Abstract
Artificial intelligence, or the discipline of developing computational algorithms able to perform tasks that requires human intelligence, offers the opportunity to improve our idea and delivery of precision medicine. Here, we provide an overview of artificial intelligence approaches for the analysis of large-scale RNA-sequencing datasets in cancer. We present the major solutions to disentangle inter- and intra-tumor heterogeneity of transcriptome profiles for an effective improvement of patient management. We outline the contributions of learning algorithms to the needs of cancer genomics, from identifying rare cancer subtypes to personalizing therapeutic treatments.
1. Introduction
Artificial intelligence (AI) is becoming a fundamental asset for healthcare and life science research. Despite being in its infancy, research activities employing AI are changing our understanding and vision of science. The European Commission has recently estimated that 13% of global venture capital investments (i.e., ~5 billion of Euros) are for start-ups dedicated to AI application in medicine [1]. This commitment reflects the interest in the potential of AI to improve healthcare. Precision medicine is a new approach to health. In the last decade, the generation of Big Data through genome sequencing (i.e., genomic Big Data), the collection of clinical data, and the growth of bioinformatics has made it possible to identify the genetic causes responsible for onset and progression of diseases and to support the clinical management of patients. Despite the high expectations, personalized therapeutic treatments still remain limited. A breakdown is the lack of AI infrastructure and models capable of supporting the constant generation of genomic Big Data [2]. Consequently, the challenge remains how to interpret the variety of information contained in these data [3].
The need for AI models is even more evident in complex diseases such as cancer. The heterogeneity that characterizes Big Data is amplified in cancer, where diversity not only manifests itself across individuals (i.e., inter-tumor) but also within each tumor (i.e., intra-tumor) [4]. So far, cancer sequencing projects have made available genomic profiles for thousands of biological samples, corresponding to petabytes of genetic information [5]. With the introduction of single-cell technologies, the complexity of genomic information has grown rapidly. This heterogeneity represents the major hurdle to achieve effective precision oncology. Therefore, AI is the pivotal tool to exploit the information available in genomic Big Data and ultimately “deliver” a medicine of precision. The COVID-19 pandemic has opened up new possibilities for AI development. The pandemic has increased the use of AI in biomedical research: from remotely monitoring patients, to predicting the spread of the SARS-CoV-2 coronavirus or in developing new drugs [6,7]. The pandemic has also brought about new clinical practices, primarily the use of mRNA vaccines. This technological leap forward gives the possibility of accelerating the delivery of similar therapies to cancer [8].
Transcriptomics generally refers to the high-throughput profiling of all RNA species produced by cells. Among genomic Big Data, transcriptomics has seen an explosive growth in recent years [9]. RNA sequencing (RNA-seq) profiles dynamic biological processes that are active in a population of cells or in single cells. Assessing the complexity of these profiles could inform the discovery of new biomarkers and therapeutic targets. Since RNA-seq screenings are becoming part of precision medicine trials [10,11], AI mining of these data is thus required to determine novel clinical targets.
In this paper, we provide an overview of AI approaches applied to high-volume bulk and single-cell RNA-seq in cancer genomics and precision oncology. We do not intend to provide a comprehensive characterization of all published AI methods and their technical details. By contrast, we illustrate the major AI solutions to disentangle the heterogeneity of cancer transcriptomes for an effective improvement of patient management. We explain distinct strategies to face the “heterogeneity challenge”. We then outline some of the major contributions of applying AI to the needs of cancer genomics, from identifying rare cancer subtypes to personalizing treatment for individuals.
2. AI in the Era of Transcriptomic Big Data
From the first drafts of the human genome [12], 20 years ago, the number of scientific works employing sequencing data has exponentially increased (Figure 1). RNA-seq has become a widespread tool to profile cancer transcriptome at both population and single-cell level.
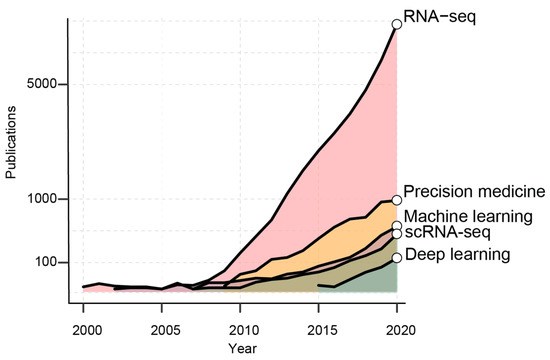
Figure 1.
The graph shows the number of PubMed publications per years containing the reported keywords.
So far, genomic screenings have made available more than 106,585 RNA-seq samples (Table 1) and this number is constantly increasing.

Table 1.
The table reports the number of publicly available bulk and single-cell RNA-seq experiments. Data stored in reported repository are frozen at 15 March 2021.
The availability of these data seizes the opportunity to boost the development of novel diagnostic tools and targeted treatments. Indeed, the implementation of AI models has increased in the last 10 years, as machine-learning [13] (ML) and, recently, as deep-learning [14] methods (DL, Figure 1). These learning methods effectively leverage the variability of Big Data to achieve consistent predictions without the need of modeling the system of interest [15]. In the flavor of supervised, semi-supervised and unsupervised, AI algorithms can be employed to capture dependencies, make predictions and recognize patterns in heterogeneous datasets [13]. AI approaches are commonly used to solve regression, classification, dimensionality reduction and clustering tasks. Being part of AI, ML and DL aim at performing tasks that normally require human intelligence. ML and DL accomplish similar tasks with distinct mathematical approaches. While ML algorithms still need human guidance to improve their predictions, DL methods can autonomously determine the accuracy of a prediction. Overall, DL is part of ML where algorithms are generally based on artificial neural networks (NNs), the closest representation of the human brain [16] (Figure 2).
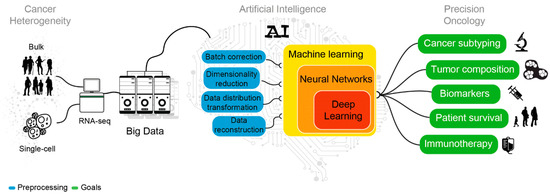
Figure 2.
Sketch representing the analyses needed to decipher cancer heterogeneity and achieve an effective precision oncology.
In cancer transcriptomics, ML and DL models have been applied to classify different cancer subtypes and cell populations [17,18,19,20], characterize tumor immune microenvironment [21,22,23,24,25], discover new prognostic biomarkers [26,27,28], assess and predict disease recurrence and patient survival [29,30,31,32], identify new putative actionable vulnerabilities [33,34], and predict tumor antigen immunogenicity [35] (Figure 2).
Disentangling Big Data heterogeneity is the major challenge that researchers have to face to gain novel scientific insights. When learning from real transcriptomic data, the heterogeneity increases due to the variable expression of genes across samples driven by genetic, environmental, demographic, and technical factors [36]. In cancer, the complexity is additionally hampered by the intra- and inter-tumoral heterogeneity of samples [37]. Nevertheless, the constant sequencing of transcriptomes, and thus the increasing volume of data, represents on its own a solid ground for the application of learning approaches to disentangle the noise from the true biological signal.
However, for an effective application of AI algorithms to cancer transcriptomics, the availability of highly-curated datasets is fundamental [38]. Defining an appropriate training dataset with well-defined features is the first important step to ensure better performance of AI models, particularly when integrating data from different sources [38,39]. In this view, data repositories (e.g., refine.bio, RNAseqDB) have been developed to uniformly process and normalize cancer transcriptomes from publicly available sources [40,41]. The use of harmonized and standardized data becomes crucial for the successful translation of AI predictions to the clinical practice.
Finally, to boost the application of AI in precision oncology, it is of primary importance to provide the scientific community with the code and data behind the AI model. This practice is fundamental to ensure the reproducibility and transparency of results [42]. Nowadays, researchers have the opportunity to favor method shareability by implementing them in popular machine learning frameworks, such as PyTorch [43] and Keras [44], and uploading the trained models on dedicated repositories like Kipoi [45].
3. Managing the Heterogeneity of Cancer Transcriptomes
Heterogeneity of cancer transcriptomes can arise from both technical and biological confounders [36]. Several strategies have been developed to increase signal-to-noise ratio in large-scale RNA-seq datasets. In this view, a crucial step for the effectiveness of AI algorithms is data preprocessing [46]. To remove the effect of confounders that can lead to false data dependencies, techniques such as batch-correction, dimensionality reduction, data discretization and feature selection are normally employed. These strategies can be used independently or in combination, either as a core or a result of AI. Below, we explore the tight link between these approaches and learning strategies. All methods that we report are listed in Table 2 and summarized in Figure 3.
Table 2.
The table reports all learning approaches reported in the main text with respect to each section.
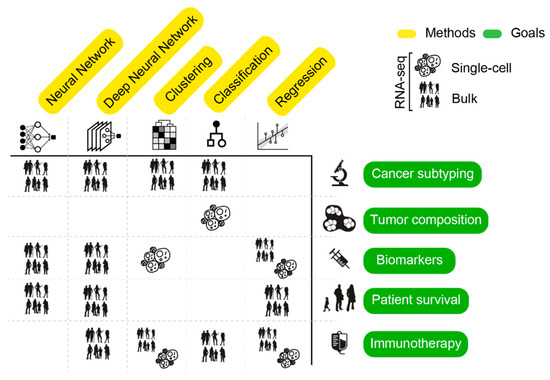
Figure 3.
Graphical summary of AI approaches (columns) applied to solve tasks (rows) presented in this review. Cells show the RNA-seq data type used for the analysis. The “immunotherapy” task includes assessment of tumor microenvironment and identification of neoepitopes.
3.1. Batch-Correction of Technical Heterogeneity
Technical heterogeneity rises during the experimental generation of sequencing data. In particular, cancer transcriptomes can be profiled (i) from distinct sample types; (ii) using different protocols and platforms; and (iii) processed in unconnected laboratories by specific users in separate times. This “batch-specific” heterogeneity confounds the real biological signal of large-scale datasets [79]. Therefore, an effective removal of batch-effects is an essential step during data integration. Conventionally, batch-effects in bulk RNA-seq are resolved using ML regression models [80]. With the advent of single-cell RNA-seq, different non-linear, transfer-learning, supervised and unsupervised DL approaches have been successfully proposed. For instance, NNs trained to minimize discrepancies between distributions of replicates have been shown to attenuate technical confounders [47]. Autoencoders, or unsupervised NNs, that gradually remove batch-effect over iterations have shown to amplify biological signals by transferring information across batches [48]. Similarly, unsupervised deep embedding NNs that simultaneously learn gene expression representations and cluster assignments demonstrated a great removal of batch-effects while preserving biological heterogeneity [49]. Supervised mutual nearest neighbor detection within cell types revealed an improved clustering of cell types across batches [50]. Overall, DL approaches showed the best reduction of batch-effects can be achieved while learning from clustering data across iterations.
3.2. Dimensionality Reduction Approaches
Heterogeneity emerges from the high dimensionality of transcriptomic datasets, which profile thousands of RNA isoforms. These profiles result in lists of fixed-length vectors of real values, namely features, which are highly variable in a specific range. The extremely high number of heterogeneous features prevents direct identification of biological similarities across samples under a phenotype of interest. In this view, dimensionality reduction is a useful pre-processing approach to remove confounders, speed up learning methods and improve their accuracy in detecting similarities [81]. These techniques perform dimensionality reduction by either extracting novel features or selecting the best informative features from the original dataset.
3.2.1. Feature Extraction
Principal Component Analysis (PCA) is conventionally used for dimensionality reduction and exploratory analyses of transcriptomic profiles [82]. PCA aims at extracting a limited number of new features that maximize the variability present in the original data through a linear approach. In precision oncology, different PCA-based approaches showed to enhance the consistency of cancer subtyping [83], the discovery of putative novel therapeutic targets [84], and the identification of prognostic gene signatures [85].
Being a linear approach, the accuracy of PCA is limited when dealing with large-scale RNA-seq datasets [86]. To overcome this issue, non-linear methods, such as T-distributed Stochastic Neighbor Embedding (t-SNE) and Uniform Manifold Approximation and Projection (UMAP), have been recently employed to capture variability of cancer transcriptomes. t-SNE and UMAP aim at deconvoluting relationships between neighbors in high-volume datasets [87,88], with different implementations [89,90]. These unsupervised non-linear dimensionality reduction approaches showed to be effective in separating cell types in scRNA-seq datasets. Recently, these methods have also been shown to capture the heterogeneity of large-scale bulk RNA-seq [91]. Applied to thousands of cancer transcriptomes, t-SNE revealed small gene signatures correlating with long-term survival in the majority of tumor types [92].
Feature extraction can also be performed employing DL approaches. For instance, convolutional NNs can automatically reduce data dimensionality and perform classification tasks. Convolutional NN methods have been successfully implemented in digital pathology. Interestingly, these algorithms have been capable of inferring cancer transcriptomic profiles from histological images [93]. Applied to bulk RNA-seq datasets, CNN revealed a high accuracy to classify cancer subtypes [51] and predict cancer progression [52]. Similarly, DL methods have been designed to improve our understanding of single-cell heterogeneity. Deep generative models, which combine probabilistic models and NNs, have recently been shown to enhance dimensionality reduction of scRNA-seq datasets by preserving their global structure, thus improving the interpretation of results. Applied to a large-scale melanoma dataset, this DL method accurately discriminated tumor cells from microenvironment components [53].
3.2.2. Feature Selection
Feature selection (FS) aims at identifying the most important attributes from heterogeneous high-dimensional data [13]. The choice of an appropriate FS method is fundamental for the identification of the real biological information, especially in precision oncology when searching prognostic gene signature, biomarkers and actionable targets. In terms of AI, the use of a list of selected features reduces overfitting effects and increases model stability, and thus prediction accuracy. It has been shown that NN approaches improve their accuracy when coupled with FS methods [94]. Similarly, regression models based on FS resulted in an improved classification of breast cancer subtypes from bulk RNA-seq datasets [17]. A review of the main feature selection approaches for bulk and single cell transcriptomic data has been recently presented [95].
Despite being pivotal for increasing the accuracy of AI predictions, the presence of extensive correlations between variables in large-scale datasets could reduce the stability of selected features [96]. To mitigate this issue, DL-based feature selection algorithms have been introduced. For instance, NNs have been successfully applied to identify small gene signatures as oncogenic biomarkers from a large-scale pan-cancer RNA-seq dataset [54]. Similarly, the use of polynomial and radial kernels that are pivotal for ML algorithms has been shown to achieve higher accuracy than conventional FS approaches in selecting oncogenic gene signatures from bulk RNA-seq data [55].
3.3. Data Distribution Transformation
Since AI methods learn from inputs to predict outputs, the different scale and distribution of features in the training data can impact on model performance, particularly if the algorithm is based on distance measures [97]. For instance, features with a considerable spread of values may result in large error gradients causing NN instability [98]. This holds particularly true for cancer transcriptomes where distinct genes whose expression fluctuates in a small interval can drive the phenotype rather than single genes with large expression spread [99]. Feature centering and scaling is a critical preprocessing step to assure that all variables proportionally contribute to the AI model. These steps are widely used to normalize bulk and single-cell RNA-seq data [100,101]. Feature scaling helps to detect informative gene signature and altered processes from expression data. For instance, rank-based preprocessing normalization of gene expression profiles from bulk RNA-seq experiments has been shown to be effective in determining the real altered pathways in KRAS-driven cancers [56].
Similarly, data discretization is a preprocessing step through which values are divided in a finite number of classes. AI methods, such as classification and clustering, can improve in learning speed and accuracy by discretizing the distribution of numerical input values [102,103]. In this view, we recently demonstrated how discretization of expression profiles boosts the prediction of altered biological processes in large-scale transcriptomic datasets [57]. This preprocessing step allowed us to identify the role of the tumor-suppressor gene, PTEN, in modulating immune-related processes and determine the maximum expression level for which PTEN leads to a worse patient survival. Discretization of isoform-level gene expression profiles has been successfully applied to increase accuracy of glioblastoma subtype classification [58]. Overall, data discretization increases signal-to-noise ratio at the cost of a partial loss of information, which is mitigated by the large quantity of data. Applied to transcriptomic Big Data, this technique offers the chance to extract relevant information while accounting for their intrinsic heterogeneity. However, due to information loss, the choice of an appropriate discretization strategy impacts on the design and performance of the AI model, therefore remaining a non-trivial task [104].
3.4. Data Reconstruction: The Sparsity Issue
Features that have many zero values are commonly referred to as sparse, and their presence can lead to overfitting and reduced performances in AI models. The presence of sparse features characterizes single-cell RNA-seq datasets due to experimental limitations [105,106]. Data reconstruction aims at transforming incomplete input values into a corresponding complete set [107]. Several reconstruction methods have been developed to overcome this technical heterogeneity of single-cell transcriptomic profiles. Most of them are autoencoder-based DL algorithms, which use probabilistic data generative processes to reconstruct the observed profiles from low-dimensional or latent space representations [59,60,61,105]. The use of these data reconstruction tools has been demonstrated to enhance performance in recovering biologically meaningful states, improving data clustering and, consequently, differential expression analysis.
4. AI Mining of Cancer Transcriptomes
Cancer manifests its genetic heterogeneity with the presence of distinct histological subtypes and tumor microenvironment (TME) compositions across tumors and within the same disease. Inter- and intra-tumor heterogeneity have different clinical implications, making their accurate identification pivotal for therapeutic decisions [37]. As previously mentioned, AI models applied to transcriptomic Big Data have increased the accuracy of cancer classification, biomarker discovery, disease recurrence and patient survival forecast, and understanding of immune regulation.
4.1. Assessing Inter-Tumor Heterogeneity: Classification of Cancer Subtypes
One of the most used AI approaches to assess inter-tumor heterogeneity and improve the identification of distinct molecular subtypes using transcriptomic data is unsupervised learning clustering. This technique aims at identifying groups of samples with similar biological features by partitioning data according to similarity measures [108]. Different studies have shown the utility of clustering approaches in detecting molecular features (i.e., gene expression signature) responsible for patient prognosis and management. For example, non-negative matrix factorization clustering of gene expression data has been successfully exploited to improve ovarian cancer subtyping [62]. This approach identified distinct molecular subtypes associated with different patient survival and residual disease. Recently, topic modeling has been proposed to enhance the detection of more subtle inter-tumor heterogeneity. Developed for natural language processing, this probabilistic clustering algorithm aims at discovering the hidden “topics” that reflect the biological heterogeneity and enhancing its comprehensive interpretation [109]. Applied to breast and lung cancer RNA-seq datasets, topic modeling outperformed standard clustering algorithms in identifying subtype-specific molecular features and their corresponding clinical outcomes [20].
The use of prior information can be a useful solution to train AI models more effectively [13]. For this reason, when prior knowledge about subtype features is available, supervised methods can be exploited for a more accurate cancer subtyping [17,18,63,65,66]. For instance, feature selection of differentially expressed genes and scoring of system-level properties (e.g., protein-protein interaction network centrality, gene essentiality, gene evolutionary origin, pathway information) has been employed to select gene signatures to train support vector machine (SVM) predictors of cancer recurrence and prognosis [110,111,112,113]. Similarly, the integration of pathway enrichment scores as input of random forest improved breast cancer classification is relative to single-gene signature-based methods [63]. Recently, partition around medoids clustering of metabolism-related gene set activity scores has been used to identify prostate cancer subtypes associated with patient prognosis and therapy response [64].
However, the heterogeneous composition of large-scale datasets can lead to AI models that are biased toward specific subtypes, thus impacting patient management [65]. A Naïve Bayes classifier based on binary rules that define gene expression dependencies within individual samples has been proposed to improve the identification of patient-specific tumor subtypes [65]. This sample-specific approach revealed an improved identification of breast cancer subtypes, regardless of the biological (i.e., tumor cellularity) and technical (i.e., sequencing technology) heterogeneity in the dataset. Similarly, single-sample feature selection of gene signatures combined with multiclass logistic regression achieved the best performance to classify breast cancer subtypes on 4731 RNA-seq expression profiles [17].
DL approaches have shown advantages over supervised ML methods for their ability of automatically extracting features from input data [114]. An autoencoder-based DL approach combining supervised classification and unsupervised clustering revealed the presence of novel breast and bladder cancer subtypes associated with different prognosis [66]. Convolutional NNs have been employed to infer tumor’s primary tissue of origin of metastasis and to guide management of patients with cancer of unknown primary [67]. Again, integrating biological information (i.e., gene set enrichment analysis) in NNs resulted in an improved classification of individual colorectal and breast cancer subtypes relative to canonical ML approaches [18]. A further advantage of embedding prior biological information in AI models is the easier clinical interpretability of the features defining different subtypes, which can foster the development of novel therapeutic strategies.
4.2. Deciphering Intra-Tumor Heterogeneity
4.2.1. Defining Cell Types and Clones
Transcriptomic profiling of single cells has allowed direct access to intra-tumor heterogeneity through the identification of cell types and clones composing the tumor mass. Learning clustering represents the commonest approach to identify gene signatures representative of specific cell types [21,22,68,69]. As mentioned above, scRNA-seq data are highly heterogeneous, noisy and sparse. This makes clustering analysis particularly challenging. To face this issue, dimensionality reduction approaches (e.g., principal coordinate analysis (PCoA), t-SNE) are generally employed as preprocessing steps of clustering analysis [21,22,68]. These approaches, followed by a manual revision of the identified gene signatures, have been successfully applied to identify cell types associated with different proliferative states and therapy responses in nasopharyngeal tumors and osteosarcomas [21,22]. Similar to cancer subtyping, the integration of prior knowledge in learning algorithms can be useful to improve the interpretation of intra-tumor heterogeneity. Cell clustering using gene set features derived from enrichment analysis improved glioblastoma subtyping, revealing novel metabolism-associated groups associated with distinct prognostic and therapeutic properties [69]. Similarly, clustering including information about somatic alterations has been shown to improve the accuracy of subclone detection and prediction of subclonal neoantigens in breast cancer and melanoma, respectively [70].
4.2.2. Assessment of TME
The heterogeneous composition of the tumor mass increases the complexity of cancer transcriptomes, making the systematic characterization of TME fundamental for the development of personalized therapies. The fine quantification of tumor-infiltrating immune cells can help to guide the selection and understand the effect of immunotherapeutic approaches. For these reasons, ML methods have been proposed to deconvolute cell-type abundance from bulk transcriptomic profiles of mixed populations. Among others, a ML approach based on non-negative matrix factorization has shown to accurately define cell-type-specific expression signatures exploiting tissue heterogeneity in more than 2300 cancer transcriptomes [24]. In absence of physical cell isolation, this method demonstrated to successfully separate the contribution of malignant cells from immune cells and fibroblasts in both head and neck tumors and melanomas. Similarly, least square regressions have been employed to isolate cell-type-specific contributions while accounting for the presence of uncharacterized cells in melanoma samples [74]. The use of gene sets rather than single genes in a curve-fitting approach has been shown to be effective in defining expression profiles of 60 different cell types from 9947 RNA-seq profiles across 37 cancer types [25].
The application of learning approaches to single-cell transcriptomic profiles of physically isolated cells has improved the characterization of TME composition and interactions. PCA dimensionality reduction followed by joint embedding and clustering approach elucidated the cellular composition of osteosarcoma, showing that TME-based chemotherapy may reduce osteoclast differentiation to osteosarcoma [22]. UMAP-based clustering analysis of scRNA-seq data unveiled the existence of novel subtypes of B cells associated with tumor progression [75].
Finally, the combination of bulk and single-cell transcriptome profiling of tumors can improve the characterization of TME and the selection of personalized therapeutic treatments. For instance, supervised clustering approach of 2269 bulk and 10,434 single-cell colorectal transcriptomes identified a TME-associated chemotherapy resistant gene signature enabling tumor subtyping with potential therapeutic response [76].
Overall, deconvolution AI algorithms represent a powerful tool for improving our understanding of TME composition and the delivery of personalized medicine.
4.3. Biomarker Identification
To deliver an effective personalized medicine, the precise identification of patient-specific genetic markers that drive the disease is fundamental. To foster the discovery of novel cancer vulnerabilities, ML and DL have been applied to large-scale transcriptomic profiles and, often, integrated with pharmacogenomics (i.e., drug sensitivity) data. These approaches commonly employ protein-protein interaction network-based feature selection analyses to identify gene signatures associated with drug response, which are then used to train ML classifiers. Recently, an example of these ML frameworks based on ridge regressions has been shown to accurately identify gene signature associated with drug response of colorectal and bladder cancer patients [26]. A similar ML framework employing Cox Proportional Hazards (Cox-PH) regression has been used to determine functional protein-protein interaction subnetworks as prognostic biomarkers in different cancer types [27]. NN classifiers such as restricted Boltzmann machines have been successfully exploited to identify biomarker gene regulatory networks, with available targeting drugs, associated with lung cancer development [71].
The integration of multiple AI techniques can be a handy solution to improve the identification of cancer vulnerabilities. Recently, a combination of three learning algorithms resulted in identifying histone deacetylase inhibitors as potential therapeutic targets for multiple soft tissue sarcomas [33]. In particular, the framework employed (i) NNs to determine gene expression signatures of soft tissue sarcomas relative to healthy tissues, (ii) random forest to identify novel diagnostic markers, and (iii) k-nearest neighbor algorithm to determine prognostic genes.
The analysis of single-cell transcriptomic data can enhance the detection of gene signatures that can discriminate between somatic cells and the other cell types composing the tumor mass. Recently, the combination of dimensionality reduction performed by projected matrix decomposition and clustering through non-negative matrix factorization identified gene signatures of healthy brain cells [28]. Applied to bulk glioblastoma RNA-seq data, these gene signatures successfully predicted patient survival. Similarly, feature selection through maximum relevance minimum redundancy analysis followed by SVM classification revealed glioblastoma-specific biomarkers associated with cancer aggressiveness [72]. As described for bulk RNA-seq, the integration of prior knowledge (e.g., protein-protein, ligand-receptor, regulatory interactions) can also improve biomarker discovery using single-cell transcriptomic profiles. Dimensionality reduction via diffusion map and shared-nearest-neighbor clustering of glioblastoma cells identified potential prognostic biomarkers [73]. Overall, the identification of gene sets as biomarkers rather than single genes provides more comprehensive information on the relevant biological processes responsible for the disease, enlarging the list of novel putative drug targets.
4.4. Prediction of Patient Survival
Stratification of patients into groups with different survival probabilities using prognostic biomarkers is pivotal to prioritize treatments and avoid unnecessary therapies [115]. Traditionally, the effect of gene expression on patients’ survival is measured using linear Cox-PH regression models [116]. However, the high-dimensionality of transcriptomic large-scale datasets impacts on the performance of Cox-PH models leading to overfitting issues [30]. For this reason, ML extensions of the Cox-PH model employing random forest have been developed [117]. Recently, NN approaches have been shown to outperform classical survival methods [29,30,31,32]. These algorithms exploit feature selection through NNs to obtain a subset of surrogate prognostic features. The surrogate features are then used in the Cox-PH model to predict the hazard ratios. Applied to transcriptomic data of kidney cancer, surrogate features defined by NNs have been shown to capture real biological processes (i.e., p 53 signaling pathway) responsible for different prognosis of patients [30]. The integration of prior information about drug treatments in NN prognostic models has been shown to improve therapeutic indications according to the predicted effect of treatment options on individual patients [31]. The use of autoencoders in the feature selection step has also been proposed [32].
To address the scarcity of training samples available for specific cancer types, Cox-PH NN generalizations can be exploited using transfer learning approaches [14]. Transfer learning is a ML technique by which a model trained on one setting is exploited on another related setting [118]. Transfer learning has been employed to assess patient survival in pan-cancer RNA-seq datasets [29]. The model showed a higher prognostic performance than competing methods and exploiting risk score backpropagation [119] allowed to assess the biological pathways that impact on patient’s survival outcome in the tested cancer types.
Together, these results show that NN extensions of Cox-PH modeling improve the identification of prognostic gene signature responsible for cancer progression, and thus of putative novel biomarkers.
4.5. Identification of Neoepitopes
Neoepitopes are tumor-specific peptides that are presented by antigen-presenting cells through the major histocompatibility complex and recognized by the immune system [35]. Several promising immunotherapeutic anticancer approaches (e.g., vaccines, chimeric antigen receptor and T-cell receptor engineered T cells) rely on the identification of suitable target antigens. However, one of the major obstacles for the broader applicability of such therapies is the lack of targetable tumor-specific antigens for many cancer types [120]. Furthermore, in vitro selection of antigens remains an expensive and difficult task. So far, genomic studies analyzed thousands of sequencing data to identify somatic alterations driving tumor progression, but only somatic mutations have been exploited for their potential to generate novel peptides that can stimulate the immune system. Recent studies have shown that the aberrant alternative splicing that characterizes many cancer types has a stronger potential of generating neoepitopes [121].
NNs have been developed to improve peptide-prediction accuracy and MHC-ligand identification [122] from somatic mutations. To date, these approaches have been proved to be also effective in predicting immunogenic peptides derived from somatic splicing defects in melanoma, B-cell lymphoma and leukemia cell lines, even if lacking clinically relevant validations [123]. An ensemble of ML classifiers (i.e., Naïve Bayes, random forest and SVM) has recently been shown to improve immunogenetic predictions of neoantigens and proposed for ranking their potential effectiveness [77]. Nevertheless, the lack of clinically validated neoantigens on large-scale cohorts limits the efficient training of AI algorithms [124]. To solve this issue, a multimodal recurrent NNs approach integrating mass spectrometry data has been proposed [78]. Overall, the identification of neoantigens from large-scale transcriptomic dataset is still in its infancy and presents considerable opportunities for AI improvements.
5. Conclusions
Despite the results achieved so far, the application of AI to cancer transcriptome Big Data for valuable precision oncology is still limited. The complexity of cancer heterogeneity remains the major challenge to disentangle. On the one hand, AI represents the most powerful tool to extract the real biological information from large-scale transcriptomic datasets. As national and international sequencing consortia generate sequencing data, the ability of DL algorithms to capture the hidden relationships responsible for a phenotype without requiring a human supervision will become pivotal for our understanding of diseases and guide personalized therapeutic interventions. On the other hand, AI data mining poses several challenges. Harnessing Big Data carries with it the ‘curse of dimensionality’ phenomenon, or the need of more data when information increases [125,126]. When dimensionality grows, data becomes sparse. Any sample is likely to be more separated from its neighbors at the increase of the space dimensionality. Hence, having data fully representative of the heterogeneity of a phenotype will become more and more complicated as the variables of interest will increase. This holds particularly true for cancer types that are rare and heterogeneous. Dimensionality reduction methods are a solution to mitigate the curse of dimensionality. Similarly, data discretization approaches can help to reduce dimensionality supporting the paradigm of “less is more”. Despite being powerful tools, AI approaches require tailor-made designs to achieve good performances and biologically relevant results. The “black-box” nature of learning algorithms needs to be fully exploited to reach a comprehensive understanding of the cancer phenotype of interest. Improving the interpretability of results of AI models remains an important challenge [127], especially when selecting for therapeutic treatments. However, the integration of prior biological knowledge into the algorithms can guide toward this direction. Combining data from multi omics approaches will provide a deeper understanding of cancer heterogeneity. However, new AI methods will be required to face the resulting curse of dimensionality. Of note, part of cancer transcriptomic data originates from preclinical research employing cell lines and mouse models. Despite the undeniable value of these data, molecular differences between these models and patient tumors call for caution in extending results to the human system [128,129]. Therefore, approaches aiming at delineating the similarities and differences between preclinical and clinical transcriptomes are required for an effective application of AI to improve the patient’s quality of life [130,131].
The demand of AI in precision oncology will go hand in hand with the need of doctors and experts that will be able to translate results into real precision therapeutic decisions and participate actively in the development of learning strategies. In this light, a precision AI-driven oncology will become effectively available on demand.
Author Contributions
Conceptualization, M.D.G., S.P. (Serena Peirone) and M.C.; data curation, M.D.G., S.P. (Serena Peirone), S.P. (Sarah Perrone) and F.P.; writing—original draft preparation, M.D.G., S.P. (Serena Peirone), S.P. (Sarah Perrone), F.P., F.V. and M.C.; writing—review and editing, M.D.G., S.P. (Serena Peirone), E.T., F.F. and M.C.; visualization, M.C.; supervision, M.C.; project administration, M.C.; funding acquisition, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
The research leading to these results has received funding from AIRC under MFAG 2017—ID. 20566 project—P.I. Cereda Matteo. Funding for open access charge: Italian Association for Cancer Research. MC is supported by the “Compagnia di San Paolo” institutional grant.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
A tutorial to implement AI algorithms in the R scripting language for sample classification and gene signature discovery is available at http://www.ceredalab/AI/index.html and at https://github.com/matteocereda/AI, accessed on 24 April 2021.
Acknowledgments
We sincerely thank Giuseppe Basso (1948–2021) for his guidance, teachings, and dedication to foster a research aimed at the needs of patients.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Watch, A.I. Jrc Science for Policy Report. Available online: https://publications.jrc.ec.europa.eu/repository/bitstream/JRC120214/jrc120214_ai_in_medicine_and_healthcare_report-aiwatch_v50.pdf (accessed on 28 February 2021).
- Fröhlich, H.; Balling, R.; Beerenwinkel, N.; Kohlbacher, O.; Kumar, S.; Lengauer, T.; Maathuis, M.H.; Moreau, Y.; Murphy, S.A.; Przytycka, T.M.; et al. From Hype to Reality: Data Science Enabling Personalized Medicine. BMC Med. 2018, 16, 150. [Google Scholar] [CrossRef]
- Big Biological Impacts from Big Data. Available online: https://www.sciencemag.org/features/2014/06/big-biological-impacts-big-data (accessed on 28 February 2021).
- Cereda, M.; Mourikis, T.P.; Ciccarelli, F.D. Genetic Redundancy, Functional Compensation, and Cancer Vulnerability. Trends Cancer Res. 2016, 2, 160–162. [Google Scholar] [CrossRef]
- Marx, V. Biology: The Big Challenges of Big Data. Nature 2013, 498, 255–260. [Google Scholar] [CrossRef]
- McCall, B. COVID-19 and Artificial Intelligence: Protecting Health-Care Workers and Curbing the Spread. Lancet Digit. Health 2020, 2, e166–e167. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial Intelligence in COVID-19 Drug Repurposing. Lancet Digit. Health 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, Y.; Zhang, Z.; Han, L. Maximizing the Utility of Cancer Transcriptomic Data. Trends Cancer Res. 2018, 4, 823–837. [Google Scholar] [CrossRef]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-Generation Personalised Medicine for High-Risk Paediatric Cancer Patients—The INFORM Pilot Study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef]
- Tirtei, E.; Cereda, M.; De Luna, E.; Quarello, P.; Asaftei, S.D.; Fagioli, F. Omic Approaches to Pediatric Bone Sarcomas. Pediatric Blood Cancer 2020, 67, e28072. [Google Scholar] [CrossRef]
- McPherson, J.D.; Marra, M.; Hillier, L.; Waterston, R.H.; Chinwalla, A.; Wallis, J.; Sekhon, M.; Wylie, K.; Mardis, E.R.; Wilson, R.K.; et al. A Physical Map of the Human Genome. Nature 2001, 409, 934–941. [Google Scholar]
- Libbrecht, M.W.; Noble, W.S. Machine Learning Applications in Genetics and Genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep Learning: New Computational Modelling Techniques for Genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.E.; Peña, J.-M.; Jayamohan, J.; Jérusalem, A. Mechanistic Models versus Machine Learning, a Fight Worth Fighting for the Biological Community? Biol. Lett. 2018, 14. [Google Scholar] [CrossRef]
- Crick, F. The Recent Excitement about Neural Networks. Nature 1989, 337, 129–132. [Google Scholar] [CrossRef]
- Cascianelli, S.; Molineris, I.; Isella, C.; Masseroli, M.; Medico, E. Machine Learning for RNA Sequencing-Based Intrinsic Subtyping of Breast Cancer. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A Novel Deep Learning-Based Framework for Cancer Molecular Subtype Classification. Oncogenesis 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, Z.; Yu, X.; Zhang, Z. RNA-Seq-Based Breast Cancer Subtypes Classification Using Machine Learning Approaches. Comput. Intell. Neurosci. 2020, 2020, 4737969. [Google Scholar] [CrossRef] [PubMed]
- Valle, F.; Osella, M.; Caselle, M. A Topic Modeling Analysis of TCGA Breast and Lung Cancer Transcriptomic Data. Cancers 2020, 12, 3799. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-P.; Yin, J.-H.; Li, W.-F.; Li, H.-J.; Chen, D.-P.; Zhang, C.-J.; Lv, J.-W.; Wang, Y.-Q.; Li, X.-M.; Li, J.-Y.; et al. Single-Cell Transcriptomics Reveals Regulators Underlying Immune Cell Diversity and Immune Subtypes Associated with Prognosis in Nasopharyngeal Carcinoma. Cell Res. 2020, 30, 1024–1042. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-Cell RNA Landscape of Intratumoral Heterogeneity and Immunosuppressive Microenvironment in Advanced Osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Bao, X.; Shi, R.; Zhao, T.; Wang, Y.; Anastasov, N.; Rosemann, M.; Fang, W. Integrated Analysis of Single-Cell RNA-Seq and Bulk RNA-Seq Unravels Tumour Heterogeneity plus M2-like Tumour-Associated Macrophage Infiltration and Aggressiveness in TNBC. Cancer Immunol. Immunother. 2021, 70, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Lee, H.; Kim, D.; Han, S.K.; Ha, D.; Shin, K.; Kim, S. Network-Based Machine Learning in Colorectal and Bladder Organoid Models Predicts Anti-Cancer Drug Efficacy in Patients. Nat. Commun. 2020, 11, 5485. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Yao, C.Q.; Sabine, V.S.; Grzadkowski, M.; Stimper, V.; Starmans, M.H.W.; Wang, J.; Nguyen, F.; Moon, N.C.; Lin, X.; et al. Pathway-Based Subnetworks Enable Cross-Disease Biomarker Discovery. Nat. Commun. 2018, 9, 4746. [Google Scholar] [CrossRef]
- Wu, W.; Ma, X. Joint Learning Dimension Reduction and Clustering of Single-Cell RNA-Sequencing Data. Bioinformatics 2020, 36. [Google Scholar] [CrossRef]
- Qiu, Y.L.; Zheng, H.; Devos, A.; Selby, H.; Gevaert, O. A Meta-Learning Approach for Genomic Survival Analysis. Nat. Commun. 2020, 11, 6350. [Google Scholar] [CrossRef]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-Nnet: An Artificial Neural Network Method for Prognosis Prediction of High-Throughput Omics Data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized Treatment Recommender System Using a Cox Proportional Hazards Deep Neural Network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef]
- Huang, Z.; Johnson, T.S.; Han, Z.; Helm, B.; Cao, S.; Zhang, C.; Salama, P.; Rizkalla, M.; Yu, C.Y.; Cheng, J.; et al. Deep Learning-Based Cancer Survival Prognosis from RNA-Seq Data: Approaches and Evaluations. BMC Med. Genom. 2020, 13, 41. [Google Scholar] [CrossRef]
- Van IJzendoorn, D.G.P.; Szuhai, K.; Briaire-de Bruijn, I.H.; Kostine, M.; Kuijjer, M.L.; Bovée, J.V.M.G. Machine Learning Analysis of Gene Expression Data Reveals Novel Diagnostic and Prognostic Biomarkers and Identifies Therapeutic Targets for Soft Tissue Sarcomas. PLoS Comput. Biol. 2019, 15, e1006826. [Google Scholar] [CrossRef]
- Tabl, A.A.; Alkhateeb, A.; ElMaraghy, W.; Rueda, L.; Ngom, A. A Machine Learning Approach for Identifying Gene Biomarkers Guiding the Treatment of Breast Cancer. Front. Genet. 2019, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhu, C.; Liu, Q. Toward in Silico Identification of Tumor Neoantigens in Immunotherapy. Trends Mol. Med. 2019, 25, 980–992. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Storey, J.D. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Cereda, M.; Gambardella, G.; Benedetti, L.; Iannelli, F.; Patel, D.; Basso, G.; Guerra, R.F.; Mourikis, T.P.; Puccio, I.; Sinha, S.; et al. Patients with Genetically Heterogeneous Synchronous Colorectal Cancer Carry Rare Damaging Germline Mutations in Immune-Related Genes. Nat. Commun. 2016, 7, 12072. [Google Scholar] [CrossRef]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A Primer on Deep Learning in Genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep Learning for Computational Biology. Mol. Syst. Biol. 2016, 12. [Google Scholar] [CrossRef]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying Cancer and Normal RNA Sequencing Data from Different Sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef] [PubMed]
- Refine.bio. Available online: https://www.refine.bio (accessed on 15 April 2021).
- Jones, D.T. Setting the Standards for Machine Learning in Biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 659–660. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. arXiv Prepr. 2019, arXiv:1912.01703. [Google Scholar]
- Gulli, A.; Pal, S. Deep Learning with Keras: Implement Neural Networks with Keras on Theano and TensorFlow; Packt Publishing: Birmingham, UK, 2017; ISBN 9781787128422. [Google Scholar]
- Avsec, Ž.; Kreuzhuber, R.; Israeli, J.; Xu, N.; Cheng, J.; Shrikumar, A.; Banerjee, A.; Kim, D.S.; Beier, T.; Urban, L.; et al. The Kipoi Repository Accelerates Community Exchange and Reuse of Predictive Models for Genomics. Nat. Biotechnol. 2019, 37, 592–600. [Google Scholar] [CrossRef] [PubMed]
- García, S.; Ramírez-Gallego, S.; Luengo, J.; Benítez, J.M.; Herrera, F. Big Data Preprocessing: Methods and Prospects. Big Data Anal. 2016, 1, 9. [Google Scholar] [CrossRef]
- Shaham, U.; Stanton, K.P.; Zhao, J.; Li, H.; Raddassi, K.; Montgomery, R.; Kluger, Y. Removal of Batch Effects Using Distribution-Matching Residual Networks. Bioinformatics 2017, 33, 2539–2546. [Google Scholar] [CrossRef]
- Wang, T.; Johnson, T.S.; Shao, W.; Lu, Z.; Helm, B.R.; Zhang, J.; Huang, K. BERMUDA: A Novel Deep Transfer Learning Method for Single-Cell RNA Sequencing Batch Correction Reveals Hidden High-Resolution Cellular Subtypes. Genome Biol. 2019, 20, 165. [Google Scholar] [CrossRef]
- Li, X.; Wang, K.; Lyu, Y.; Pan, H.; Zhang, J.; Stambolian, D.; Susztak, K.; Reilly, M.P.; Hu, G.; Li, M. Deep Learning Enables Accurate Clustering with Batch Effect Removal in Single-Cell RNA-Seq Analysis. Nat. Commun. 2020, 11, 2338. [Google Scholar] [CrossRef]
- Yang, Y.; Li, G.; Qian, H.; Wilhelmsen, K.C.; Shen, Y.; Li, Y. SMNN: Batch Effect Correction for Single-Cell RNA-Seq Data via Supervised Mutual Nearest Neighbor Detection. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Elbashir, M.K.; Ezz, M.; Mohammed, M.; Saloum, S.S. Lightweight Convolutional Neural Network for Breast Cancer Classification Using RNA-Seq Gene Expression Data. IEEE Access 2019, 7, 185338–185348. [Google Scholar] [CrossRef]
- López-García, G.; Jerez, J.M.; Franco, L.; Veredas, F.J. Transfer Learning with Convolutional Neural Networks for Cancer Survival Prediction Using Gene-Expression Data. PLoS ONE 2020, 15, e0230536. [Google Scholar] [CrossRef]
- Ding, J.; Condon, A.; Shah, S.P. Interpretable Dimensionality Reduction of Single Cell Transcriptome Data with Deep Generative Models. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Park, S.; Shin, B.; Shim, W.S.; Choi, Y.; Kang, K.; Kang, K. Wx: A Neural Network-Based Feature Selection Algorithm for Transcriptomic Data. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Liu, S.; Xu, C.; Zhang, Y.; Liu, J.; Yu, B.; Liu, X.; Dehmer, M. Feature Selection of Gene Expression Data for Cancer Classification Using Double RBF-Kernels. BMC Bioinform. 2018, 19, 396. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA Interference Reveals That Oncogenic KRAS-Driven Cancers Require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Lauria, A.; Peirone, S.; Giudice, M.D.; Priante, F.; Rajan, P.; Caselle, M.; Oliviero, S.; Cereda, M. Identification of Altered Biological Processes in Heterogeneous RNA-Sequencing Data by Discretization of Expression Profiles. Nucleic Acids Res. 2020, 48, 1730–1747. [Google Scholar] [CrossRef]
- Jung, S.; Bi, Y.; Davuluri, R.V. Evaluation of Data Discretization Methods to Derive Platform Independent Isoform Expression Signatures for Multi-Class Tumor Subtyping. BMC Genom. 2015, 16 (Suppl. 11), S3. [Google Scholar] [CrossRef] [PubMed]
- Talwar, D.; Mongia, A.; Sengupta, D.; Majumdar, A. AutoImpute: Autoencoder Based Imputation of Single-Cell RNA-Seq Data. Sci. Rep. 2018, 8, 16329. [Google Scholar] [CrossRef] [PubMed]
- Arisdakessian, C.; Poirion, O.; Yunits, B.; Zhu, X.; Garmire, L.X. DeepImpute: An Accurate, Fast, and Scalable Deep Neural Network Method to Impute Single-Cell RNA-Seq Data. Genome Biol. 2019, 20, 211. [Google Scholar] [CrossRef] [PubMed]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-Cell RNA-Seq Denoising Using a Deep Count Autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Armasu, S.M.; Kalli, K.R.; Maurer, M.J.; Heinzen, E.P.; Keeney, G.L.; Cliby, W.A.; Oberg, A.L.; Kaufmann, S.H.; Goode, E.L. Pooled Clustering of High-Grade Serous Ovarian Cancer Gene Expression Leads to Novel Consensus Subtypes Associated with Survival and Surgical Outcomes. Clin. Cancer Res. 2017, 23, 4077–4085. [Google Scholar] [CrossRef]
- Alcaraz, N.; List, M.; Batra, R.; Vandin, F.; Ditzel, H.J.; Baumbach, J. De Novo Pathway-Based Biomarker Identification. Nucleic Acids Res. 2017, 45, e151. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, R.; Liang, F.; Zhang, L.; Liang, X. Identification of Metabolism-Associated Prostate Cancer Subtypes and Construction of a Prognostic Risk Model. Front. Oncol. 2020, 10, 598801. [Google Scholar] [CrossRef]
- Paquet, E.R.; Hallett, M.T. Absolute Assignment of Breast Cancer Intrinsic Molecular Subtype. J. Natl. Cancer Inst. 2015, 107, 357. [Google Scholar] [CrossRef]
- Chen, R.; Yang, L.; Goodison, S.; Sun, Y. Deep-Learning Approach to Identifying Cancer Subtypes Using High-Dimensional Genomic Data. Bioinformatics 2020, 36, 1476–1483. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, Z.; Namburi, S.; Pattison, A.; Posner, A.; Balachander, S.; Paisie, C.A.; Reddi, H.V.; Rueter, J.; Gill, A.J.; et al. CUP-AI-Dx: A Tool for Inferring Cancer Tissue of Origin and Molecular Subtype Using RNA Gene-Expression Data and Artificial Intelligence. EBioMedicine 2020, 61, 103030. [Google Scholar] [CrossRef]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.-J.; Shah, P.; et al. A Single-Cell Landscape of High-Grade Serous Ovarian Cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-Based Classification of Glioblastoma Uncovers a Mitochondrial Subtype with Therapeutic Vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, B.; Minn, A.; Zhang, N.R. DENDRO: Genetic Heterogeneity Profiling and Subclone Detection by Single-Cell RNA Sequencing. Genome Biol. 2020, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Nguyen, N.D.; Talos, F.; Wang, D. ECMarker: Interpretable Machine Learning Model Identifies Gene Expression Biomarkers Predicting Clinical Outcomes and Reveals Molecular Mechanisms of Human Disease in Early Stages. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Cheng, Q.; Li, J.; Fan, F.; Cao, H.; Dai, Z.-Y.; Wang, Z.-Y.; Feng, S.-S. Identification and Analysis of Glioblastoma Biomarkers Based on Single Cell Sequencing. Front. Bioeng. Biotechnol. 2020, 8, 167. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, M.; Wang, Q.; Zhang, J.; Zhou, T.; Sun, X. Single-Cell Transcriptome-Based Multilayer Network Biomarker for Predicting Prognosis and Therapeutic Response of Gliomas. Brief. Bioinform. 2020, 21, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous Enumeration of Cancer and Immune Cell Types from Bulk Tumor Gene Expression Data. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tan, Y.; Sun, F.; Hou, L.; Zhang, C.; Ge, T.; Yu, H.; Wu, C.; Zhu, Y.; Duan, L.; et al. Single-Cell Transcriptome and Antigen-Immunoglobin Analysis Reveals the Diversity of B Cells in Non-Small Cell Lung Cancer. Genome Biol. 2020, 21, 152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tian, X.; Ji, L.; Zhang, X.; Cao, Y.; Shen, C.; Hu, Y.; Wong, J.W.H.; Fang, J.-Y.; Hong, J.; et al. A Tumor Microenvironment-Specific Gene Expression Signature Predicts Chemotherapy Resistance in Colorectal Cancer Patients. NPJ Precis Oncol. 2021, 5, 7. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate Genome-Level Prediction of Neoantigens by Harnessing Sequence and Amino Acid Immunogenicity Information. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA Class II Antigen Presentation through Integrated Deep Learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A Benchmark of Batch-Effect Correction Methods for Single-Cell RNA Sequencing Data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. NAR Genom. Bioinform. 2020, 2. [Google Scholar] [CrossRef]
- Velliangiri, S.; Alagumuthukrishnan, S.; Thankumar Joseph, S.I. A Review of Dimensionality Reduction Techniques for Efficient Computation. Procedia Comput. Sci. 2019, 165, 104–111. [Google Scholar] [CrossRef]
- Abid, A.; Zhang, M.J.; Bagaria, V.K.; Zou, J. Exploring Patterns Enriched in a Dataset with Contrastive Principal Component Analysis. Nat. Commun. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Raj-Kumar, P.K.; Liu, J.; Hooke, J.A.; Kovatich, A.J.; Kvecher, L.; Shriver, C.D.; Hu, H. PCA-PAM50 Improves Consistency between Breast Cancer Intrinsic and Clinical Subtyping Reclassifying a Subset of Luminal A Tumors as Luminal B. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Taguchi, Y.-H.; Iwadate, M.; Umeyama, H. SFRP1 Is a Possible Candidate for Epigenetic Therapy in Non-Small Cell Lung Cancer. BMC Med. Genom. 2016, 9 (Suppl. 1), 28. [Google Scholar] [CrossRef]
- Chen, D.-T.; Hsu, Y.-L.; Fulp, W.J.; Coppola, D.; Haura, E.B.; Yeatman, T.J.; Cress, W.D. Prognostic and Predictive Value of a Malignancy-Risk Gene Signature in Early-Stage Non-Small Cell Lung Cancer. J. Natl. Cancer Inst. 2011, 103, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Walsh, J.R.; Long, J.; Davis, C.B.; Henstock, P.; Hodge, M.R.; Maciejewski, M.; Mu, X.J.; Ra, S.; Zhao, S.; et al. Standard Machine Learning Approaches Outperform Deep Representation Learning on Phenotype Prediction from Transcriptomics Data. BMC Bioinform. 2020, 21, 119. [Google Scholar] [CrossRef]
- Van der Maaten, L. Visualizing Data Using T-SNE. Available online: https://www.jmlr.org/papers/volume9/vandermaaten08a/vandermaaten08a.pdf?fbclid=IwAR0Bgg1eA5TFmqOZeCQXsIoL6PKrVXUFaskUKtg6yBhVXAFFvZA6yQiYx-M (accessed on 6 March 2021).
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv Prepr. 2018, arXiv:1802.03426. [Google Scholar]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality Reduction for Visualizing Single-Cell Data Using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Kobak, D.; Linderman, G.C. Initialization Is Critical for Preserving Global Data Structure in Both T-SNE and UMAP. Nat. Biotechnol. 2021, 39, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; Hsiao, C.J.; Stephens, M. Visualizing the Structure of RNA-Seq Expression Data Using Grade of Membership Models. PLoS Genet. 2017, 13, e1006599. [Google Scholar] [CrossRef]
- Mandel, J.; Avula, R.; Prochownik, E.V. Sequential Analysis of Transcript Expression Patterns Improves Survival Prediction in Multiple Cancers. BMC Cancer 2020, 20, 297. [Google Scholar] [CrossRef]
- Schmauch, B.; Romagnoni, A.; Pronier, E.; Saillard, C.; Maillé, P.; Calderaro, J.; Kamoun, A.; Sefta, M.; Toldo, S.; Zaslavskiy, M.; et al. A Deep Learning Model to Predict RNA-Seq Expression of Tumours from Whole Slide Images. Nat. Commun. 2020, 11, 3877. [Google Scholar] [CrossRef]
- Chen, Z.; Pang, M.; Zhao, Z.; Li, S.; Miao, R.; Zhang, Y.; Feng, X.; Feng, X.; Zhang, Y.; Duan, M.; et al. Feature Selection May Improve Deep Neural Networks for the Bioinformatics Problems. Bioinformatics 2019, 36, 1542–1552. [Google Scholar] [CrossRef]
- Liang, S.; Ma, A.; Yang, S.; Wang, Y.; Ma, Q. A Review of Matched-Pairs Feature Selection Methods for Gene Expression Data Analysis. Comput. Struct. Biotechnol. J. 2018, 16, 88–97. [Google Scholar] [CrossRef]
- Khaire, U.M.; Dhanalakshmi, R. Stability of Feature Selection Algorithm: A Review. J. King Saud Univ. Comput. Inf. Sci. 2019. [Google Scholar] [CrossRef]
- Bishop, C.M. Neural Networks for Pattern Recognition; Oxford University Press: New York, NY, USA, 1995; ISBN 9780198538646. [Google Scholar]
- Bengio, Y.; Simard, P.; Frasconi, P. Learning Long-Term Dependencies with Gradient Descent Is Difficult. IEEE Trans. Neural Netw. 1994, 5, 157–166. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and Analytical Challenges in Single-Cell Transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Witten, I.H.; Frank, E.; Hall, M.A. Data Mining: Practical Machine Learning Tools and Techniques; Elsevier Science: Amsterdam, The Netherlands, 2011; ISBN 9780123748560. [Google Scholar]
- Ramírez-Gallego, S.; García, S.; Mouriño-Talín, H.; Martínez-Rego, D.; Bolón-Canedo, V.; Alonso-Betanzos, A.; Benítez, J.M.; Herrera, F. Data Discretization: Taxonomy and Big Data Challenge. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2016, 6, 5–21. [Google Scholar] [CrossRef]
- Gallo, C.A.; Cecchini, R.L.; Carballido, J.A.; Micheletto, S.; Ponzoni, I. Discretization of Gene Expression Data Revised. Brief. Bioinform. 2015, 17, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Angerer, P.; Simon, L.; Tritschler, S.; Wolf, F.A.; Fischer, D.; Theis, F.J. Single Cells Make Big Data: New Challenges and Opportunities in Transcriptomics. Curr. Opin. Syst. Biol. 2017, 4, 85–91. [Google Scholar] [CrossRef]
- Chai, X.; Gu, H.; Li, F.; Duan, H.; Hu, X.; Lin, K. Deep Learning for Irregularly and Regularly Missing Data Reconstruction. Sci. Rep. 2020, 10, 3302. [Google Scholar] [CrossRef] [PubMed]
- Jaskowiak, P.A.; Costa, I.G.; Campello, R.J.G.B. Clustering of RNA-Seq Samples: Comparison Study on Cancer Data. Methods 2018, 132, 42–49. [Google Scholar] [CrossRef]
- Liu, L.; Tang, L.; Dong, W.; Yao, S.; Zhou, W. An Overview of Topic Modeling and Its Current Applications in Bioinformatics. Springerplus 2016, 5, 1608. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, M.; Zhu, H.; Xu, J. A 15-Gene Signature for Prediction of Colon Cancer Recurrence and Prognosis Based on SVM. Gene 2017, 604, 33–40. [Google Scholar] [CrossRef]
- Mourikis, T.P.; Benedetti, L.; Foxall, E.; Temelkovski, D.; Nulsen, J.; Perner, J.; Cereda, M.; Lagergren, J.; Howell, M.; Yau, C.; et al. Patient-Specific Cancer Genes Contribute to Recurrently Perturbed Pathways and Establish Therapeutic Vulnerabilities in Esophageal Adenocarcinoma. Nat. Commun. 2019, 10, 3101. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, B. Semi-Supervised Learning Improves Gene Expression-Based Prediction of Cancer Recurrence. Bioinformatics 2011, 27, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Mohaiminul Islam, M.; Huang, S.; Ajwad, R.; Chi, C.; Wang, Y.; Hu, P. An Integrative Deep Learning Framework for Classifying Molecular Subtypes of Breast Cancer. Comput. Struct. Biotechnol. J. 2020, 18, 2185–2199. [Google Scholar]
- Kalia, M. Biomarkers for Personalized Oncology: Recent Advances and Future Challenges. Metabolism 2015, 64, S16–S21. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; ISBN 9781475732948. [Google Scholar]
- Ishwaran, H.; Kogalur, U.B.; Blackstone, E.H.; Lauer, M.S. Random Survival Forests. Ann. Appl. Stat. 2008, 2, 841–860. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; The MIT Press: Cambridge, MA, USA, 2016; ISBN 9780262035613. [Google Scholar]
- Yousefi, S.; Amrollahi, F.; Amgad, M.; Dong, C.; Lewis, J.E.; Song, C.; Gutman, D.A.; Halani, S.H.; Velazquez Vega, J.E.; Brat, D.J.; et al. Predicting Clinical Outcomes from Large Scale Cancer Genomic Profiles with Deep Survival Models. Sci. Rep. 2017, 7, 11707. [Google Scholar] [CrossRef] [PubMed]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA Splicing in Cancer Immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.-V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; Improved Prediction of Binding to MHC Class I Molecules Integrating Information from Multiple Receptor and Peptide Length Datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef]
- Smart, A.C.; Margolis, C.A.; Pimentel, H.; He, M.X.; Miao, D.; Adeegbe, D.; Fugmann, T.; Wong, K.-K.; Van Allen, E.M. Intron Retention Is a Source of Neoepitopes in Cancer. Nat. Biotechnol. 2018, 36, 1056–1058. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best Practices for Bioinformatic Characterization of Neoantigens for Clinical Utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Curse of Dimensionality. Encycl. Database Syst. 2009, 545–546. [Google Scholar]
- Altman, N.; Krzywinski, M. The Curse(s) of Dimensionality. Nat. Methods 2018, 15, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Jackson, S.A. Machine Learning and Complex Biological Data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Neumann, A.; Timmermann, B.; Meinke, S.; Heyd, F. Differential Interleukin-2 Transcription Kinetics Render Mouse but Not Human T Cells Vulnerable to Splicing Inhibition Early after Activation. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Artemaki, P.I.; Letsos, P.A.; Zoupa, I.C.; Katsaraki, K.; Karousi, P.; Papageorgiou, S.G.; Pappa, V.; Scorilas, A.; Kontos, C.K. The Multifaceted Role and Utility of MicroRNAs in Indolent B-Cell Non-Hodgkin Lymphomas. Biomedicines 2021, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.; Chen, Y.; Jones, A.; Shibue, T.; Hahn, W.C.; Boehm, J.S.; Vazquez, F.; Tsherniak, A.; McFarland, J.M. Global Computational Alignment of Tumor and Cell Line Transcriptional Profiles. Nat. Commun. 2021, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A First-Generation Pediatric Cancer Dependency Map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).