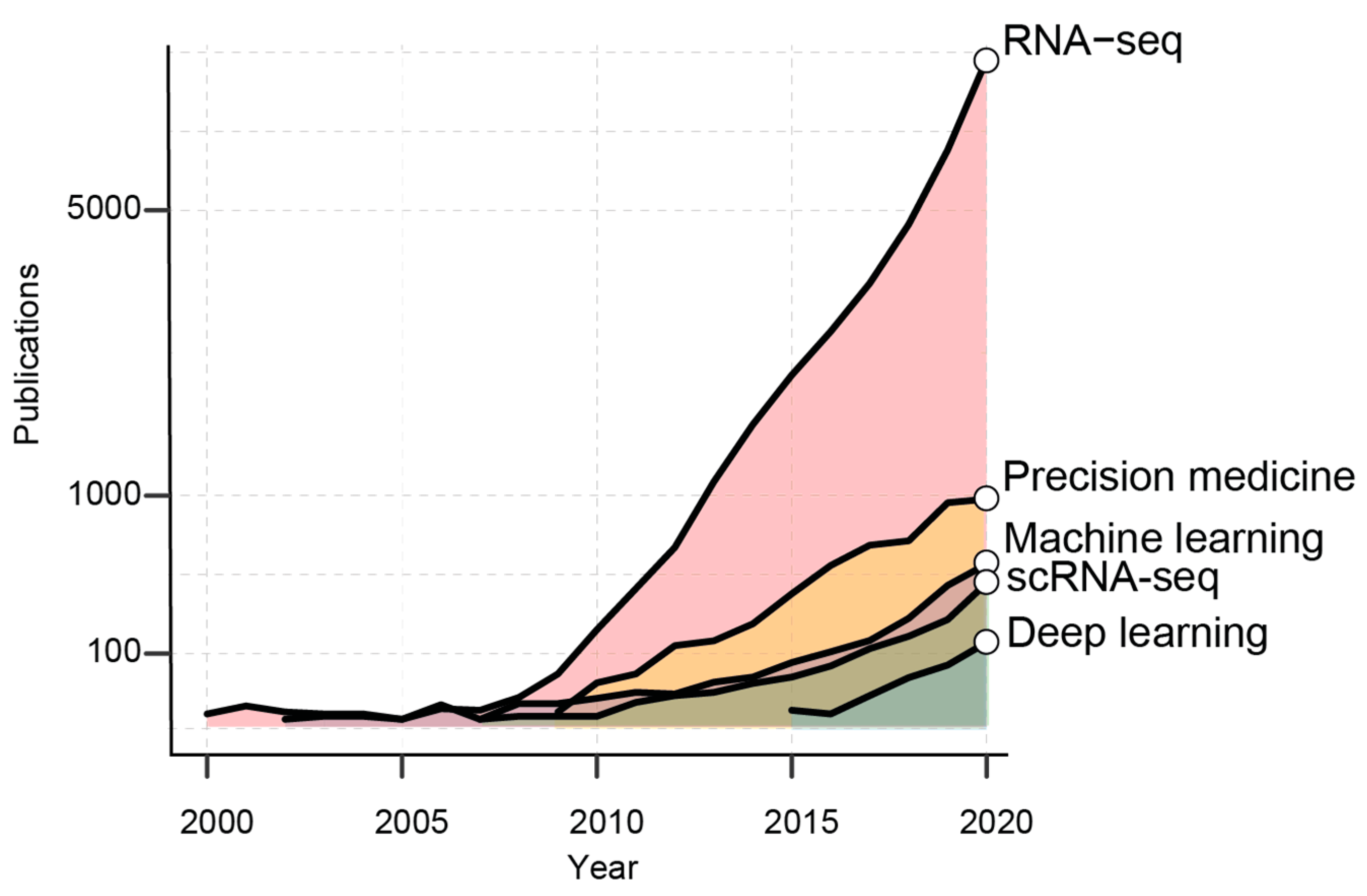
1. Introduction
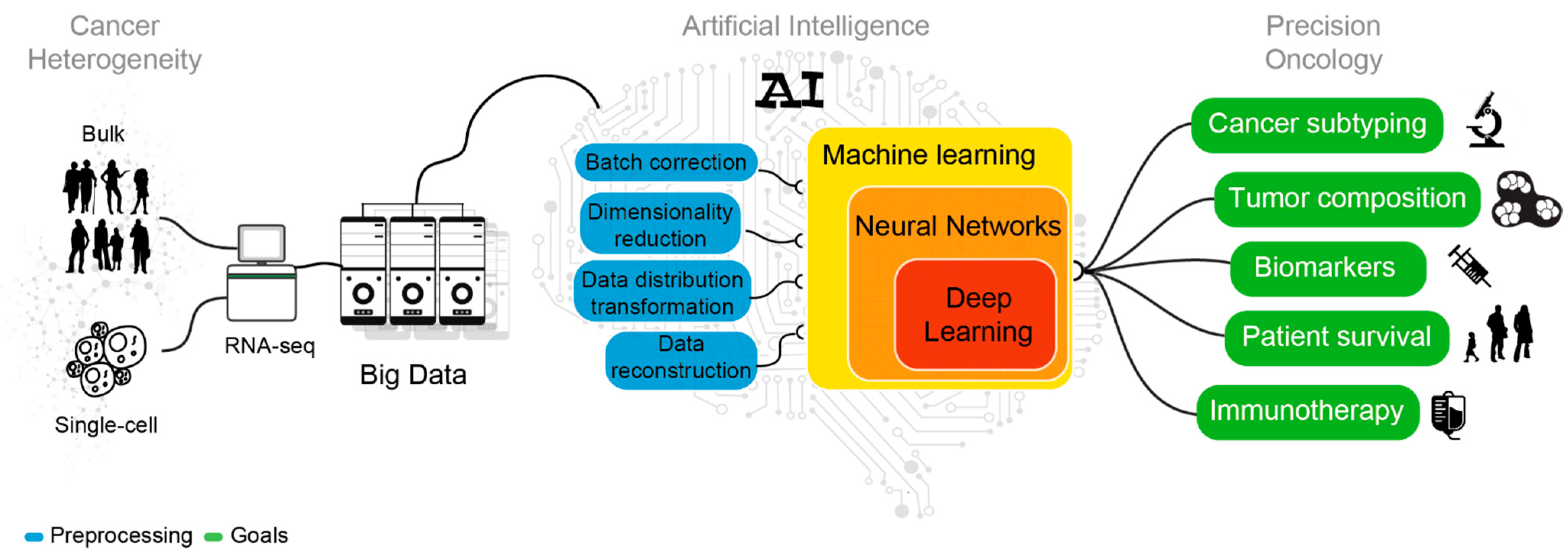
2. AI in the Era of Transcriptomic Big Data
3. Managing the Heterogeneity of Cancer Transcriptomes
3.1. Batch-Correction of Technical Heterogeneity
3.2. Dimensionality Reduction Approaches
3.2.1. Feature Extraction
3.2.2. Feature Selection
3.3. Data Distribution Transformation
3.4. Data Reconstruction: The Sparsity Issue
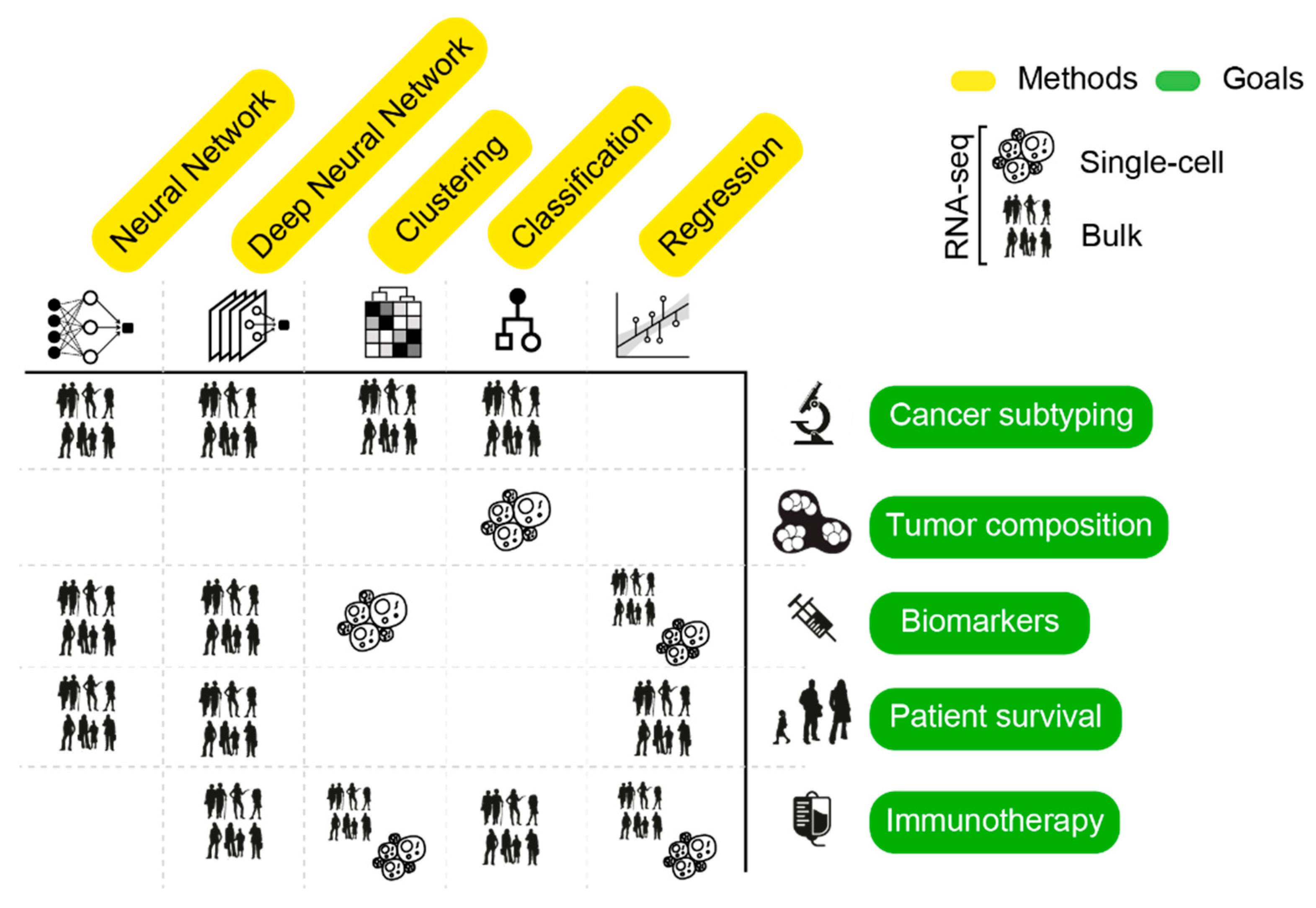
4. AI Mining of Cancer Transcriptomes
4.1. Assessing Inter-Tumor Heterogeneity: Classification of Cancer Subtypes
4.2. Deciphering Intra-Tumor Heterogeneity
4.2.1. Defining Cell Types and Clones
4.2.2. Assessment of TME
4.3. Biomarker Identification
4.4. Prediction of Patient Survival
4.5. Identification of Neoepitopes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watch, A.I. Jrc Science for Policy Report. Available online: https://publications.jrc.ec.europa.eu/repository/bitstream/JRC120214/jrc120214_ai_in_medicine_and_healthcare_report-aiwatch_v50.pdf (accessed on 28 February 2021).
- Fröhlich, H.; Balling, R.; Beerenwinkel, N.; Kohlbacher, O.; Kumar, S.; Lengauer, T.; Maathuis, M.H.; Moreau, Y.; Murphy, S.A.; Przytycka, T.M.; et al. From Hype to Reality: Data Science Enabling Personalized Medicine. BMC Med. 2018, 16, 150. [Google Scholar] [CrossRef]
- Big Biological Impacts from Big Data. Available online: https://www.sciencemag.org/features/2014/06/big-biological-impacts-big-data (accessed on 28 February 2021).
- Cereda, M.; Mourikis, T.P.; Ciccarelli, F.D. Genetic Redundancy, Functional Compensation, and Cancer Vulnerability. Trends Cancer Res. 2016, 2, 160–162. [Google Scholar] [CrossRef]
- Marx, V. Biology: The Big Challenges of Big Data. Nature 2013, 498, 255–260. [Google Scholar] [CrossRef]
- McCall, B. COVID-19 and Artificial Intelligence: Protecting Health-Care Workers and Curbing the Spread. Lancet Digit. Health 2020, 2, e166–e167. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial Intelligence in COVID-19 Drug Repurposing. Lancet Digit. Health 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, Y.; Zhang, Z.; Han, L. Maximizing the Utility of Cancer Transcriptomic Data. Trends Cancer Res. 2018, 4, 823–837. [Google Scholar] [CrossRef]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-Generation Personalised Medicine for High-Risk Paediatric Cancer Patients—The INFORM Pilot Study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef]
- Tirtei, E.; Cereda, M.; De Luna, E.; Quarello, P.; Asaftei, S.D.; Fagioli, F. Omic Approaches to Pediatric Bone Sarcomas. Pediatric Blood Cancer 2020, 67, e28072. [Google Scholar] [CrossRef]
- McPherson, J.D.; Marra, M.; Hillier, L.; Waterston, R.H.; Chinwalla, A.; Wallis, J.; Sekhon, M.; Wylie, K.; Mardis, E.R.; Wilson, R.K.; et al. A Physical Map of the Human Genome. Nature 2001, 409, 934–941. [Google Scholar]
- Libbrecht, M.W.; Noble, W.S. Machine Learning Applications in Genetics and Genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep Learning: New Computational Modelling Techniques for Genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.E.; Peña, J.-M.; Jayamohan, J.; Jérusalem, A. Mechanistic Models versus Machine Learning, a Fight Worth Fighting for the Biological Community? Biol. Lett. 2018, 14. [Google Scholar] [CrossRef]
- Crick, F. The Recent Excitement about Neural Networks. Nature 1989, 337, 129–132. [Google Scholar] [CrossRef]
- Cascianelli, S.; Molineris, I.; Isella, C.; Masseroli, M.; Medico, E. Machine Learning for RNA Sequencing-Based Intrinsic Subtyping of Breast Cancer. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A Novel Deep Learning-Based Framework for Cancer Molecular Subtype Classification. Oncogenesis 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, Z.; Yu, X.; Zhang, Z. RNA-Seq-Based Breast Cancer Subtypes Classification Using Machine Learning Approaches. Comput. Intell. Neurosci. 2020, 2020, 4737969. [Google Scholar] [CrossRef] [PubMed]
- Valle, F.; Osella, M.; Caselle, M. A Topic Modeling Analysis of TCGA Breast and Lung Cancer Transcriptomic Data. Cancers 2020, 12, 3799. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-P.; Yin, J.-H.; Li, W.-F.; Li, H.-J.; Chen, D.-P.; Zhang, C.-J.; Lv, J.-W.; Wang, Y.-Q.; Li, X.-M.; Li, J.-Y.; et al. Single-Cell Transcriptomics Reveals Regulators Underlying Immune Cell Diversity and Immune Subtypes Associated with Prognosis in Nasopharyngeal Carcinoma. Cell Res. 2020, 30, 1024–1042. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-Cell RNA Landscape of Intratumoral Heterogeneity and Immunosuppressive Microenvironment in Advanced Osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Bao, X.; Shi, R.; Zhao, T.; Wang, Y.; Anastasov, N.; Rosemann, M.; Fang, W. Integrated Analysis of Single-Cell RNA-Seq and Bulk RNA-Seq Unravels Tumour Heterogeneity plus M2-like Tumour-Associated Macrophage Infiltration and Aggressiveness in TNBC. Cancer Immunol. Immunother. 2021, 70, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Lee, H.; Kim, D.; Han, S.K.; Ha, D.; Shin, K.; Kim, S. Network-Based Machine Learning in Colorectal and Bladder Organoid Models Predicts Anti-Cancer Drug Efficacy in Patients. Nat. Commun. 2020, 11, 5485. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Yao, C.Q.; Sabine, V.S.; Grzadkowski, M.; Stimper, V.; Starmans, M.H.W.; Wang, J.; Nguyen, F.; Moon, N.C.; Lin, X.; et al. Pathway-Based Subnetworks Enable Cross-Disease Biomarker Discovery. Nat. Commun. 2018, 9, 4746. [Google Scholar] [CrossRef]
- Wu, W.; Ma, X. Joint Learning Dimension Reduction and Clustering of Single-Cell RNA-Sequencing Data. Bioinformatics 2020, 36. [Google Scholar] [CrossRef]
- Qiu, Y.L.; Zheng, H.; Devos, A.; Selby, H.; Gevaert, O. A Meta-Learning Approach for Genomic Survival Analysis. Nat. Commun. 2020, 11, 6350. [Google Scholar] [CrossRef]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-Nnet: An Artificial Neural Network Method for Prognosis Prediction of High-Throughput Omics Data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized Treatment Recommender System Using a Cox Proportional Hazards Deep Neural Network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef]
- Huang, Z.; Johnson, T.S.; Han, Z.; Helm, B.; Cao, S.; Zhang, C.; Salama, P.; Rizkalla, M.; Yu, C.Y.; Cheng, J.; et al. Deep Learning-Based Cancer Survival Prognosis from RNA-Seq Data: Approaches and Evaluations. BMC Med. Genom. 2020, 13, 41. [Google Scholar] [CrossRef]
- Van IJzendoorn, D.G.P.; Szuhai, K.; Briaire-de Bruijn, I.H.; Kostine, M.; Kuijjer, M.L.; Bovée, J.V.M.G. Machine Learning Analysis of Gene Expression Data Reveals Novel Diagnostic and Prognostic Biomarkers and Identifies Therapeutic Targets for Soft Tissue Sarcomas. PLoS Comput. Biol. 2019, 15, e1006826. [Google Scholar] [CrossRef]
- Tabl, A.A.; Alkhateeb, A.; ElMaraghy, W.; Rueda, L.; Ngom, A. A Machine Learning Approach for Identifying Gene Biomarkers Guiding the Treatment of Breast Cancer. Front. Genet. 2019, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhu, C.; Liu, Q. Toward in Silico Identification of Tumor Neoantigens in Immunotherapy. Trends Mol. Med. 2019, 25, 980–992. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Storey, J.D. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Cereda, M.; Gambardella, G.; Benedetti, L.; Iannelli, F.; Patel, D.; Basso, G.; Guerra, R.F.; Mourikis, T.P.; Puccio, I.; Sinha, S.; et al. Patients with Genetically Heterogeneous Synchronous Colorectal Cancer Carry Rare Damaging Germline Mutations in Immune-Related Genes. Nat. Commun. 2016, 7, 12072. [Google Scholar] [CrossRef]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A Primer on Deep Learning in Genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep Learning for Computational Biology. Mol. Syst. Biol. 2016, 12. [Google Scholar] [CrossRef]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying Cancer and Normal RNA Sequencing Data from Different Sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef] [PubMed]
- Refine.bio. Available online: https://www.refine.bio (accessed on 15 April 2021).
- Jones, D.T. Setting the Standards for Machine Learning in Biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 659–660. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. arXiv Prepr. 2019, arXiv:1912.01703. [Google Scholar]
- Gulli, A.; Pal, S. Deep Learning with Keras: Implement Neural Networks with Keras on Theano and TensorFlow; Packt Publishing: Birmingham, UK, 2017; ISBN 9781787128422. [Google Scholar]
- Avsec, Ž.; Kreuzhuber, R.; Israeli, J.; Xu, N.; Cheng, J.; Shrikumar, A.; Banerjee, A.; Kim, D.S.; Beier, T.; Urban, L.; et al. The Kipoi Repository Accelerates Community Exchange and Reuse of Predictive Models for Genomics. Nat. Biotechnol. 2019, 37, 592–600. [Google Scholar] [CrossRef] [PubMed]
- García, S.; Ramírez-Gallego, S.; Luengo, J.; Benítez, J.M.; Herrera, F. Big Data Preprocessing: Methods and Prospects. Big Data Anal. 2016, 1, 9. [Google Scholar] [CrossRef]
- Shaham, U.; Stanton, K.P.; Zhao, J.; Li, H.; Raddassi, K.; Montgomery, R.; Kluger, Y. Removal of Batch Effects Using Distribution-Matching Residual Networks. Bioinformatics 2017, 33, 2539–2546. [Google Scholar] [CrossRef]
- Wang, T.; Johnson, T.S.; Shao, W.; Lu, Z.; Helm, B.R.; Zhang, J.; Huang, K. BERMUDA: A Novel Deep Transfer Learning Method for Single-Cell RNA Sequencing Batch Correction Reveals Hidden High-Resolution Cellular Subtypes. Genome Biol. 2019, 20, 165. [Google Scholar] [CrossRef]
- Li, X.; Wang, K.; Lyu, Y.; Pan, H.; Zhang, J.; Stambolian, D.; Susztak, K.; Reilly, M.P.; Hu, G.; Li, M. Deep Learning Enables Accurate Clustering with Batch Effect Removal in Single-Cell RNA-Seq Analysis. Nat. Commun. 2020, 11, 2338. [Google Scholar] [CrossRef]
- Yang, Y.; Li, G.; Qian, H.; Wilhelmsen, K.C.; Shen, Y.; Li, Y. SMNN: Batch Effect Correction for Single-Cell RNA-Seq Data via Supervised Mutual Nearest Neighbor Detection. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Elbashir, M.K.; Ezz, M.; Mohammed, M.; Saloum, S.S. Lightweight Convolutional Neural Network for Breast Cancer Classification Using RNA-Seq Gene Expression Data. IEEE Access 2019, 7, 185338–185348. [Google Scholar] [CrossRef]
- López-García, G.; Jerez, J.M.; Franco, L.; Veredas, F.J. Transfer Learning with Convolutional Neural Networks for Cancer Survival Prediction Using Gene-Expression Data. PLoS ONE 2020, 15, e0230536. [Google Scholar] [CrossRef]
- Ding, J.; Condon, A.; Shah, S.P. Interpretable Dimensionality Reduction of Single Cell Transcriptome Data with Deep Generative Models. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Park, S.; Shin, B.; Shim, W.S.; Choi, Y.; Kang, K.; Kang, K. Wx: A Neural Network-Based Feature Selection Algorithm for Transcriptomic Data. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Liu, S.; Xu, C.; Zhang, Y.; Liu, J.; Yu, B.; Liu, X.; Dehmer, M. Feature Selection of Gene Expression Data for Cancer Classification Using Double RBF-Kernels. BMC Bioinform. 2018, 19, 396. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA Interference Reveals That Oncogenic KRAS-Driven Cancers Require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Lauria, A.; Peirone, S.; Giudice, M.D.; Priante, F.; Rajan, P.; Caselle, M.; Oliviero, S.; Cereda, M. Identification of Altered Biological Processes in Heterogeneous RNA-Sequencing Data by Discretization of Expression Profiles. Nucleic Acids Res. 2020, 48, 1730–1747. [Google Scholar] [CrossRef]
- Jung, S.; Bi, Y.; Davuluri, R.V. Evaluation of Data Discretization Methods to Derive Platform Independent Isoform Expression Signatures for Multi-Class Tumor Subtyping. BMC Genom. 2015, 16 (Suppl. 11), S3. [Google Scholar] [CrossRef] [PubMed]
- Talwar, D.; Mongia, A.; Sengupta, D.; Majumdar, A. AutoImpute: Autoencoder Based Imputation of Single-Cell RNA-Seq Data. Sci. Rep. 2018, 8, 16329. [Google Scholar] [CrossRef] [PubMed]
- Arisdakessian, C.; Poirion, O.; Yunits, B.; Zhu, X.; Garmire, L.X. DeepImpute: An Accurate, Fast, and Scalable Deep Neural Network Method to Impute Single-Cell RNA-Seq Data. Genome Biol. 2019, 20, 211. [Google Scholar] [CrossRef] [PubMed]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-Cell RNA-Seq Denoising Using a Deep Count Autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Armasu, S.M.; Kalli, K.R.; Maurer, M.J.; Heinzen, E.P.; Keeney, G.L.; Cliby, W.A.; Oberg, A.L.; Kaufmann, S.H.; Goode, E.L. Pooled Clustering of High-Grade Serous Ovarian Cancer Gene Expression Leads to Novel Consensus Subtypes Associated with Survival and Surgical Outcomes. Clin. Cancer Res. 2017, 23, 4077–4085. [Google Scholar] [CrossRef]
- Alcaraz, N.; List, M.; Batra, R.; Vandin, F.; Ditzel, H.J.; Baumbach, J. De Novo Pathway-Based Biomarker Identification. Nucleic Acids Res. 2017, 45, e151. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, R.; Liang, F.; Zhang, L.; Liang, X. Identification of Metabolism-Associated Prostate Cancer Subtypes and Construction of a Prognostic Risk Model. Front. Oncol. 2020, 10, 598801. [Google Scholar] [CrossRef]
- Paquet, E.R.; Hallett, M.T. Absolute Assignment of Breast Cancer Intrinsic Molecular Subtype. J. Natl. Cancer Inst. 2015, 107, 357. [Google Scholar] [CrossRef]
- Chen, R.; Yang, L.; Goodison, S.; Sun, Y. Deep-Learning Approach to Identifying Cancer Subtypes Using High-Dimensional Genomic Data. Bioinformatics 2020, 36, 1476–1483. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, Z.; Namburi, S.; Pattison, A.; Posner, A.; Balachander, S.; Paisie, C.A.; Reddi, H.V.; Rueter, J.; Gill, A.J.; et al. CUP-AI-Dx: A Tool for Inferring Cancer Tissue of Origin and Molecular Subtype Using RNA Gene-Expression Data and Artificial Intelligence. EBioMedicine 2020, 61, 103030. [Google Scholar] [CrossRef]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.-J.; Shah, P.; et al. A Single-Cell Landscape of High-Grade Serous Ovarian Cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-Based Classification of Glioblastoma Uncovers a Mitochondrial Subtype with Therapeutic Vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, B.; Minn, A.; Zhang, N.R. DENDRO: Genetic Heterogeneity Profiling and Subclone Detection by Single-Cell RNA Sequencing. Genome Biol. 2020, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Nguyen, N.D.; Talos, F.; Wang, D. ECMarker: Interpretable Machine Learning Model Identifies Gene Expression Biomarkers Predicting Clinical Outcomes and Reveals Molecular Mechanisms of Human Disease in Early Stages. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Cheng, Q.; Li, J.; Fan, F.; Cao, H.; Dai, Z.-Y.; Wang, Z.-Y.; Feng, S.-S. Identification and Analysis of Glioblastoma Biomarkers Based on Single Cell Sequencing. Front. Bioeng. Biotechnol. 2020, 8, 167. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, M.; Wang, Q.; Zhang, J.; Zhou, T.; Sun, X. Single-Cell Transcriptome-Based Multilayer Network Biomarker for Predicting Prognosis and Therapeutic Response of Gliomas. Brief. Bioinform. 2020, 21, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous Enumeration of Cancer and Immune Cell Types from Bulk Tumor Gene Expression Data. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tan, Y.; Sun, F.; Hou, L.; Zhang, C.; Ge, T.; Yu, H.; Wu, C.; Zhu, Y.; Duan, L.; et al. Single-Cell Transcriptome and Antigen-Immunoglobin Analysis Reveals the Diversity of B Cells in Non-Small Cell Lung Cancer. Genome Biol. 2020, 21, 152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tian, X.; Ji, L.; Zhang, X.; Cao, Y.; Shen, C.; Hu, Y.; Wong, J.W.H.; Fang, J.-Y.; Hong, J.; et al. A Tumor Microenvironment-Specific Gene Expression Signature Predicts Chemotherapy Resistance in Colorectal Cancer Patients. NPJ Precis Oncol. 2021, 5, 7. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate Genome-Level Prediction of Neoantigens by Harnessing Sequence and Amino Acid Immunogenicity Information. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA Class II Antigen Presentation through Integrated Deep Learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A Benchmark of Batch-Effect Correction Methods for Single-Cell RNA Sequencing Data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. NAR Genom. Bioinform. 2020, 2. [Google Scholar] [CrossRef]
- Velliangiri, S.; Alagumuthukrishnan, S.; Thankumar Joseph, S.I. A Review of Dimensionality Reduction Techniques for Efficient Computation. Procedia Comput. Sci. 2019, 165, 104–111. [Google Scholar] [CrossRef]
- Abid, A.; Zhang, M.J.; Bagaria, V.K.; Zou, J. Exploring Patterns Enriched in a Dataset with Contrastive Principal Component Analysis. Nat. Commun. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Raj-Kumar, P.K.; Liu, J.; Hooke, J.A.; Kovatich, A.J.; Kvecher, L.; Shriver, C.D.; Hu, H. PCA-PAM50 Improves Consistency between Breast Cancer Intrinsic and Clinical Subtyping Reclassifying a Subset of Luminal A Tumors as Luminal B. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Taguchi, Y.-H.; Iwadate, M.; Umeyama, H. SFRP1 Is a Possible Candidate for Epigenetic Therapy in Non-Small Cell Lung Cancer. BMC Med. Genom. 2016, 9 (Suppl. 1), 28. [Google Scholar] [CrossRef]
- Chen, D.-T.; Hsu, Y.-L.; Fulp, W.J.; Coppola, D.; Haura, E.B.; Yeatman, T.J.; Cress, W.D. Prognostic and Predictive Value of a Malignancy-Risk Gene Signature in Early-Stage Non-Small Cell Lung Cancer. J. Natl. Cancer Inst. 2011, 103, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Walsh, J.R.; Long, J.; Davis, C.B.; Henstock, P.; Hodge, M.R.; Maciejewski, M.; Mu, X.J.; Ra, S.; Zhao, S.; et al. Standard Machine Learning Approaches Outperform Deep Representation Learning on Phenotype Prediction from Transcriptomics Data. BMC Bioinform. 2020, 21, 119. [Google Scholar] [CrossRef]
- Van der Maaten, L. Visualizing Data Using T-SNE. Available online: https://www.jmlr.org/papers/volume9/vandermaaten08a/vandermaaten08a.pdf?fbclid=IwAR0Bgg1eA5TFmqOZeCQXsIoL6PKrVXUFaskUKtg6yBhVXAFFvZA6yQiYx-M (accessed on 6 March 2021).
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv Prepr. 2018, arXiv:1802.03426. [Google Scholar]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality Reduction for Visualizing Single-Cell Data Using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Kobak, D.; Linderman, G.C. Initialization Is Critical for Preserving Global Data Structure in Both T-SNE and UMAP. Nat. Biotechnol. 2021, 39, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; Hsiao, C.J.; Stephens, M. Visualizing the Structure of RNA-Seq Expression Data Using Grade of Membership Models. PLoS Genet. 2017, 13, e1006599. [Google Scholar] [CrossRef]
- Mandel, J.; Avula, R.; Prochownik, E.V. Sequential Analysis of Transcript Expression Patterns Improves Survival Prediction in Multiple Cancers. BMC Cancer 2020, 20, 297. [Google Scholar] [CrossRef]
- Schmauch, B.; Romagnoni, A.; Pronier, E.; Saillard, C.; Maillé, P.; Calderaro, J.; Kamoun, A.; Sefta, M.; Toldo, S.; Zaslavskiy, M.; et al. A Deep Learning Model to Predict RNA-Seq Expression of Tumours from Whole Slide Images. Nat. Commun. 2020, 11, 3877. [Google Scholar] [CrossRef]
- Chen, Z.; Pang, M.; Zhao, Z.; Li, S.; Miao, R.; Zhang, Y.; Feng, X.; Feng, X.; Zhang, Y.; Duan, M.; et al. Feature Selection May Improve Deep Neural Networks for the Bioinformatics Problems. Bioinformatics 2019, 36, 1542–1552. [Google Scholar] [CrossRef]
- Liang, S.; Ma, A.; Yang, S.; Wang, Y.; Ma, Q. A Review of Matched-Pairs Feature Selection Methods for Gene Expression Data Analysis. Comput. Struct. Biotechnol. J. 2018, 16, 88–97. [Google Scholar] [CrossRef]
- Khaire, U.M.; Dhanalakshmi, R. Stability of Feature Selection Algorithm: A Review. J. King Saud Univ. Comput. Inf. Sci. 2019. [Google Scholar] [CrossRef]
- Bishop, C.M. Neural Networks for Pattern Recognition; Oxford University Press: New York, NY, USA, 1995; ISBN 9780198538646. [Google Scholar]
- Bengio, Y.; Simard, P.; Frasconi, P. Learning Long-Term Dependencies with Gradient Descent Is Difficult. IEEE Trans. Neural Netw. 1994, 5, 157–166. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and Analytical Challenges in Single-Cell Transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Witten, I.H.; Frank, E.; Hall, M.A. Data Mining: Practical Machine Learning Tools and Techniques; Elsevier Science: Amsterdam, The Netherlands, 2011; ISBN 9780123748560. [Google Scholar]
- Ramírez-Gallego, S.; García, S.; Mouriño-Talín, H.; Martínez-Rego, D.; Bolón-Canedo, V.; Alonso-Betanzos, A.; Benítez, J.M.; Herrera, F. Data Discretization: Taxonomy and Big Data Challenge. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2016, 6, 5–21. [Google Scholar] [CrossRef]
- Gallo, C.A.; Cecchini, R.L.; Carballido, J.A.; Micheletto, S.; Ponzoni, I. Discretization of Gene Expression Data Revised. Brief. Bioinform. 2015, 17, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Angerer, P.; Simon, L.; Tritschler, S.; Wolf, F.A.; Fischer, D.; Theis, F.J. Single Cells Make Big Data: New Challenges and Opportunities in Transcriptomics. Curr. Opin. Syst. Biol. 2017, 4, 85–91. [Google Scholar] [CrossRef]
- Chai, X.; Gu, H.; Li, F.; Duan, H.; Hu, X.; Lin, K. Deep Learning for Irregularly and Regularly Missing Data Reconstruction. Sci. Rep. 2020, 10, 3302. [Google Scholar] [CrossRef] [PubMed]
- Jaskowiak, P.A.; Costa, I.G.; Campello, R.J.G.B. Clustering of RNA-Seq Samples: Comparison Study on Cancer Data. Methods 2018, 132, 42–49. [Google Scholar] [CrossRef]
- Liu, L.; Tang, L.; Dong, W.; Yao, S.; Zhou, W. An Overview of Topic Modeling and Its Current Applications in Bioinformatics. Springerplus 2016, 5, 1608. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, M.; Zhu, H.; Xu, J. A 15-Gene Signature for Prediction of Colon Cancer Recurrence and Prognosis Based on SVM. Gene 2017, 604, 33–40. [Google Scholar] [CrossRef]
- Mourikis, T.P.; Benedetti, L.; Foxall, E.; Temelkovski, D.; Nulsen, J.; Perner, J.; Cereda, M.; Lagergren, J.; Howell, M.; Yau, C.; et al. Patient-Specific Cancer Genes Contribute to Recurrently Perturbed Pathways and Establish Therapeutic Vulnerabilities in Esophageal Adenocarcinoma. Nat. Commun. 2019, 10, 3101. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, B. Semi-Supervised Learning Improves Gene Expression-Based Prediction of Cancer Recurrence. Bioinformatics 2011, 27, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Mohaiminul Islam, M.; Huang, S.; Ajwad, R.; Chi, C.; Wang, Y.; Hu, P. An Integrative Deep Learning Framework for Classifying Molecular Subtypes of Breast Cancer. Comput. Struct. Biotechnol. J. 2020, 18, 2185–2199. [Google Scholar]
- Kalia, M. Biomarkers for Personalized Oncology: Recent Advances and Future Challenges. Metabolism 2015, 64, S16–S21. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; ISBN 9781475732948. [Google Scholar]
- Ishwaran, H.; Kogalur, U.B.; Blackstone, E.H.; Lauer, M.S. Random Survival Forests. Ann. Appl. Stat. 2008, 2, 841–860. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; The MIT Press: Cambridge, MA, USA, 2016; ISBN 9780262035613. [Google Scholar]
- Yousefi, S.; Amrollahi, F.; Amgad, M.; Dong, C.; Lewis, J.E.; Song, C.; Gutman, D.A.; Halani, S.H.; Velazquez Vega, J.E.; Brat, D.J.; et al. Predicting Clinical Outcomes from Large Scale Cancer Genomic Profiles with Deep Survival Models. Sci. Rep. 2017, 7, 11707. [Google Scholar] [CrossRef] [PubMed]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA Splicing in Cancer Immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.-V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; Improved Prediction of Binding to MHC Class I Molecules Integrating Information from Multiple Receptor and Peptide Length Datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef]
- Smart, A.C.; Margolis, C.A.; Pimentel, H.; He, M.X.; Miao, D.; Adeegbe, D.; Fugmann, T.; Wong, K.-K.; Van Allen, E.M. Intron Retention Is a Source of Neoepitopes in Cancer. Nat. Biotechnol. 2018, 36, 1056–1058. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best Practices for Bioinformatic Characterization of Neoantigens for Clinical Utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Curse of Dimensionality. Encycl. Database Syst. 2009, 545–546. [Google Scholar]
- Altman, N.; Krzywinski, M. The Curse(s) of Dimensionality. Nat. Methods 2018, 15, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Jackson, S.A. Machine Learning and Complex Biological Data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Neumann, A.; Timmermann, B.; Meinke, S.; Heyd, F. Differential Interleukin-2 Transcription Kinetics Render Mouse but Not Human T Cells Vulnerable to Splicing Inhibition Early after Activation. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Artemaki, P.I.; Letsos, P.A.; Zoupa, I.C.; Katsaraki, K.; Karousi, P.; Papageorgiou, S.G.; Pappa, V.; Scorilas, A.; Kontos, C.K. The Multifaceted Role and Utility of MicroRNAs in Indolent B-Cell Non-Hodgkin Lymphomas. Biomedicines 2021, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.; Chen, Y.; Jones, A.; Shibue, T.; Hahn, W.C.; Boehm, J.S.; Vazquez, F.; Tsherniak, A.; McFarland, J.M. Global Computational Alignment of Tumor and Cell Line Transcriptional Profiles. Nat. Commun. 2021, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A First-Generation Pediatric Cancer Dependency Map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Repository | URL | Bulk | Single-Cell |
|---|---|---|---|
| GDC | portal.gdc.cancer.gov | 27,894 | 18 |
| ENCODE | www.encodeproject.org | 2323 | 7 |
| GEO | www.ncbi.nlm.nih.gov/geo | 30,510 | 2346 |
| SRA | www.ncbi.nlm.nih.gov/sra | 1874 | 6428 |
| St. Jude | www.stjude.cloud | 3215 | - |
| ICGC | dcc.icgc.org | 12,840 | - |
| GTEx | www.gtexportal.org/home | 17,382 | - |
| DepMap | depmap.org/portal | 1376 | - |
| Human Cell Atlas | data.humancellatlas.org | - | 289 |
| Single Cell Portal | singlecell.broadinstitute.org | - | 83 |
| Section | Method | RNA-Seq Experiment | Authors |
|---|---|---|---|
| Batch-correction of technical heterogeneity | Residual neural network | single-cell | Shaham et al., 2017 [47] |
| autoencoder | single-cell | T. Wang et al., 2019 [48] | |
| Autoencoder and iterative clustering | single-cell | Li et al., 2020 [49] | |
| Supervised mutual nearest neighbor | single-cell | Yang et al., 2020 [50] | |
| Feature extraction | Convolutional neural network | bulk | Elbashir et al., 2019 [51] |
| Convolutional neural network | bulk | López-García et al., 2020 [52] | |
| Deep generative models | single-cell | Ding et al., 2018 [53] | |
| Wx, neural network | bulk | Park et al., 2019 [54] | |
| Double Radial Basis Function Kernels | bulk | Liu et al., 2018 [55] | |
| Data distribution transformation | Rank-based normalization | bulk | Barbie et al., 2009 [56] |
| GSECA, Gene Set Enrichment Class Analysis | bulk | Lauria et al., 2020 [57] | |
| Equal-width, equal-frequency binning, k-means clustering | bulk | Jung et al., 2015 [58] | |
| Data reconstruction: the sparsity issue | AutoImpute, autoencoder | single-cell | Talwar et al., 2018 [59] |
| DeepImpute, autoencoder | single-cell | Arisdakessian et al., 2019 [60] | |
| DCA, autoencoder | single-cell | Eraslan et al., 2019 [61] | |
| Assessing inter-tumor heterogeneity: classification of cancer subtypes | Non-negative matrix factorization | bulk | Wang et al., 2017 [62] |
| Topic modeling | bulk | Valle et al., 2020 [20] | |
| Random forest | bulk | Alcaraz et al., 2017 [63] | |
| Partition around medoids | bulk | Zhang et al., 2020 [64] | |
| Naïve Bayes classifier | bulk | Paquet et al., 2015 [65] | |
| Multiclass logistic regression | bulk | Cascianelli et al., 2020 [17] | |
| DeepType, neural network | bulk | Chen et al., 2020 [66] | |
| CUP-AI-Dx, convolutional neural network | bulk | Zhao et al., 2020 [67] | |
| DeepCC, neural network | bulk | Gao et al., 2019 [18] | |
| Defining cell types and clones | Density clustering | single-cell | Izar et al., 2020 [68] |
| Graph-based clustering | single-cell | Chen et al., 2020 [21], Zhou et al., 2020 [22] | |
| Consensus clustering | single-cell | Garofano et al., 2021 [69] | |
| DENDRO, kernel-based clustering | single-cell | Zhou et al., 2020 [70] | |
| Biomarker identification | Interaction network and ridge regression | bulk | Kong et al., 2020 [26] |
| SIMMS, Interaction network and Cox Proportional Hazards | bulk | Haider et al., 2018 [27] | |
| ECMarker, Boltzman machines | bulk | Jin et al., 2020 [71] | |
| Integration of ML techniques | bulk | van IJzendoorn et al., 2019 [33] | |
| DRjCC, non-negative matrix factorization | single-cell | Wu et al., 2020 [28] | |
| maximum relevance minimum redundancy, Support vector machine | single-cell | Cheng et al., 2020 [72] | |
| Diffusion map, shared nearest-neighbor clustering and Cox Proportional Hazards | single-cell | Zhang et al., 2020 [73] | |
| Prediction of patient survival | Cox-nnet, neural network and Cox Proportional Hazards | bulk | Ching et al., 2018 [30] |
| DeepSurv, neural network and Cox Proportional Hazards | bulk | Katzman et al., 2018 [31] | |
| AECOX, autoencoder and Cox Proportional Hazards, | bulk | Huang et al., 2020 [32] | |
| Neural network and Cox Proportional Hazards | bulk | Qiu et al., 2020 [29] | |
| Assessment of tumor microenvironment | CIBERSORTx, support vector regression | single-cell/bulk | Newman et al., 2015 [24] |
| EPIC, least square regression | single-cell/bulk | Racle et al., 2017 [74] | |
| xCell, non-linear regression | bulk | Aran et al., 2017 [25] | |
| Graph-based clustering | single-cell | Chen et al., 2020 [75] | |
| K-means clustering | single-cell/bulk | Zhu et al., 2021 [76] | |
| Identification of neoepitopes | Neopepsee, Naïve Bayes, random forest, support vector machine | bulk | Kim et al., 2018 [77] |
| MARIA, multimodal recurrent neural network | bulk | Chen et al., 2019 [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Giudice, M.; Peirone, S.; Perrone, S.; Priante, F.; Varese, F.; Tirtei, E.; Fagioli, F.; Cereda, M. Artificial Intelligence in Bulk and Single-Cell RNA-Sequencing Data to Foster Precision Oncology. Int. J. Mol. Sci. 2021, 22, 4563. https://doi.org/10.3390/ijms22094563
Del Giudice M, Peirone S, Perrone S, Priante F, Varese F, Tirtei E, Fagioli F, Cereda M. Artificial Intelligence in Bulk and Single-Cell RNA-Sequencing Data to Foster Precision Oncology. International Journal of Molecular Sciences. 2021; 22(9):4563. https://doi.org/10.3390/ijms22094563
Chicago/Turabian StyleDel Giudice, Marco, Serena Peirone, Sarah Perrone, Francesca Priante, Fabiola Varese, Elisa Tirtei, Franca Fagioli, and Matteo Cereda. 2021. "Artificial Intelligence in Bulk and Single-Cell RNA-Sequencing Data to Foster Precision Oncology" International Journal of Molecular Sciences 22, no. 9: 4563. https://doi.org/10.3390/ijms22094563
APA StyleDel Giudice, M., Peirone, S., Perrone, S., Priante, F., Varese, F., Tirtei, E., Fagioli, F., & Cereda, M. (2021). Artificial Intelligence in Bulk and Single-Cell RNA-Sequencing Data to Foster Precision Oncology. International Journal of Molecular Sciences, 22(9), 4563. https://doi.org/10.3390/ijms22094563