
Targeting HR Repair as a Synthetic Lethal Approach to Increase DNA Damage Sensitivity by a RAD52 Inhibitor in BRCA2-Deficient Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
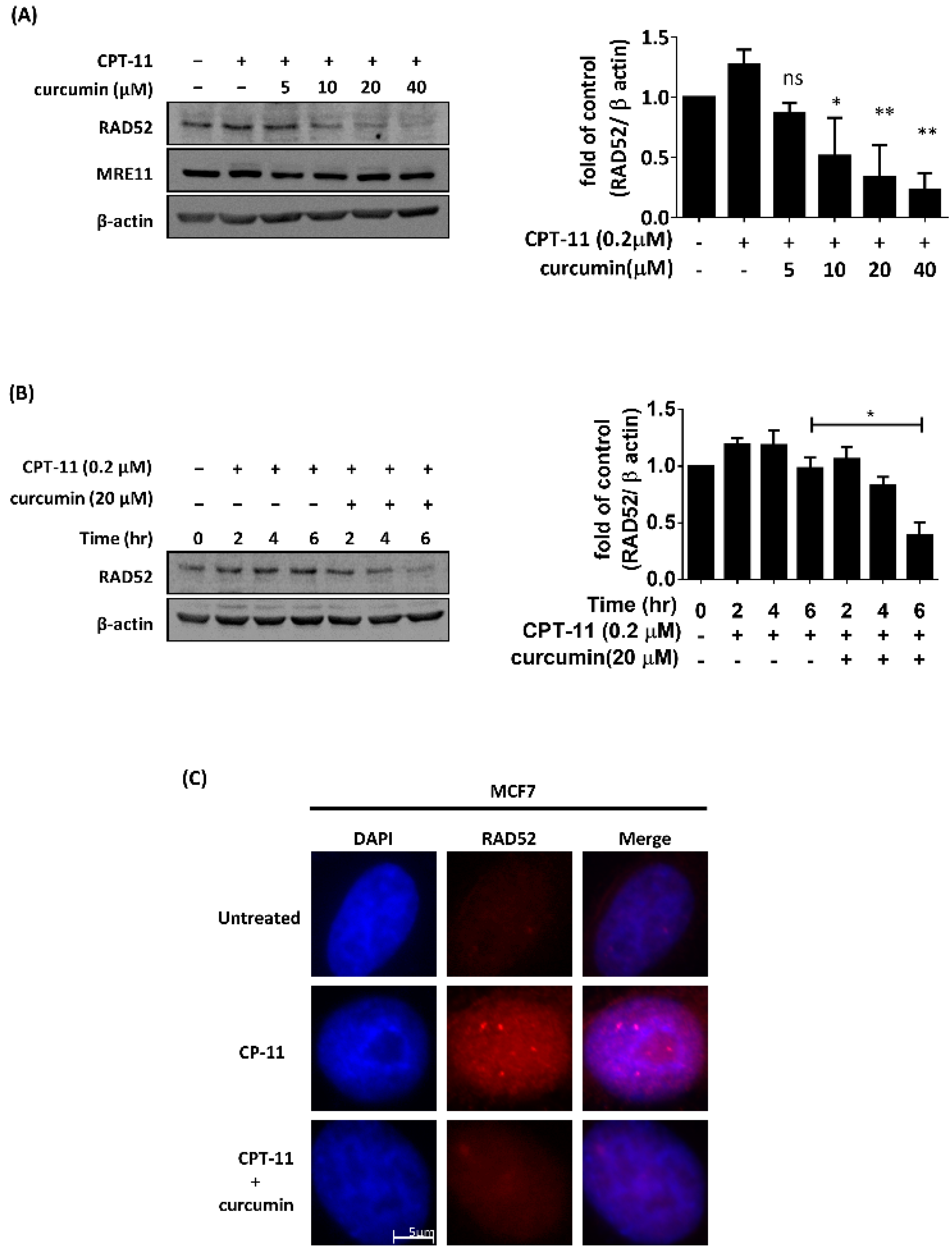
2.1. Curcumin Impaired the Expression and Foci Formation of RAD52 Induced by CPT-11 in MCF7 Cells
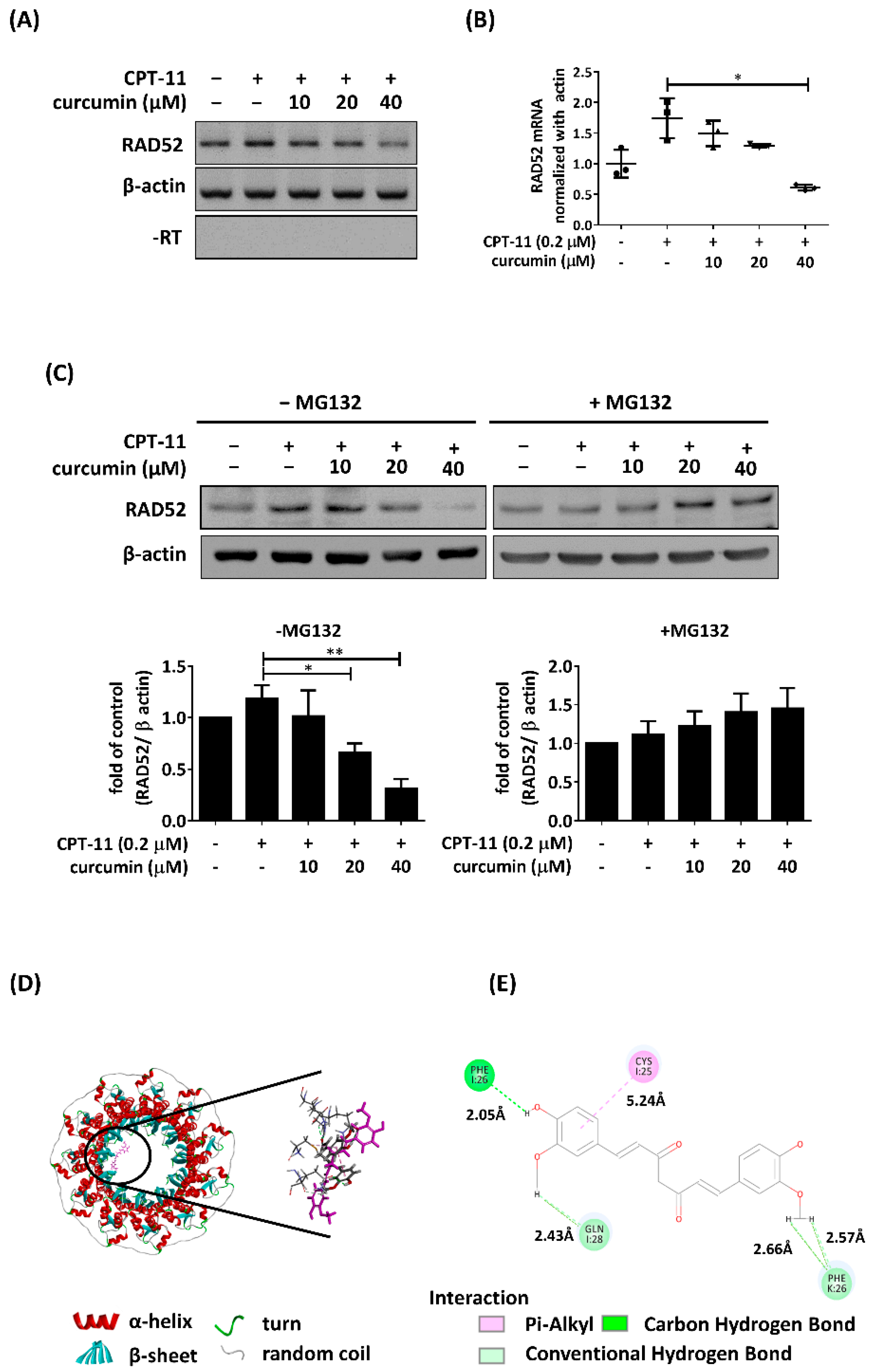
2.2. Curcumin Inhibited the Expression of RAD52 Recombinase Following CPT-11 Treatment
2.3. Curcumin Specifically Sensitized BRCA2-Deficient Cells to CPT-11
2.4. Curcumin Impaired the Repair System of BRCA2-Deficient Cells, Resulting in Genomic Instability
2.5. Curcumin Inhibited HR to Sensitize BRCA2-Deficient Cells
2.6. Curcumin Decreased the Sensitivity to CPT-11 in Capan1 Cells That Overexpress Exogenous BRCA2
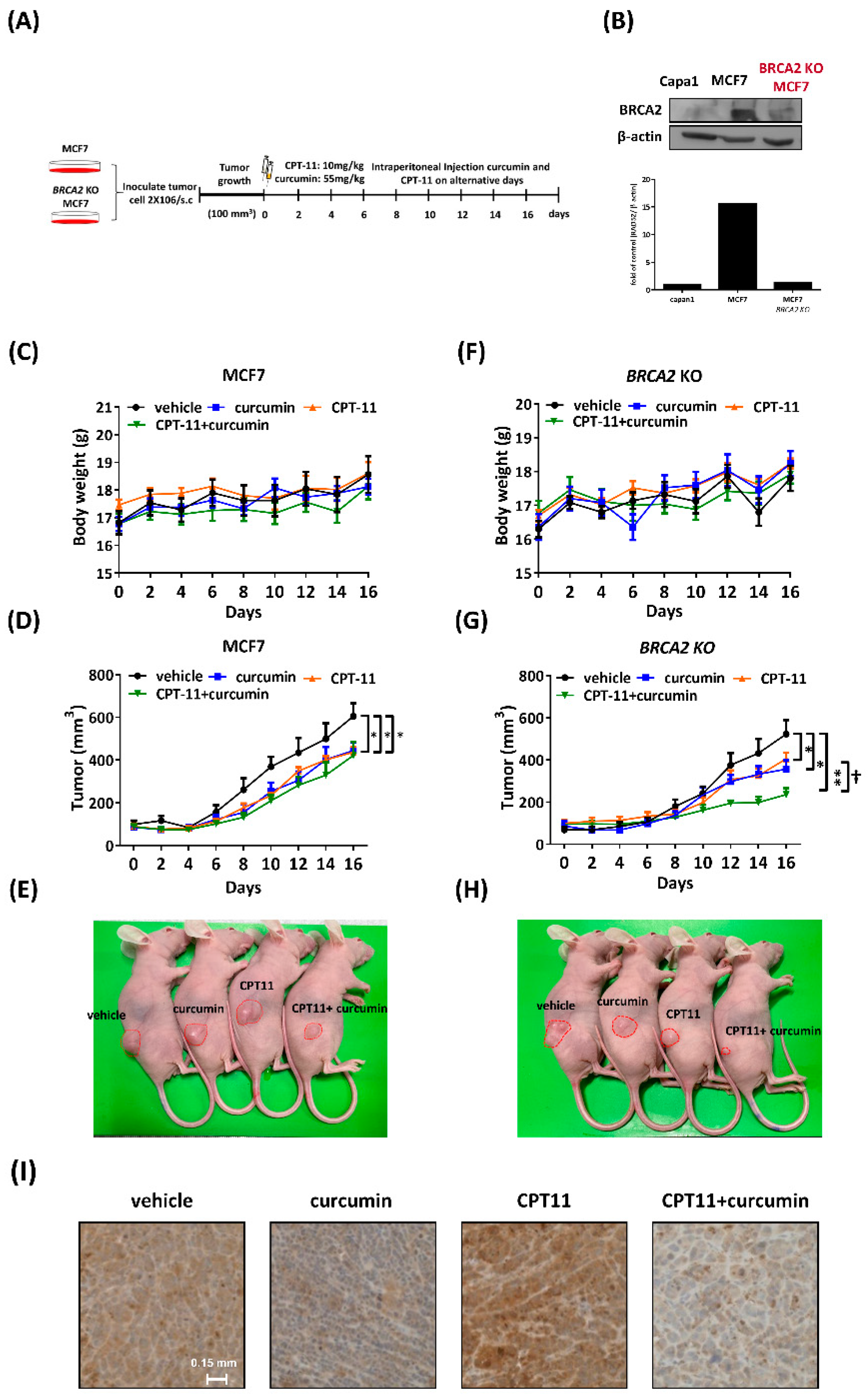
2.7. Curcumin Sensitized BRCA2-Knockout MCF7 Cells to Chemotherapy In Vivo
3. Discussion
4. Materials and Methods
4.1. Drugs, Chemicals, and Plasmids
4.2. Cell Culture and Reagents
4.3. BRCA2-Knockdown Cell Line Generation
4.4. BRCA2 Transfection
4.5. Homologous Recombination Reporter Assay
4.6. Clonogenic Assay
4.7. Immunoblot Analysis
4.8. Immunofluorescence
4.9. Comet Assay
4.10. Apoptosis Assay
4.11. Xenograft Tumor
4.12. Cell Viability Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Barrios-Gonzales, D.; Commo, F.; Olaussen, K.A.; Vagner, S.; Adam, J.; Goubar, A.; Dorvault, N.; Lazar, V.; Job, B.; et al. Molecular Characteristics of ERCC1-Negative versus ERCC1-Positive Tumors in Resected NSCLC. Clin. Cancer Res. 2011, 17, 5562–5572. [Google Scholar] [CrossRef]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Tainer, J.A. The MRE11–RAD50–NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Hengel, S.R.; Spies, M.A. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chem. Biol. 2017, 24, 1101–1119. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef]
- Kang, Z.; Yang, Q.; Li, Y. DNA Repair in Cancer. J. Oncol. 2019, 2019, 8676947. [Google Scholar] [CrossRef]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.D.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nat. Cell Biol. 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer—the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef]
- Hendrickson, E.A. RAD52: Viral Friend or Foe? Cancers 2020, 12, 399. [Google Scholar] [CrossRef] [PubMed]
- Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2010, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Powell, S.N. Molecular Pathways: Understanding the Role of Rad52 in Homologous Recombination for Therapeutic Advancement. Clin. Cancer Res. 2012, 18, 6400–6406. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Nogueira, A.; Fernandes, M.; Catarino, R.; Medeiros, R. RAD52 Functions in Homologous Recombination and Its Importance on Genomic Integrity Maintenance and Cancer Therapy. Cancers 2019, 11, 1622. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Yang, Y.; Wang, G.; Chen, X.; Ju, Y. Curcumin attenuates resistance to irinotecan via induction of apoptosis of cancer stem cells in chemoresistant colon cancer cells. Int. J. Oncol. 2018, 53, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Mortezaee, K.; Goradel, N.H.; Khanlarkhani, N.; Salehi, E.; Nashtaei, M.S.; Najafi, M.; Sahebkar, A. Curcumin as an anti-inflammatory agent: Implications to radiotherapy and chemotherapy. J. Cell. Physiol. 2019, 234, 5728–5740. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, L.-D.; Jia, Y.-G.; Yang, J.; Wang, X.; Liu, C.; Lou, J.-Y. DNA damage response of epithelial ovarian cancer cells (primary culture) to chemo-radiotherapy. J. Sichuan Univ. Med. Sci. Ed. 2014, 45, 185–191. [Google Scholar]
- Lima, C.M.R.; Sherman, C.A.; Brescia, F.J.; Brunson, C.Y.; Green, M.R. Irinotecan/gemcitabine combination chemotherapy in pancreatic cancer. Oncology 2001, 15, 46–51. [Google Scholar]
- Zhao, Q.; Guan, J.; Qin, Y.; Ren, P.; Zhang, Z.; Lv, J.; Sun, S.; Zhang, C.; Mao, W. Curcumin sensitizes lymphoma cells to DNA damage agents through regulating Rad51-dependent homologous recombination. Biomed. Pharmacother. 2018, 97, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.-Y.; Wang, H.-E.; Yu, C.-C.; Liu, H.-C.; Liu, Y.-C.; Chiang, I.-T. Curcumin Triggers DNA Damage and Inhibits Expression of DNA Repair Proteins in Human Lung Cancer Cells. Anticancer Res. 2015, 35, 3867–3873. [Google Scholar]
- Wang, S.-H.; Lin, P.-Y.; Chiu, Y.-C.; Huang, J.-S.; Kuo, Y.-T.; Wu, J.-C.; Chen, C.-C. Curcumin-Mediated HDAC Inhibition Suppresses the DNA Damage Response and Contributes to Increased DNA Damage Sensitivity. PLoS ONE 2015, 10, e0134110. [Google Scholar] [CrossRef]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef]
- Mannava, M.K.C.; Suresh, K.; Bommaka, M.K.; Konga, D.B.; Nangia, A. Curcumin-Artemisinin Coamorphous Solid: Xenograft Model Preclinical Study. Pharmaceutics 2018, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bravo, V.; Carceles-Cordon, M.; Hoshida, V.R.-B.Y.; Cordon-Cardo, C.; Galsky, M.D.; Domingo-Domenech, V.R.-B.M.C.-C.C.C.-C.J. The role of GATA2 in lethal prostate cancer aggressiveness. Nat. Rev. Urol. 2017, 14, 38–48. [Google Scholar] [CrossRef]
- Shah, S.; Prasad, S.; Knudsen, K.E. Targeting Pioneering Factor and Hormone Receptor Cooperative Pathways to Suppress Tumor Progression. Cancer Res. 2012, 72, 1248–1259. [Google Scholar] [CrossRef]
- Fararjeh, A.-F.S.; Tu, S.-H.; Chen, L.-C.; Liu, Y.-R.; Lin, Y.-K.; Chang, H.-L.; Chang, H.-W.; Wu, C.-H.; Hwang-Verslues, W.W.; Ho, Y.-S. The impact of the effectiveness of GATA3 as a prognostic factor in breast cancer. Hum. Pathol. 2018, 80, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Abuzenadah, A.; Al-Saedi, S.; Karim, S.; Al-Qahtani, M. Role of Overexpressed Transcription Factor FOXO1 in Fatal Cardiovascular Septal Defects in Patau Syndrome: Molecular and Therapeutic Strategies. Int. J. Mol. Sci. 2018, 19, 3547. [Google Scholar] [CrossRef] [PubMed]
- Sacher, M.; Pfander, B.; Hoege, C.; Jentsch, S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 2006, 8, 1284–1290. [Google Scholar] [CrossRef]
- Ohuchi, T.; Seki, M.; Branzei, D.; Maeda, D.; Ui, A.; Ogiwara, H.; Tada, S.; Enomoto, T. Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair 2008, 7, 879–889. [Google Scholar] [CrossRef]
- Plate, I.; Albertsen, L.; Lisby, M.; Hallwyl, S.C.; Feng, Q.; Seong, C.; Rothstein, R.; Sung, P.; Mortensen, U.H. Rad52 multimerization is important for its nuclear localization in Saccharomyces cerevisiae. DNA Repair 2008, 7, 57–66. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saotome, M.; Saito, K.; Yasuda, T.; Ohtomo, H.; Sugiyama, S.; Nishimura, Y.; Kurumizaka, H.; Kagawa, W. Structural Basis of Homology-Directed DNA Repair Mediated by RAD52. iScience 2018, 3, 50–62. [Google Scholar] [CrossRef]
- Hanamshet, K.; Mazina, O.M.; Mazin, A.V. Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes 2016, 7, 63. [Google Scholar] [CrossRef]
- Khade, N.V.; Sugiyama, T. Roles of C-Terminal Region of Yeast and Human Rad52 in Rad51-Nucleoprotein Filament Formation and ssDNA Annealing. PLoS ONE 2016, 11, e0158436. [Google Scholar] [CrossRef]
- Kagawa, W.; Arai, N.; Ichikawa, Y.; Saito, K.; Sugiyama, S.; Saotome, M.; Shibata, T.; Kurumizaka, H. Functional analyses of the C-terminal half of the Saccharomyces cerevisiae Rad52 protein. Nucleic Acids Res. 2014, 42, 941–951. [Google Scholar] [CrossRef]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar] [CrossRef] [PubMed]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Q.; Zhang, Y.; Huang, K.; Sun, R.; Zhao, Q. Compound F779-0434 causes synthetic lethality in BRCA2-deficient cancer cells by disrupting RAD52–ssDNA association. RSC Adv. 2018, 8, 18859–18869. [Google Scholar] [CrossRef]
- Leu, Y.-L.; Wang, T.-H.; Wu, C.-C.; Huang, K.-Y.; Jiang, Y.-W.; Hsu, Y.-C.; Chen, C.-Y. Hydroxygenkwanin Suppresses Non-Small Cell Lung Cancer Progression by Enhancing EGFR Degradation. Molecules 2020, 25, 941. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, W.-C.; Chen, C.-Y.; Chern, C.-Y.; Wang, C.-A.; Lee, W.-C.; Chi, Y.-C.; Cheng, S.-F.; Kuo, Y.-T.; Chiu, Y.-C.; Tseng, S.-T.; et al. Targeting HR Repair as a Synthetic Lethal Approach to Increase DNA Damage Sensitivity by a RAD52 Inhibitor in BRCA2-Deficient Cancer Cells. Int. J. Mol. Sci. 2021, 22, 4422. https://doi.org/10.3390/ijms22094422
Tseng W-C, Chen C-Y, Chern C-Y, Wang C-A, Lee W-C, Chi Y-C, Cheng S-F, Kuo Y-T, Chiu Y-C, Tseng S-T, et al. Targeting HR Repair as a Synthetic Lethal Approach to Increase DNA Damage Sensitivity by a RAD52 Inhibitor in BRCA2-Deficient Cancer Cells. International Journal of Molecular Sciences. 2021; 22(9):4422. https://doi.org/10.3390/ijms22094422
Chicago/Turabian StyleTseng, Wei-Che, Chi-Yuan Chen, Ching-Yuh Chern, Chu-An Wang, Wen-Chih Lee, Ying-Chih Chi, Shu-Fang Cheng, Yi-Tsen Kuo, Ya-Chen Chiu, Shih-Ting Tseng, and et al. 2021. "Targeting HR Repair as a Synthetic Lethal Approach to Increase DNA Damage Sensitivity by a RAD52 Inhibitor in BRCA2-Deficient Cancer Cells" International Journal of Molecular Sciences 22, no. 9: 4422. https://doi.org/10.3390/ijms22094422
APA StyleTseng, W.-C., Chen, C.-Y., Chern, C.-Y., Wang, C.-A., Lee, W.-C., Chi, Y.-C., Cheng, S.-F., Kuo, Y.-T., Chiu, Y.-C., Tseng, S.-T., Lin, P.-Y., Liou, S.-J., Li, Y.-C., & Chen, C.-C. (2021). Targeting HR Repair as a Synthetic Lethal Approach to Increase DNA Damage Sensitivity by a RAD52 Inhibitor in BRCA2-Deficient Cancer Cells. International Journal of Molecular Sciences, 22(9), 4422. https://doi.org/10.3390/ijms22094422