
Transcriptome Analysis of Seed Weight Plasticity in Brassica napus

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
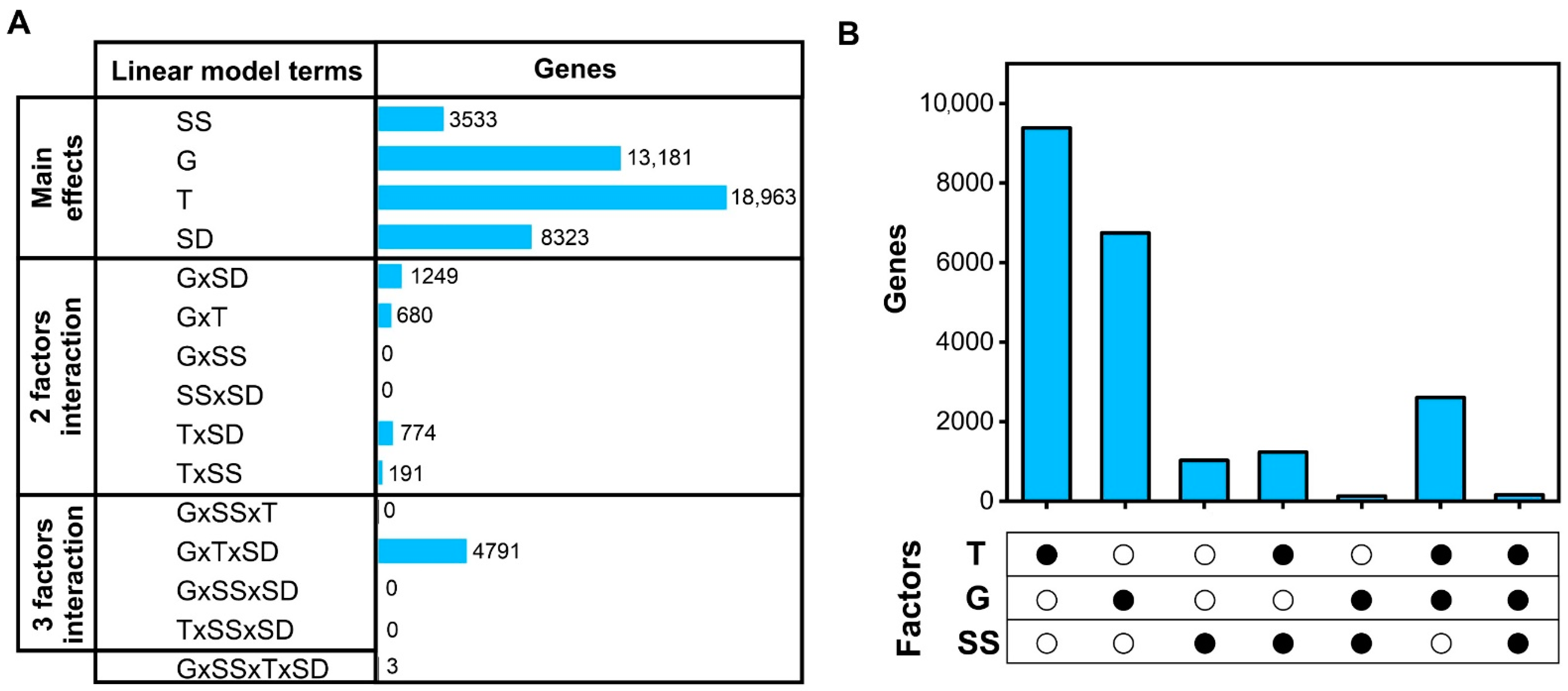
2.1. Multivariate Analysis of RNA-Seq Data Uncovers the Main Factors Affecting Seed Transcriptomes in Rapeseed under Field Conditions
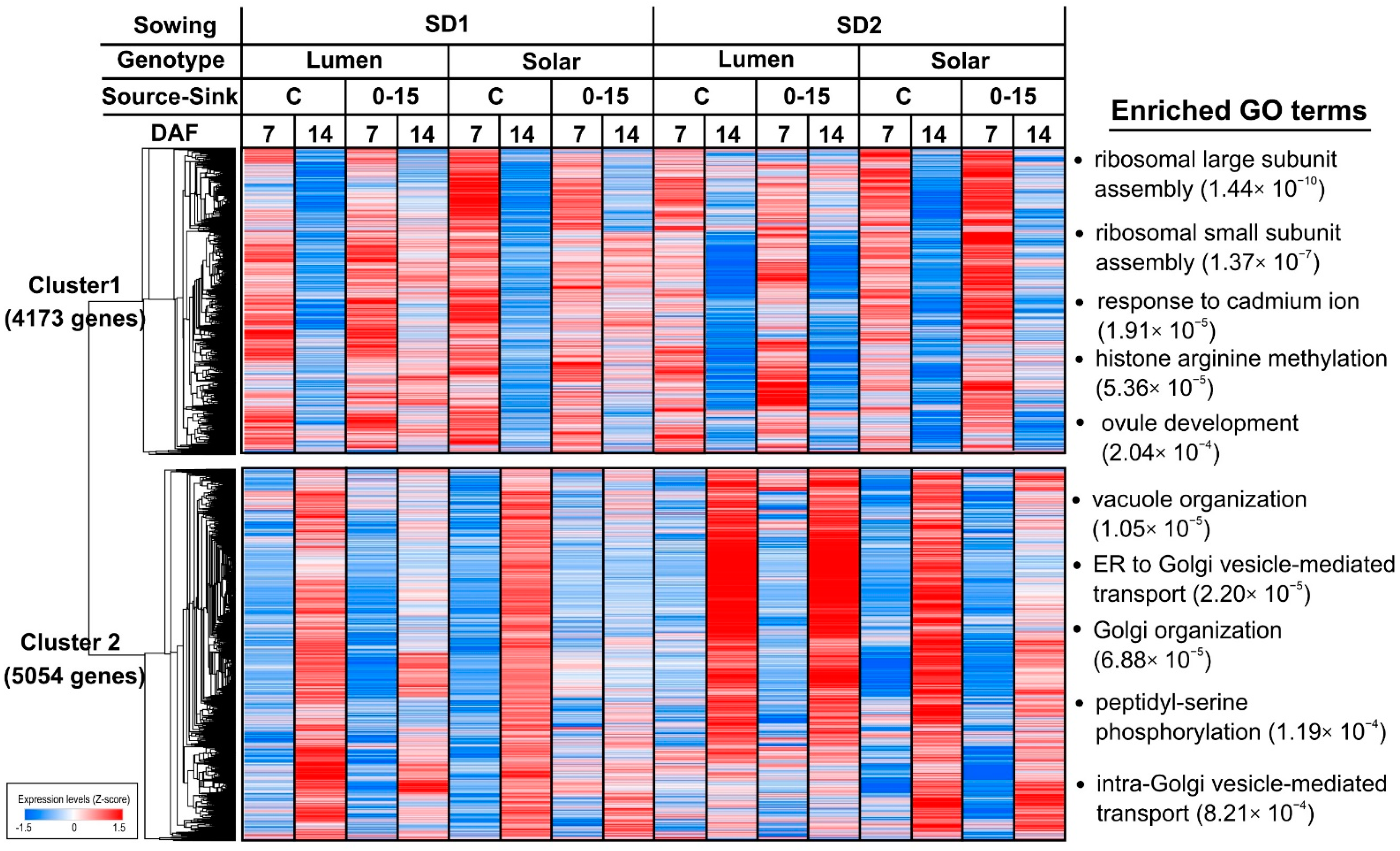
2.2. Genes Regulated by Development Time under Field Conditions Are Highly Enriched in Biological Processes Related to Seed Filling
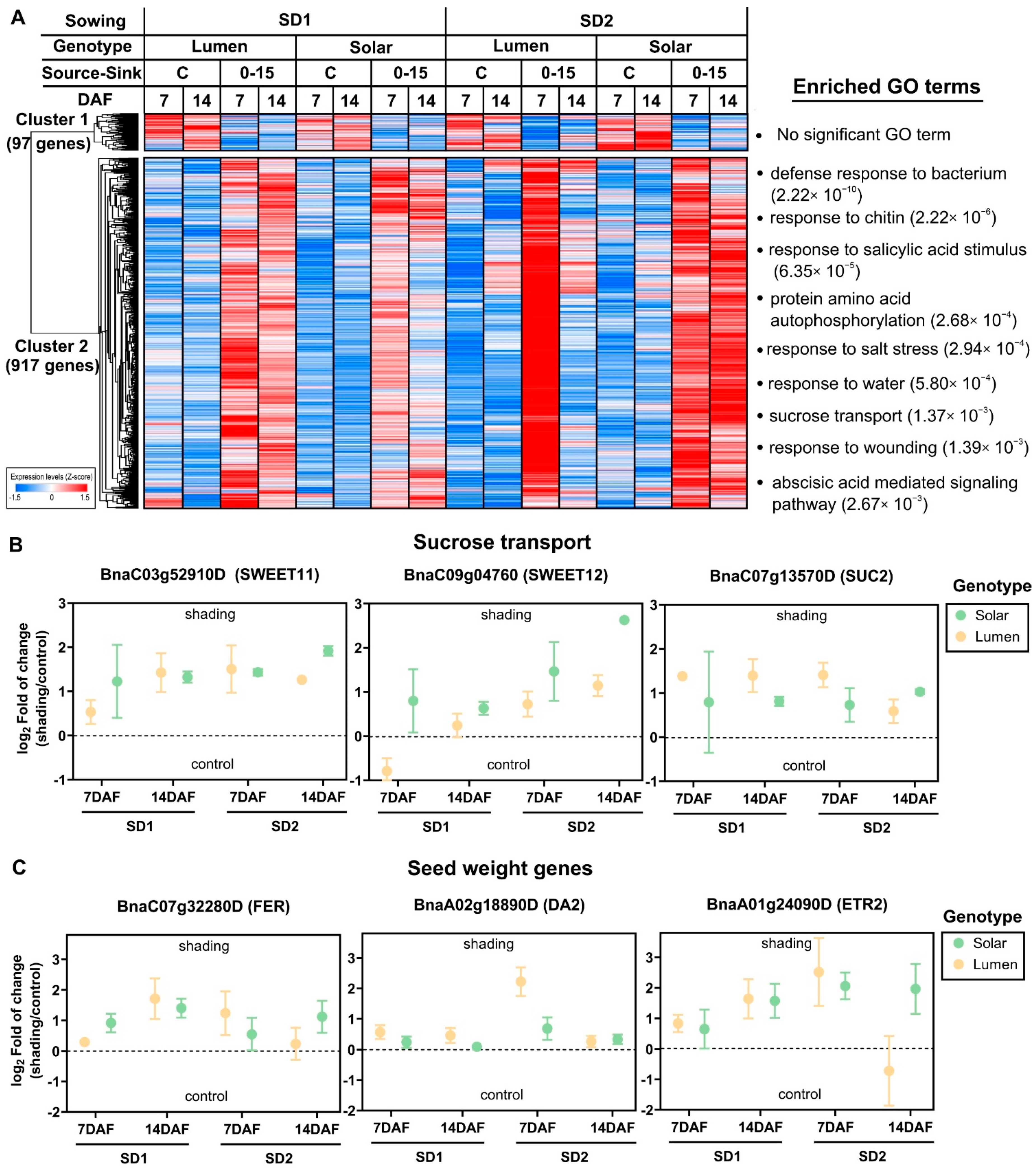
2.3. Reduction in the Source–Sink Ratio at Flowering Induces the Expression of Genes Involved in Stress Response and Seed Weight
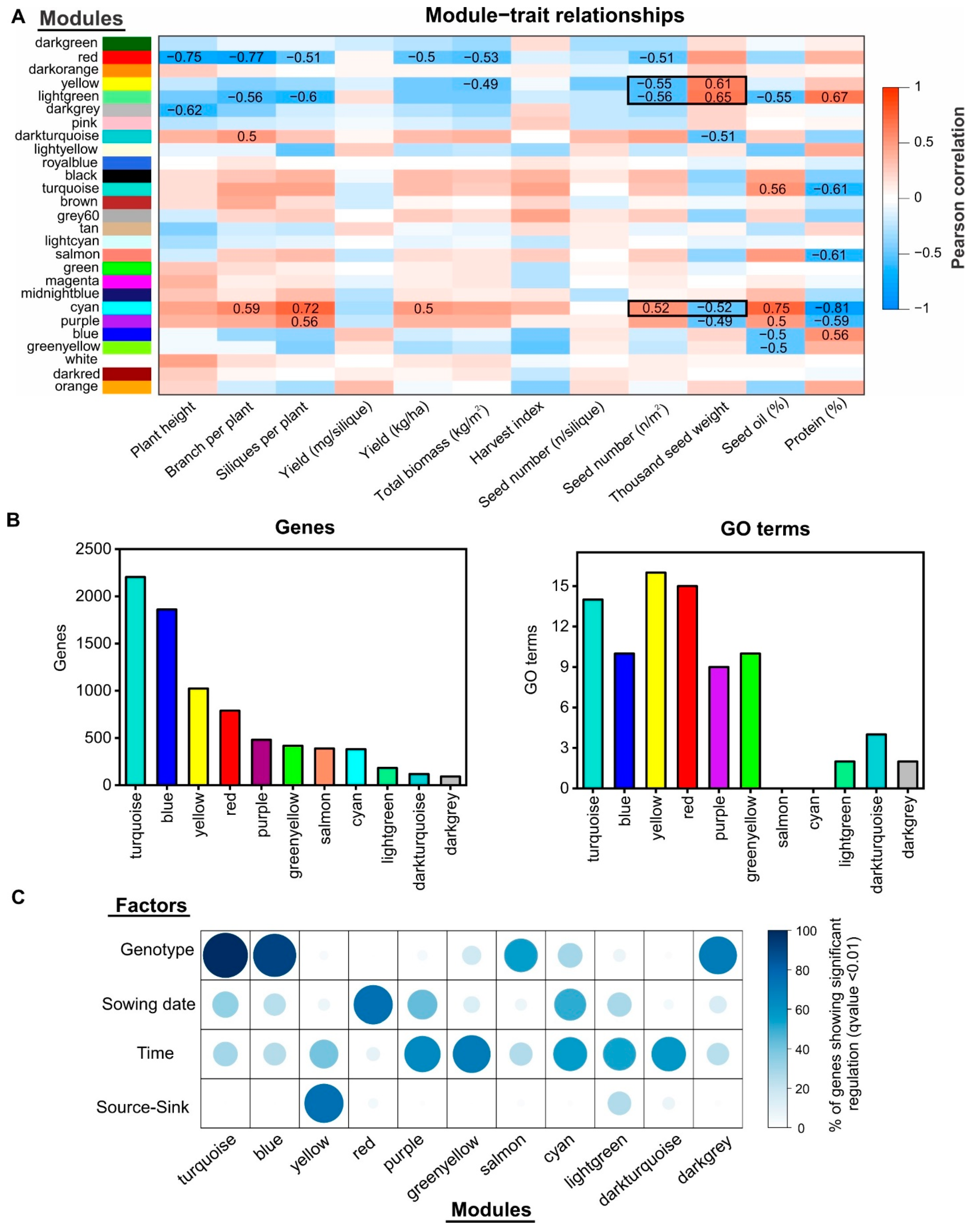
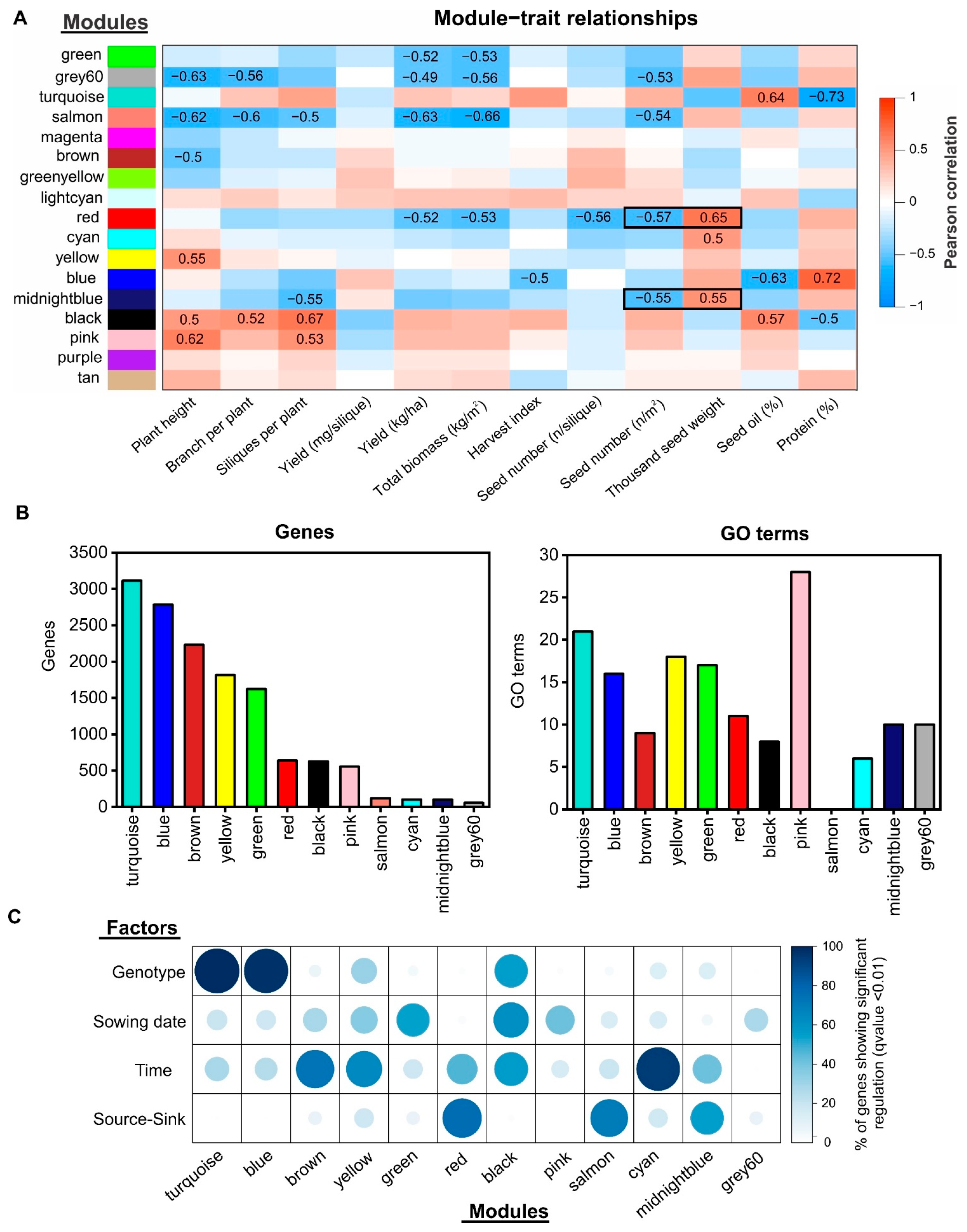
2.4. Gene Co-Expression Network Analysis Uncovers Novel Gene Modules Related to Seed Yield and Quality under Field Conditions
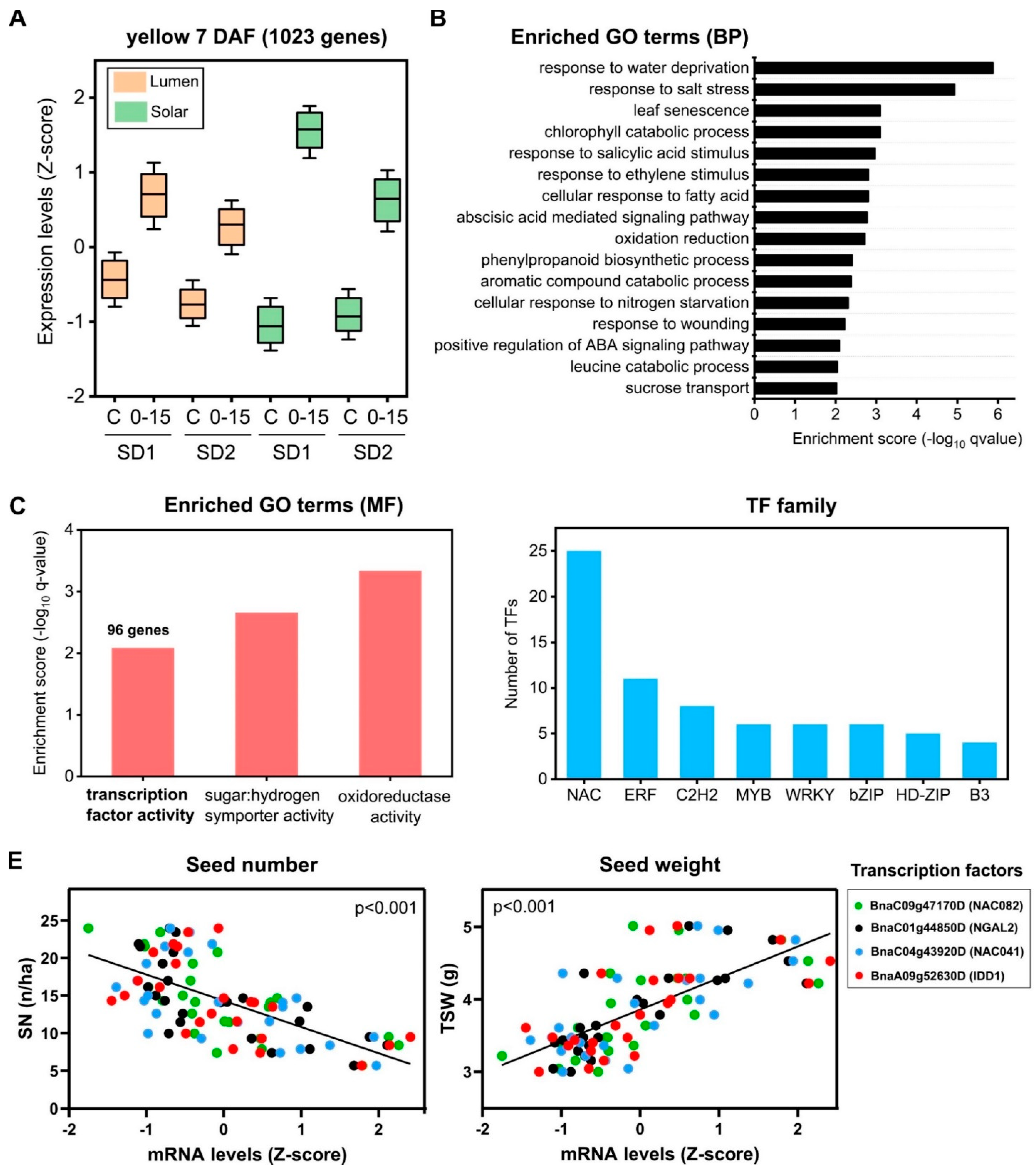
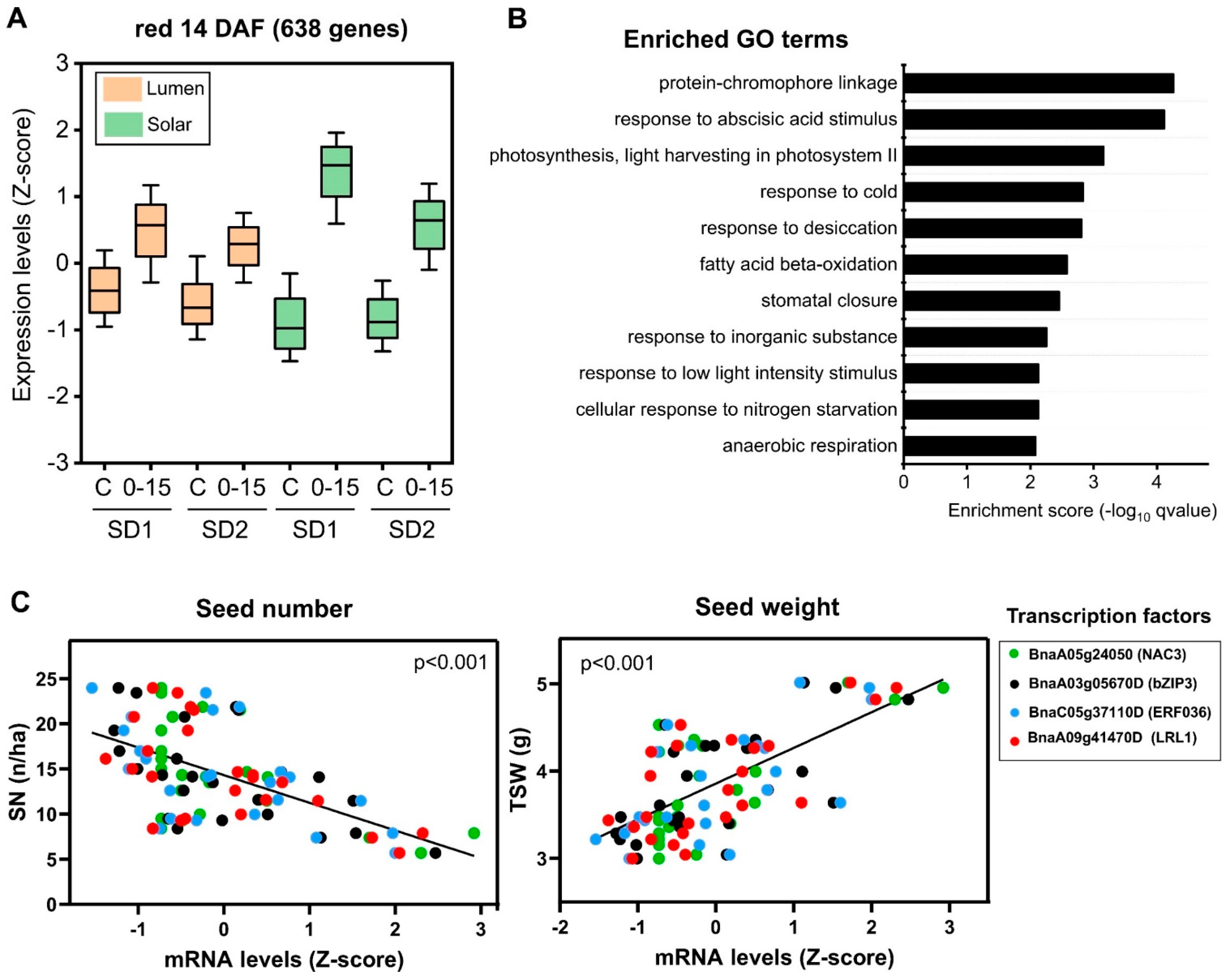
2.5. Identification of Regulatory Factors Associated with the Compensation of SW and SN Decreases by Source–Sink Restriction at Flowering
3. Discussion
3.1. Identification of Genes Related to Seed Weight Plasticity in Rapeseed
3.2. Transcription Factors Associated with SW and SN Response under Field Conditions
4. Materials and Methods
4.1. Field Experiment, Treatments, and Crop Management
4.2. Phenology and Physiological Plant Sampling
4.3. Seed Sampling and RNA Isolation
4.4. RNA-Seq Analysis
4.5. Gene Co-Expression Network Construction
4.6. Gene Ontology Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savadi, S. Molecular Regulation of Seed Development and Strategies for Engineering Seed Size in Crop Plants. Plant Growth Regul. 2018, 84, 401–422. [Google Scholar] [CrossRef]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield Trends Are Insufficient to Double Global Crop Production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef]
- Universidade Federal de Ouro Preto. UFOP Report on Global Market Supply 2017/2018; Universidade Federal de Ouro Preto: Ouro Preto, Brazil, 2019. [Google Scholar]
- FAO. Food Outlook—Biannual Report on Global Food Markets; FAO: Rome, Italy, 2020. [Google Scholar]
- FAOSTAT Crops Production 2021. 2021. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 23 March 2021).
- Peltonen-Sainio, P.; Kangas, A.; Salo, Y.; Jauhiainen, L. Grain Number Dominates Grain Weight in Temperate Cereal Yield Determination: Evidence Based on 30 Years of Multi-Location Trials. Field Crops Res. 2007, 100, 179–188. [Google Scholar] [CrossRef]
- Beddington, J. Food Security: Contributions from Science to a New and Greener Revolution. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 61–71. [Google Scholar] [CrossRef]
- Rondanini, D.P.; Gomez, N.V.; Agosti, M.B.; Miralles, D.J. Global Trends of Rapeseed Grain Yield Stability and Rapeseed-to-Wheat Yield Ratio in the Last Four Decades. Eur. J. Agron. 2012, 37, 56–65. [Google Scholar] [CrossRef]
- Sadras, V.O. Evolutionary Aspects of the Trade-off between Seed Size and Number in Crops. Field Crops Res. 2007, 100, 125–138. [Google Scholar] [CrossRef]
- Slafer, G.A.; Savin, R.; Sadras, V.O. Coarse and Fine Regulation of Wheat Yield Components in Response to Genotype and Environment. Field Crops Res. 2014, 157, 71–83. [Google Scholar] [CrossRef]
- Peltonen-Sainio, P.; Jauhiainen, L.; Sadras, V.O. Phenotypic Plasticity of Yield and Agronomic Traits in Cereals and Rapeseed at High Latitudes. Field Crops Res. 2011, 124, 261–269. [Google Scholar] [CrossRef]
- Rondanini, D.P.; Mantese, A.I.; Savin, R.; Hall, A.J. Water Content Dynamics of Achene, Pericarp and Embryo in Sunflower: Associations with Achene Potential Size and Dry-Down. Eur. J. Agron. 2009, 30, 53–62. [Google Scholar] [CrossRef]
- Castillo, F.M.; Vásquez, S.C.; Calderini, D.F. Does the Pre-Flowering Period Determine the Potential Grain Weight of Sunflower? Field Crops Res. 2017, 212, 23–33. [Google Scholar] [CrossRef]
- Labra, M.H.; Struik, P.C.; Evers, J.B.; Calderini, D.F. Plasticity of Seed Weight Compensates Reductions in Seed Number of Oilseed Rape in Response to Shading at Flowering. Eur. J. Agron. 2017, 84, 113–124. [Google Scholar] [CrossRef]
- Kirkegaard, J.A.; Lilley, J.M.; Brill, R.D.; Ware, A.H.; Walela, C.K. The Critical Period for Yield and Quality Determination in Canola (Brassica Napus L.). Field Crops Res. 2018, 222, 180–188. [Google Scholar] [CrossRef]
- Verdejo, J.; Calderini, D.F. Plasticity of Seed Weight in Winter and Spring Rapeseed Is Higher in a Narrow but Different Window after Flowering. Field Crops Res. 2020, 250, 107777. [Google Scholar] [CrossRef]
- Li, N.; Li, Y. Ubiquitin-Mediated Control of Seed Size in Plants. Front. Plant Sci. 2014, 5, 332. [Google Scholar] [CrossRef]
- Jiang, W.-B.; Huang, H.-Y.; Hu, Y.-W.; Zhu, S.-W.; Wang, Z.-Y.; Lin, W.-H. Brassinosteroid Regulates Seed Size and Shape in Arabidopsis. Plant Physiol. 2013, 162, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, Y. Maternal Control of Seed Size in Plants. J. Exp. Bot. 2015, 66, 1087–1097. [Google Scholar] [CrossRef]
- Sun, X.; Shantharaj, D.; Kang, X.; Ni, M. Transcriptional and Hormonal Signaling Control of Arabidopsis Seed Development. Curr. Opin. Plant Biol. 2010, 13, 611–620. [Google Scholar] [CrossRef]
- Wang, L.; Ruan, Y.-L. Regulation of Cell Division and Expansion by Sugar and Auxin Signaling. Front. Plant Sci. 2013, 4. [Google Scholar] [CrossRef]
- Niu, Y.; Wu, L.; Li, Y.; Huang, H.; Qian, M.; Sun, W.; Zhu, H.; Xu, Y.; Fan, Y.; Mahmood, U.; et al. Deciphering the Transcriptional Regulatory Networks That Control Size, Color, and Oil Content in Brassica Rapa Seeds. Biotechnol. Biofuels 2020, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Freeling, M. The Brassica Genome. Front. Plant Sci. 2013, 4, 148. [Google Scholar] [CrossRef] [PubMed]
- Calderini, D.F.; Castillo, F.M.; Arenas-M, A.; Molero, G.; Reynolds, M.P.; Craze, M.; Bowden, S.; Milner, M.J.; Wallington, E.J.; Dowle, A.; et al. Overcoming the Trade-off between Grain Weight and Number in Wheat by the Ectopic Expression of Expansin in Developing Seeds Leads to Increased Yield Potential. New Phytol. 2021, 230, 629–640. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential Analysis of RNA-Seq Incorporating Quantification Uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, Y.; Zhang, B. Efficient Test and Visualization of Multi-Set Intersections. Sci. Rep. 2015, 5, 16923. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape Plugin to Assess Overrepresentation of Gene Ontology Categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus/ (accessed on 9 March 2021).
- Zhang, H.; Flottmann, S. Source-Sink Manipulations Indicate Seed Yield in Canola Is Limited by Source Availability. Eur. J. Agron. 2018, 96, 70–76. [Google Scholar] [CrossRef]
- Li, N.; Song, D.; Peng, W.; Zhan, J.; Shi, J.; Wang, X.; Liu, G.; Wang, H. Maternal Control of Seed Weight in Rapeseed (Brassica Napus L.): The Causal Link between the Size of Pod (Mother, Source) and Seed (Offspring, Sink). Plant Biotechnol. J. 2019, 17, 736–749. [Google Scholar] [CrossRef]
- Xia, T.; Li, N.; Dumenil, J.; Li, J.; Kamenski, A.; Bevan, M.W.; Gao, F.; Li, Y. The Ubiquitin Receptor DA1 Interacts with the E3 Ubiquitin Ligase DA2 to Regulate Seed and Organ Size in Arabidopsis. Plant Cell 2013, 25, 3347–3359. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Gao, M.J.; Lydiate, D.J.; Li, X.; Lui, H.; Gjetvaj, B.; Hegedus, D.D.; Rozwadowski, K. Repression of Seed Maturation Genes by a Trihelix Transcriptional Repressor in Arabidopsis Seedlings. Plant Cell 2009, 21, 54–71. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, L.; Xu, R.; Cui, R.; Hao, J.; Sun, C.; Li, Y. Transcription Factors SOD7/NGAL2 and DPA4/NGAL3 Act Redundantly to Regulate Seed Size by Directly Repressing KLU Expression in Arabidopsis Thaliana. Plant Cell 2015, 27, 620–632. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, N.; Zhang, Q.; Zhou, G.; Tian, H.; Hussain, S.; Ahmed, S.; Wang, T.; Wang, S. Genome Editing to Integrate Seed Size and Abiotic Stress Tolerance Traits in Arabidopsis Reveals a Role for DPA4 and SOD7 in the Regulation of Inflorescence Architecture. Int. J. Mol. Sci. 2019, 20, 2695. [Google Scholar] [CrossRef]
- Feurtado, J.A.; Huang, D.; Wicki-Stordeur, L.; Hemstock, L.E.; Potentier, M.S.; Tsang, E.W.T.; Cutler, A.J. The Arabidopsis C2H2 Zinc Finger INDETERMINATE DOMAIN1/ENHYDROUS Promotes the Transition to Germination by Regulating Light and Hormonal Signaling during Seed Maturation. Plant Cell 2011, 23, 1772–1794. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-C.; Reca, I.-B.; Kim, Y.; Park, S.; Thomashow, M.F.; Keegstra, K.; Han, K.-H. Transcription Factors That Directly Regulate the Expression of CSLA9 Encoding Mannan Synthase in Arabidopsis Thaliana. Plant Mol. Biol. 2014, 84, 577–587. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a Central Hub for Transcription Factors and Regulatory Interactions in Plants. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef]
- Wang, B.; Li, J. Understanding the Molecular Bases of Agronomic Trait Improvement in Rice. Plant Cell 2019, 31, 1416–1417. [Google Scholar] [CrossRef] [PubMed]
- Azodi, C.B.; Pardo, J.; VanBuren, R.; de los Campos, G.; Shiu, S.-H. Transcriptome-Based Prediction of Complex Traits in Maize. Plant Cell 2020, 32, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-M.; Kong, X.-D.; Chen, F.; Huang, J.-X.; Lou, X.-Y.; Zhao, J.-Y. Transcriptome Analysis of Brassica Napus Pod Using RNA-Seq and Identification of Lipid-Related Candidate Genes. BMC Genomics 2015, 16, 858. [Google Scholar] [CrossRef]
- Shen, E.; Zhu, X.; Hua, S.; Chen, H.; Ye, C.; Zhou, L.; Liu, Q.; Zhu, Q.-H.; Fan, L.; Chen, X. Genome-Wide Identification of Oil Biosynthesis-Related Long Non-Coding RNAs in Allopolyploid Brassica Napus. BMC Genomics 2018, 19, 745. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, C.; Tang, F.; Yang, B.; Zhang, L.; Liu, J.; Huo, Q.; Wang, S.; Li, S.; Wei, L.; et al. Identification of Candidate Genes Controlling Oil Content by Combination of Genome-Wide Association and Transcriptome Analysis in the Oilseed Crop Brassica Napus. Biotechnol. Biofuels 2019, 12, 216. [Google Scholar] [CrossRef]
- Shahid, M.; Cai, G.; Zu, F.; Zhao, Q.; Qasim, M.U.; Hong, Y.; Fan, C.; Zhou, Y. Comparative Transcriptome Analysis of Developing Seeds and Silique Wall Reveals Dynamic Transcription Networks for Effective Oil Production in Brassica Napus L. Int. J. Mol. Sci. 2019, 20, 1982. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, S.; Yuan, Y.; Wang, Y.; Zeng, L.; Batley, J.; Wang, Y.-P. Transcriptomic Comparison between Developing Seeds of Yellow- and Black-Seeded Brassica Napus Reveals That Genes Influence Seed Quality. BMC Plant Biol. 2019, 19, 203. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Dong, N.; Wang, Y.; Li, G.; Wang, L.; Guo, X.; Li, J.; Wen, Z.; Wei, W. RNA-Seq Transcriptome Analysis of the Immature Seeds of Two Brassica Napus Lines with Extremely Different Thousand-Seed Weight to Identify the Candidate Genes Related to Seed Weight. PLoS ONE 2018, 13, e0191297. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Cui, Y.; Ding, Y.; Mei, J.; Dong, H.; Zhang, W.; Wu, S.; Liang, Y.; Zhang, C.; Li, J.; et al. Time-Series Analyses of Transcriptomes and Proteomes Reveal Molecular Networks Underlying Oil Accumulation in Canola. Front. Plant Sci. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Jolivet, P.; Boulard, C.; Bellamy, A.; Valot, B.; d’Andréa, S.; Zivy, M.; Nesi, N.; Chardot, T. Oil Body Proteins Sequentially Accumulate throughout Seed Development in Brassica Napus. J. Plant Physiol. 2011, 168, 2015–2020. [Google Scholar] [CrossRef] [PubMed]
- Sosso, D.; Luo, D.; Li, Q.-B.; Sasse, J.; Yang, J.; Gendrot, G.; Suzuki, M.; Koch, K.E.; McCarty, D.R.; Chourey, P.S.; et al. Seed Filling in Domesticated Maize and Rice Depends on SWEET-Mediated Hexose Transport. Nat. Genet. 2015, 47, 1489–1493. [Google Scholar] [CrossRef]
- Yang, J.; Luo, D.; Yang, B.; Frommer, W.B.; Eom, J.-S. SWEET11 and 15 as Key Players in Seed Filling in Rice. New Phytol. 2018, 218, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.-C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.-Q.; et al. Simultaneous Changes in Seed Size, Oil Content, and Protein Content Driven by Selection of SWEET Homologues during Soybean Domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef]
- Chen, L.-Q.; Lin, I.W.; Qu, X.-Q.; Sosso, D.; McFarlane, H.E.; Londoño, A.; Samuels, A.L.; Frommer, W.B. A Cascade of Sequentially Expressed Sucrose Transporters in the Seed Coat and Endosperm Provides Nutrition for the Arabidopsis Embryo. Plant Cell 2015, 27, 607–619. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, D.; Miao, Q.; Yang, J.; Xuan, Y.; Hu, Y. Essential Role of Sugar Transporter OsSWEET11 During the Early Stage of Rice Grain Filling. Plant Cell Physiol. 2017, 58, 863–873. [Google Scholar] [CrossRef]
- Wang, J.-L.; Tang, M.-Q.; Chen, S.; Zheng, X.-F.; Mo, H.-X.; Li, S.-J.; Wang, Z.; Zhu, K.-M.; Ding, L.-N.; Liu, S.-Y.; et al. Down-Regulation of BnDA1, Whose Gene Locus Is Associated with the Seeds Weight, Improves the Seeds Weight and Organ Size in Brassica Napus. Plant Biotechnol. J. 2017, 15, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Li, Z.; Ran, Q.; Wang, H.; Zhang, J. Over-Expression of Mutated ZmDA1 or ZmDAR1 Gene Improves Maize Kernel Yield by Enhancing Starch Synthesis. Plant Biotechnol. J. 2018, 16, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, H.; Hao, C.; Wang, K.; Wang, Y.; Qin, L.; An, D.; Li, T.; Zhang, X. TaDA1, a Conserved Negative Regulator of Kernel Size, Has an Additive Effect with TaGW2 in Common Wheat (Triticum Aestivum L.). Plant Biotechnol. J. 2020, 18, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Welner, D.H.; Deeba, F.; Lo Leggio, L.; Skriver, K. Chapter 13—NAC Transcription Factors: From Structure to Function in Stress-Associated Networks. In Plant Transcription Factors; Gonzalez, D.H., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 199–212. ISBN 978-0-12-800854-6. [Google Scholar]
- Agarwal, P.; Kapoor, S.; Tyagi, A.K. Transcription Factors Regulating the Progression of Monocot and Dicot Seed Development. BioEssays 2011, 33, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.; Maji, S.; Waseem, M.; Thakur, P.; Kumar, V.; Parida, S.K.; Thakur, J.K. The Mediator Subunit OsMED15a Is a Transcriptional Co-Regulator of Seed Size/Weight-Modulating Genes in Rice. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194432. [Google Scholar] [CrossRef]
- Mathew, I.E.; Das, S.; Mahto, A.; Agarwal, P. Three Rice NAC Transcription Factors Heteromerize and Are Associated with Seed Size. Front. Plant Sci. 2016, 7, 1638. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Ohtani, M.; Mitsuda, N.; Kubo, M.; Ohme-Takagi, M.; Fukuda, H.; Demura, T. VND-INTERACTING2, a NAC Domain Transcription Factor, Negatively Regulates Xylem Vessel Formation in Arabidopsis. Plant Cell 2010, 22, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Ohbayashi, I.; Lin, C.-Y.; Shinohara, N.; Matsumura, Y.; Machida, Y.; Horiguchi, G.; Tsukaya, H.; Sugiyama, M. Evidence for a Role of ANAC082 as a Ribosomal Stress Response Mediator Leading to Growth Defects and Developmental Alterations in Arabidopsis. Plant Cell 2017, 29, 2644–2660. [Google Scholar] [CrossRef]
- Wang, W.; Ryu, K.H.; Bruex, A.; Barron, C.; Schiefelbein, J. Molecular Basis for a Cell Fate Switch in Response to Impaired Ribosome Biogenesis in the Arabidopsis Root Epidermis. Plant Cell 2020, 32, 2402–2423. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, L.; Corke, F.; Smith, C.; Bevan, M.W. Control of Final Seed and Organ Size by the DA1 Gene Family in Arabidopsis Thaliana. Genes Dev. 2008, 22, 1331–1336. [Google Scholar] [CrossRef]
- Meier, U. Growth Stages of Mono- and Dicotyledonous Plants: BBCH Monograph; Blackwell Wissenschafts-Verlag: Berlin, Germany, 2018. [Google Scholar] [CrossRef]
- Kirk, P.L. Kjeldahl Method for Total Nitrogen. Anal. Chem. 1950, 22, 354–358. [Google Scholar] [CrossRef]
- Merrill, A.L.; Watt, B.K. Energy Value of Foods: Basis and Derivation; Human Nutrition Research Branch, Agricultural Research Service: Washington, DC, USA, 1973. [Google Scholar]
- Graeber, K.; Linkies, A.; Wood, A.T.A.; Leubner-Metzger, G. A Guideline to Family-Wide Comparative State-of-the-Art Quantitative RT-PCR Analysis Exemplified with a Brassicaceae Cross-Species Seed Germination Case Study. Plant Cell 2011, 23, 2045–2063. [Google Scholar] [CrossRef]
- Yaffe, H.; Buxdorf, K.; Shapira, I.; Ein-Gedi, S.; Moyal-Ben Zvi, M.; Fridman, E.; Moshelion, M.; Levy, M. LogSpin: A Simple, Economical and Fast Method for RNA Isolation from Infected or Healthy Plants and Other Eukaryotic Tissues. BMC Res. Notes 2012, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Canales, J.; Uribe, F.; Henríquez-Valencia, C.; Lovazzano, C.; Medina, J.; Vidal, E.A. Transcriptomic Analysis at Organ and Time Scale Reveals Gene Regulatory Networks Controlling the Sulfate Starvation Response of Solanum Lycopersicum. BMC Plant Biol. 2020, 20, 385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canales, J.; Verdejo, J.; Carrasco-Puga, G.; Castillo, F.M.; Arenas-M, A.; Calderini, D.F. Transcriptome Analysis of Seed Weight Plasticity in Brassica napus. Int. J. Mol. Sci. 2021, 22, 4449. https://doi.org/10.3390/ijms22094449
Canales J, Verdejo J, Carrasco-Puga G, Castillo FM, Arenas-M A, Calderini DF. Transcriptome Analysis of Seed Weight Plasticity in Brassica napus. International Journal of Molecular Sciences. 2021; 22(9):4449. https://doi.org/10.3390/ijms22094449
Chicago/Turabian StyleCanales, Javier, José Verdejo, Gabriela Carrasco-Puga, Francisca M. Castillo, Anita Arenas-M, and Daniel F. Calderini. 2021. "Transcriptome Analysis of Seed Weight Plasticity in Brassica napus" International Journal of Molecular Sciences 22, no. 9: 4449. https://doi.org/10.3390/ijms22094449
APA StyleCanales, J., Verdejo, J., Carrasco-Puga, G., Castillo, F. M., Arenas-M, A., & Calderini, D. F. (2021). Transcriptome Analysis of Seed Weight Plasticity in Brassica napus. International Journal of Molecular Sciences, 22(9), 4449. https://doi.org/10.3390/ijms22094449