
Regulatory Light Chains in Cardiac Development and Disease


Abstract
1. Introduction
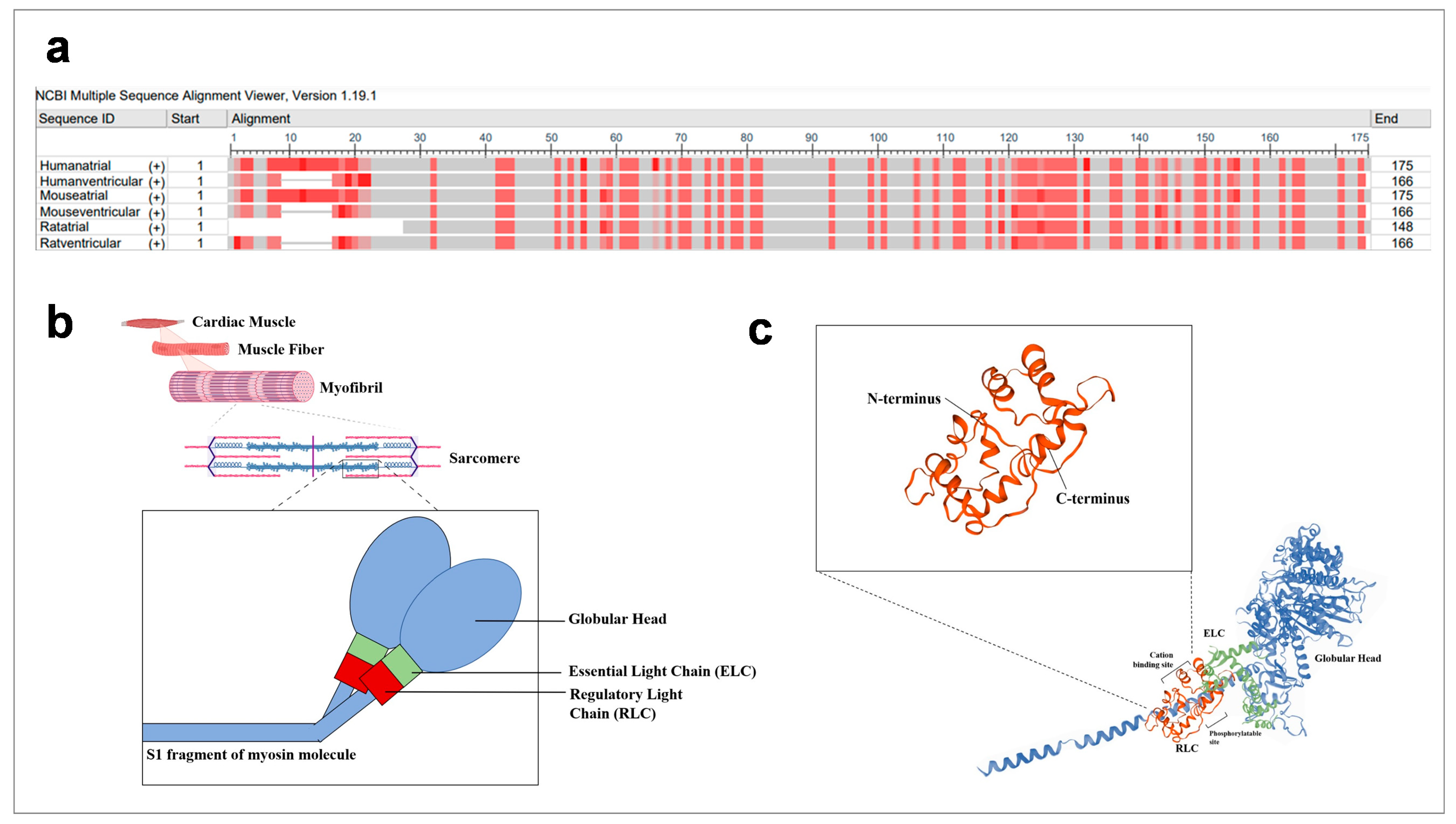
2. Structure of Regulatory Light Chains
3. Regulatory Light Chains in Cardiac Development
4. The Role of Regulatory Light Chains in Normal and Diseased Hearts
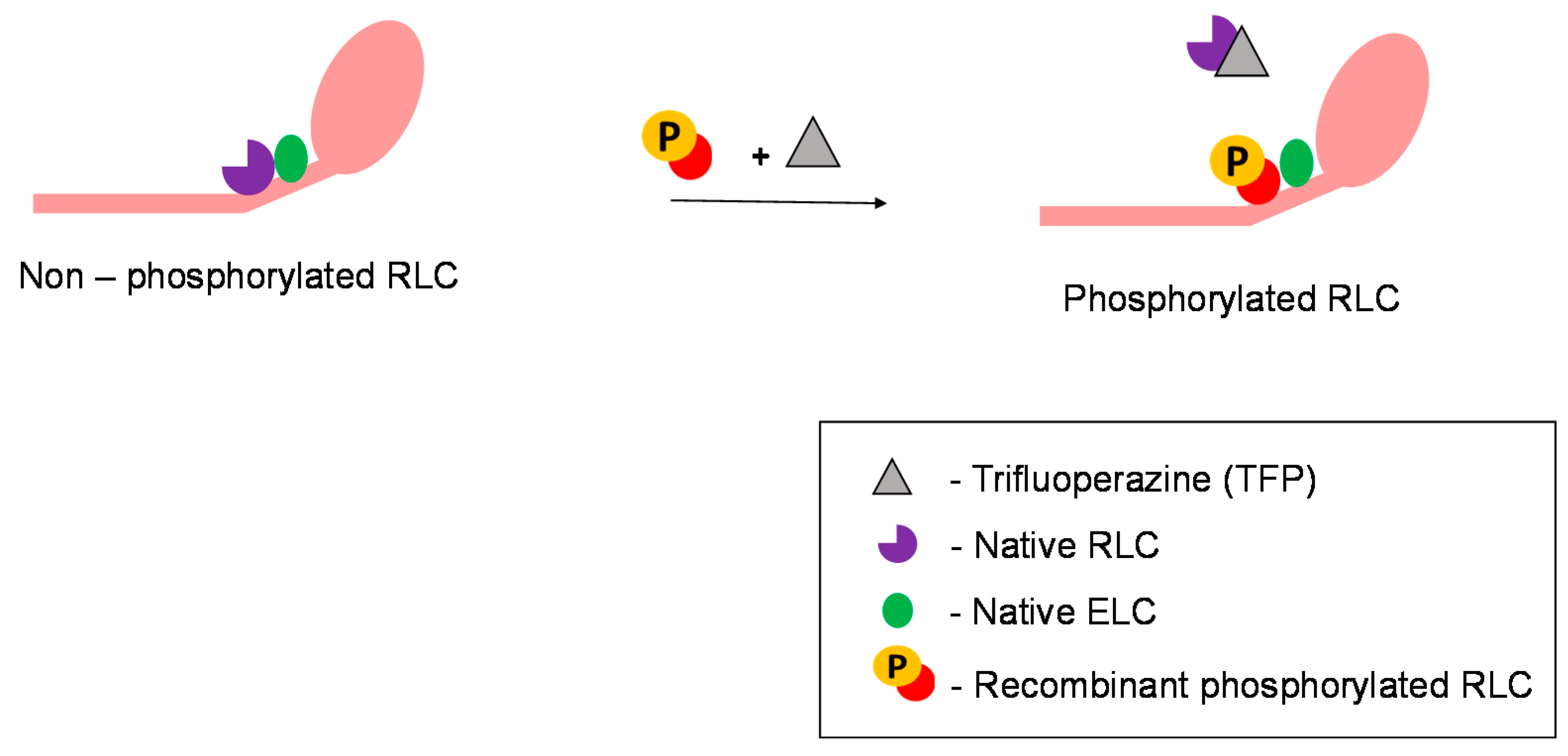
5. Experimental Strategies to Study and Exploit the Use of Regulatory Light Chains
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Toepfer, C.; Caorsi, V.; Kampourakis, T.; Sikkel, M.B.; West, T.G.; Leung, M.C.; Al-Saud, S.A.; MacLeod, K.T.; Lyon, A.R.; Marston, S.B.; et al. Myosin regulatory light chain (RLC) phosphory-lation change as a modulator of cardiac muscle contraction in disease. J. Biol. Chem. 2013, 288, 13446–13454. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chakravorty, S.; Song, W.; Ferenczi, M.A. Phosphorylation of the regulatory light chain of myosin in striated muscle: Methodological perspectives. Eur. Biophys. J. 2016, 45, 779–805. [Google Scholar] [CrossRef] [PubMed]
- Scruggs, S.B.; Solaro, R.J. The significance of regulatory light chain phosphorylation in cardiac physiology. Arch. Biochem. Biophys. 2011, 510, 129–134. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Rayment, I.; Rypniewski, W.R.; Schmidt-Base, K.; Smith, R.; Tomchick, D.R.; Benning, M.M.; Winkelmann, D.A.; Wesenberg, G.; Holden, H.M. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science 1993, 261, 50–58. [Google Scholar] [CrossRef]
- Holroyde, M.J.; Potter, J.D.; Solaro, R.J. The calcium binding properties of phosphorylated and unphosphorylated cardiac and skeletal myosins. J. Biol. Chem. 1979, 254, 6478–6482. [Google Scholar] [CrossRef]
- da Silva, A.C.; Kendrick-Jones, J.; Reinach, F.C. Determinants of ion specificity on EF-hands sites. Conversion of the Ca2+/Mg2+ site of smooth muscle myosin regulatory light chain into a Ca(2+)-specific site. J. Biol. Chem. 1995, 270, 6773–6778. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, C.R. The location of the divalent metal binding sites and the light chain subunits of vertebrate myosin. Biochemistry 1977, 16, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Szczesna, D. Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2003, 3, 187–197. [Google Scholar]
- Szczesna, D.; Zhao, J.; Jones, M.; Zhi, G.; Stull, J.; Potter, J.D. Phosphorylation of the regulatory light chains of myosin affects Ca2+sensitivity of skeletal muscle contraction. J. Appl. Physiol. 2002, 92, 1661–1670. [Google Scholar] [CrossRef]
- Janssen, P.M.L. Kinetics of cardiac muscle contraction and relaxation are linked and determined by properties of the cardiac sarcomere. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1092–H1099. [Google Scholar] [CrossRef] [PubMed]
- Grabarek, Z. Insights into modulation of calcium signaling by magnesium in calmodulin, troponin C and related EF-hand proteins. Biochim. Biophys. Acta 2011, 1813, 913–921. [Google Scholar] [CrossRef]
- Greenberg, M.J.; Watt, J.D.; Jones, M.; Kazmierczak, K.; Szczesna-Cordary, D.; Moore, J.R. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J. Mol. Cell. Cardiol. 2009, 46, 108–115. [Google Scholar] [CrossRef]
- Reinach, F.C.; Nagai, K.; Kendrick-Jones, J. Site-directed mutagenesis of the regulatory light-chain Ca2+/Mg2+ binding site and its role in hybrid myosins. Nat. Cell Biol. 1986, 322, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Scruggs, S.B.; Reisdorph, R.; Armstrong, M.L.; Warren, C.M.; Reisdorph, N.; Solaro, R.J.; Buttrick, P.M. A Novel, In-solution Separation of Endogenous Cardiac Sarcomeric Proteins and Identification of Distinct Charged Variants of Regulatory Light Chain. Mol. Cell. Proteom. 2010, 9, 1804–1818. [Google Scholar] [CrossRef]
- Pasparakis, G.; Krasnogor, N.; Cronin, L.; Davis, B.G.; Alexander, C.; Pasparakis, G. Controlled polymer synthesis—from biomimicry towards synthetic biology. Chem. Soc. Rev. 2009, 39, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, K.; Liang, J.; Yuan, C.-C.; Yadav, S.; Sitbon, Y.H.; Walz, K.; Ma, W.; Irving, T.C.; Cheah, J.X.; Gomes, A.V.; et al. Slow-twitch skeletal muscle defects accompany cardiac dysfunction in transgenic mice with a mutation in the myosin regulatory light chain. FASEB J. 2018, 33, 3152–3166. [Google Scholar] [CrossRef] [PubMed]
- Kubalak, S.W.; Miller-Hance, W.C.; O’Brien, T.X.; Dyson, E.; Chien, K.R. Chamber specification of atrial myosin light chain-2 expression precedes septation during murine cardiogenesis. J. Biol. Chem. 1994, 269, 16961–16970. [Google Scholar] [CrossRef]
- England, J.; Loughna, S. Heavy and light roles: Myosin in the morphogenesis of the heart. Cell. Mol. Life Sci. 2012, 70, 1221–1239. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Hassel, D.; Burns, C.G.; Katus, H.A.; Fishman, M.C. Cardiac myosin light chain-2: A novel essential component of thick-myofilament assembly and contractility of the heart. Circ. Res. 2006, 99, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Markman, M.M.W.; Wagenaar, G.T.M.; Ya, J.; Lamers, W.H.; Moorman, A.F.M. Myosin light chain 2a and 2v identifies the embryonic outflow tract myocardium in the developing rodent heart. Anat. Rec. 1999, 254, 135–146. [Google Scholar] [CrossRef]
- Franz, W.M.; Breves, D.; Klingel, K.; Brem, G.; Hofschneider, P.H.; Kandolf, R. Heart-specific targeting of firefly luciferase by the myosin light chain-2 promoter and developmental regulation in transgenic mice. Circ. Res. 1993, 73, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Franz, W.M.; Brem, G.; A Katus, H.; Klingel, K.; Hofschneider, P.H.; Kandolf, R. Characterization of a cardiac-selective and developmentally upregulated promoter in transgenic mice. Cardioscience 1994, 5, 235–243. [Google Scholar]
- Sedmera, D.; McQuinn, T. Embryogenesis of the Heart Muscle. Heart Fail. Clin. 2008, 4, 235–245. [Google Scholar] [CrossRef]
- Tan, C.M.J.; Lewandowski, A.J. The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagn. Ther. 2019, 47, 373–386. [Google Scholar] [CrossRef]
- Jeewandara, T. Visualizing Embryonic Heart Formation in Real Time: A Technical Summary of Imaging Early Cardiogenesis; Academic, Research Foundation of the City University of New York: New York, NY, USA, 2017. [Google Scholar]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.-L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef]
- Shahbazi, M.N. Mechanisms of human embryo development: From cell fate to tissue shape and back. Development 2020, 147, dev190629. [Google Scholar] [CrossRef]
- Kojima, Y.; Tam, O.H.; Tam, P.P. Timing of developmental events in the early mouse embryo. Semin. Cell Dev. Biol. 2014, 34, 65–75. [Google Scholar] [CrossRef]
- Desgrange, A.; Le Garrec, J.-F.; Meilhac, S.M. Left-right asymmetry in heart development and disease: Forming the right loop. Development 2018, 145, dev162776. [Google Scholar] [CrossRef]
- Savolainen, S.M.; Foley, J.F.; Elmore, S.A. Histology Atlas of the Developing Mouse Heart with Emphasis on E11.5 to E18.5. Toxicol. Pathol. 2009, 37, 395–414. [Google Scholar] [CrossRef]
- Miller-Hance, W.C.; LaCorbiere, M.; Fuller, S.J.; Evans, S.M.; Lyons, G.; Schmidt, C.; Robbins, J.; Chien, K.R. In vitro chamber specification during embryonic stem cell cardiogenesis. Expression of the ventricular myosin light chain-2 gene is independent of heart tube formation. J. Biol. Chem. 1993, 268, 25244–25252. [Google Scholar] [CrossRef]
- O’Brien, T.X.; Lee, K.J.; Chien, K.R. Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. Proc. Natl. Acad. Sci. USA 1993, 90, 5157–5161. [Google Scholar] [CrossRef] [PubMed]
- Chien, K.R.; Zhu, H.; Knowlton, K.U.; Miller-Hance, W.; van-Bilsen, M.; O’Brien, T.X.; Evans, S.M. Transcriptional regulation during cardiac growth and development. Annu. Rev. Physiol. 1993, 55, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kubalak, S.W.; Minamisawa, S.; Price, R.L.; Becker, K.D.; Hickey, R.; Ross, J.; Chien, K.R. Selective Requirement of Myosin Light Chain 2v in Embryonic Heart Function. J. Biol. Chem. 1998, 273, 1252–1256. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, F.; Lyon, R.C.; Chen, J. Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene 2015, 569, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, H.; Wang, X.; Zhan, Y.; Sheng, W.; Cai, H.; Xin, H.; Liang, Q.; Zhou, P.; Lu, C.; et al. Engineering human ventricular heart muscles based on a highly efficient system for purification of human pluripotent stem cell-derived ventricular cardiomyocytes. Stem Cell Res. Ther. 2017, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Sheikh, F.; Hollander, M.; Cai, C.; Becker, D.; Chu, P.-H.; Evans, S.; Chen, J. Embryonic atrial function is essential for mouse embryogenesis, cardiac morphogenesis and angiogenesis. Development 2003, 130, 6111–6119. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, W.; Dahme, T.; Rottbauer, W.; Ackerman, M.J.; Xu, X. Depletion of zebrafish essential and regulatory myosin light chains reduces cardiac function through distinct mechanisms. Cardiovasc. Res. 2008, 79, 97–108. [Google Scholar] [CrossRef]
- Yelon, D.; Horne, S.A.; Stainier, D.Y. Restricted Expression of Cardiac Myosin Genes Reveals Regulated Aspects of Heart Tube Assembly in Zebrafish. Dev. Biol. 1999, 214, 23–37. [Google Scholar] [CrossRef]
- Shimada, E.; Kinoshita, M.; Murata, K. Expression of cardiac myosin light chain 2 during embryonic heart development in medaka fish, Oryzias latipes, and phylogenetic relationship with other myosin light chains. Dev. Growth Differ. 2008, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ghatpande, S.; Shafiq, S.; Siddiqui, M. Ventricular Myosin Light Chain-2 Gene Expression in Developing Heart of Chicken Embryos. Biol. Res. 2001, 34, 1–6. [Google Scholar] [CrossRef]
- Gruber, P.J.; Kubalak, S.W.; Chien, K.R. Downregulation of atrial markers during cardiac chamber morphogenesis is irreversible in murine embryos. Development 1998, 125, 4427–4438. [Google Scholar]
- Dyson, E.; Sucov, H.M.; Kubalak, S.W.; Schmid-Schönbein, G.W.; DeLano, F.A.; Evans, R.M.; Ross, J., Jr.; Chien, K.R. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor alpha -/- mice. Proc. Natl. Acad. Sci. USA 1995, 92, 7386–7390. [Google Scholar] [CrossRef]
- Drakhlis, L.; Biswanath, S.; Farr, C.-M.; Lupanow, V.; Teske, J.; Ritzenhoff, K.; Franke, A.; Manstein, F.; Bolesani, E.; Kempf, H.; et al. Human heart-forming organoids recapitulate early heart and foregut development. Nat. Biotechnol. 2021, 1–10. [Google Scholar] [CrossRef]
- Zhang, L.; Nomura-Kitabayashi, A.; Sultana, N.; Cai, W.; Cai, X.; Moon, A.M.; Cai, C.-L. Mesodermal Nkx2.5 is necessary and sufficient for early second heart field development. Dev. Biol. 2014, 390, 68–79. [Google Scholar] [CrossRef]
- Terrak, M.; Wu, G.; Stafford, W.F.; Lu, R.C.; Domínguez, R. Two distinct myosin light chain structures are induced by specific variations within the bound IQ motifs—functional implications. EMBO J. 2003, 22, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Margossian, S.S.; Slayter, H.S. Electron microscopy of cardiac myosin: Its shape and properties as determined by the regulatory light chain. J. Muscle Res. Cell Motil. 1987, 8, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Watt, J.; Greenberg, M.; Jones, M.; Szczesna-Cordary, D.; Moore, J.R. Removal of the cardiac myosin regulatory light chain increases isometric force production. FASEB J. 2009, 23, 3571–3580. [Google Scholar] [CrossRef] [PubMed]
- A Hofmann, P.; Metzger, J.M.; Greaser, M.L.; Moss, R.L. Effects of partial extraction of light chain 2 on the Ca2+ sensitivities of isometric tension, stiffness, and velocity of shortening in skinned skeletal muscle fibers. J. Gen. Physiol. 1990, 95, 477–498. [Google Scholar] [CrossRef]
- Moss, R.L.; Giulian, G.G.; Greaser, M.L. Physiological effects accompanying the removal of myosin LC2 from skinned skeletal muscle fibers. J. Biol. Chem. 1982, 257, 8588–8591. [Google Scholar] [CrossRef]
- Szczesna, D.; Zhao, J.; Potter, J.D. The Regulatory Light Chains of Myosin Modulate Cross-bridge Cycling in Skeletal Muscle. J. Biol. Chem. 1996, 271, 5246–5250. [Google Scholar] [CrossRef] [PubMed]
- Corrie, J.E.T.; Brandmeier, B.D.; Ferguson, R.E.; Trentham, D.R.; Kendrick-Jones, J.; Hopkins, S.C.; Van Der Heide, U.A.; Goldman, Y.E.; Sabido-David, C.; Dale, R.E.; et al. Dynamic measurement of myosin light-chain-domain tilt and twist in muscle contraction. Nat. Cell Biol. 1999, 400, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Kaźmierczak, K.; Zhou, Z.; Aguiar-Pulido, V.; Narasimhan, G.; Szczesna-Cordary, D. Gene expression patterns in transgenic mouse models of hypertrophic cardiomyopathy caused by mutations in myosin regulatory light chain. Arch. Biochem. Biophys. 2016, 601, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Szczesna-Cordary, D.; Guzman, G.; Zhao, J.; Hernandez, O.; Wei, J.; Diaz-Perez, Z. The E22K mutation of myosin RLC that causes familial hypertrophic cardiomyopathy increases calcium sensitivity of force and ATPase in transgenic mice. J. Cell Sci. 2005, 118, 3675–3683. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, W.; Szczesna-Cordary, D. Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations. J. Muscle Res. Cell Motil. 2015, 36, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, K.; Muthu, P.; Huang, W.; Jones, M.; Wang, Y.; Szczesna-Cordary, D. Myosin regulatory light chain mutation found in hypertrophic cardiomyopathy patients increases isometric force production in transgenic mice. Biochem. J. 2012, 442, 95–103. [Google Scholar] [CrossRef]
- Abraham, T.P.; Jones, M.; Kazmierczak, K.; Liang, H.-Y.; Pinheiro, A.C.; Wagg, C.S.; Lopaschuk, G.D.; Szczesna-Cordary, D. Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice. Cardiovasc. Res. 2009, 82, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.J.; Kazmierczak, K.; Szczesna-Cordary, D.; Moore, J.R. Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc. Natl. Acad. Sci. USA 2010, 107, 17403–17408. [Google Scholar] [CrossRef]
- Granzier, H.L.; de Tombe, P.P. Myosin light chain phosphorylation to the rescue. Proc. Natl. Acad. Sci. USA 2015, 112, 9148–9149. [Google Scholar] [CrossRef] [PubMed]
- Kerrick, W.G.L.; Kazmierczak, K.; Xu, Y.; Wang, Y.; Szczesna-Cordary, D. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J. 2008, 23, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.-C.; Kazmierczak, K.; Liang, J.; Zhou, Z.; Yadav, S.; Gomes, A.V.; Irving, T.C.; Szczesna-Cordary, D. Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2338–E2347. [Google Scholar] [CrossRef] [PubMed]
- Kampourakis, T.; Ponnam, S.; Irving, M. Hypertrophic cardiomyopathy mutation R58Q in the myosin regulatory light chain perturbs thick filament-based regulation in cardiac muscle. J. Mol. Cell. Cardiol. 2018, 117, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, H.L.; Yang, Z.; Zhi, G.; Stull, J.T.; Trybus, K.M. Charge replacement near the phosphorylatable serine of the myosin regulatory light chain mimics aspects of phosphorylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1490–1494. [Google Scholar] [CrossRef]
- Metzger, J.M.; Greaser, M.L.; Moss, R.L. Variations in cross-bridge attachment rate and tension with phosphorylation of myosin in mammalian skinned skeletal muscle fibers. Implications for twitch potentiation in intact muscle. J. Gen. Physiol. 1989, 93, 855–883. [Google Scholar] [CrossRef] [PubMed]
- Kampourakis, T.; Irving, M. Phosphorylation of myosin regulatory light chain controls myosin head conformation in cardiac muscle. J. Mol. Cell. Cardiol. 2015, 85, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.S.; Hassanzadeh, S.; Winitsky, S.; Lin, H.; Satorius, C.; Vemuri, R.; Aletras, A.H.; Wen, H.; Epstein, N.D. The Overall Pattern of Cardiac Contraction Depends on a Spatial Gradient of Myosin Regulatory Light Chain Phosphorylation. Cell 2001, 107, 631–641. [Google Scholar] [CrossRef]
- Van der Velden, J.; Stienen, G.J.M. Cardiac Disorders and Pathophysiology of Sarcomeric Proteins. Physiol. Rev. 2019, 99, 381–426. [Google Scholar] [CrossRef]
- Davis, J.; Hassanzadeh, S.; Winitsky, S.; Wen, H.; Aletras, A.; Epstein, N. A Gradient of Myosin Regulatory Light-chain Phosphorylation across the Ventricular Wall Supports Cardiac Torsion. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 345–352. [Google Scholar] [CrossRef]
- Huang, J.; Shelton, J.M.; Richardson, J.A.; Kamm, K.E.; Stull, J.T. Myosin Regulatory Light Chain Phosphorylation Attenuates Cardiac Hypertrophy. J. Biol. Chem. 2008, 283, 19748–19756. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Signaling to Myosin Regulatory Light Chain in Sarcomeres. J. Biol. Chem. 2011, 286, 9941–9947. [Google Scholar] [CrossRef] [PubMed]
- Morano, I. Tuning the human heart molecular motors by myosin light chains. J. Mol. Med. 1999, 77, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Noland, T.; Kuo, J. Phosphorylation of Cardiac Myosin Light Chain 2 by Protein Kinase C and Myosin Light Chain Kinase Increases Ca2+-Stimulated Actomyosin MgATPase Activity. Biochem. Biophys. Res. Commun. 1993, 193, 254–260. [Google Scholar] [CrossRef]
- Sweeney, H.L.; Stull, J.T. Phosphorylation of myosin in permeabilized mammalian cardiac and skeletal muscle cells. Am. J. Physiol. Physiol. 1986, 250, C657–C660. [Google Scholar] [CrossRef]
- Colson, B.A.; Locher, M.R.; Bekyarova, T.; Patel, J.R.; Fitzsimons, D.P.; Irving, T.C.; Moss, R.L. Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J. Physiol. 2010, 588, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ajtai, K.; Burghardt, T.P. Ventricular myosin modifies in vitro step-size when phosphorylated. J. Mol. Cell. Cardiol. 2014, 72, 231–237. [Google Scholar] [CrossRef]
- Rayment, I.; Holden, H.M.; Whittaker, M.; Yohn, C.B.; Lorenz, M.; Holmes, K.C.; Milligan, R.A. Structure of the actin-myosin complex and its implications for muscle contraction. Science 1993, 261, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Freyzon, Y.; Trybus, K.M.; Cohen, C. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: Visualization of the pre-power stroke state. Cell 1998, 94, 559–571. [Google Scholar] [CrossRef]
- Stelzer, J.E.; Patel, J.R.; Moss, R.L. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J. Gen. Physiol. 2006, 128, 261–272. [Google Scholar] [CrossRef]
- Dias, F.A.; Walker, L.A.; Arteaga, G.M.; Walker, J.S.; Vijayan, K.; Peña, J.R.; Ke, Y.; Fogaca, R.T.; Sanbe, A.; Robbins, J.; et al. The effect of myosin regulatory light chain phosphorylation on the frequency-dependent regulation of cardiac function. J. Mol. Cell. Cardiol. 2006, 41, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Huang, J.; Battiprolu, P.K.; Hill, J.A.; Kamm, K.E.; Stull, J.T. Cardiac Myosin Light Chain Kinase is Essential for Myosin Regulatory Light Chain Phosphorylation and Normal Cardiac Function in vivo. Biophys. J. 2011, 100, 369a. [Google Scholar] [CrossRef][Green Version]
- Yadav, S.; Szczesna-Cordary, D. Pseudophosphorylation of cardiac myosin regulatory light chain: A promising new tool for treatment of cardiomyopathy. Biophys. Rev. 2017, 9, 57–64. [Google Scholar] [CrossRef]
- van der Velden, J.; Papp, Z.; Zaremba, R.; Boontje, N.M.; de Jong, J.W.; Owen, V.J.; Burton, P.B.J.; Goldmann, P.; Jaquet, K.; Stienen, G.J.M. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc. Res. 2003, 57, 37–47. [Google Scholar] [CrossRef]
- van der Velden, J.; Papp, Z.; Boontje, N.M.; Zaremba, R.; de Jong, J.W.; Janssen, P.M.L.; Hasenfuss, G.; Stienen, G.J.M. The effect of myosin light chain 2 dephosphorylation on Ca2+-sensitivity of force is enhanced in failing human hearts. Cardiovasc. Res. 2003, 57, 505–514. [Google Scholar] [CrossRef]
- Toepfer, C.N.; Sikkel, M.B.; Caorsi, V.; Vydyanath, A.; Torre, I.; Copeland, O.N.; Lyon, A.R.; Marston, S.B.; Luther, P.K.; Macleod, K.T.; et al. A post-MI power struggle: Adaptations in cardiac power occur at the sarcomere level alongside MyBP-C and RLC phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H465–H475. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Akopian, G.; Jinadasa, P.; Gluckman, T.; Terhakopian, A.; Massey, B.; Bing, R. Myocardial Infarction and Regulatory Myosin Light Chain. J. Mol. Cell. Cardiol. 1997, 29, 2641–2652. [Google Scholar] [CrossRef]
- Breithaupt, J.J.; Pulcastro, H.C.; Awinda, P.O.; DeWitt, D.C.; Tanner, B.C. Regulatory light chain phosphorylation augments length-dependent contraction in PTU-treated rats. J. Gen. Physiol. 2018, 151, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Szczesna, D.; Ghosh, D.; Li, Q.; Gomes, A.V.; Guzman, G.; Arana, C.; Zhi, G.; Stull, J.T.; Potter, J.D. Familial Hypertrophic Cardiomyopathy Mutations in the Regulatory Light Chains of Myosin Affect Their Structure, Ca2+Binding, and Phosphorylation. J. Biol. Chem. 2001, 276, 7086–7092. [Google Scholar] [CrossRef]
- van der Velden, J.; Merkus, D.; de Beer, V.; Hamdani, N.; Linke, W.A.; Boontje, N.M.; Stienen, G.J.M.; Duncker, D.J. Transmural heterogeneity of myofilament function and sarcomeric protein phosphorylation in remodeled myocardium of pigs with a recent myocardial infarction. Front. Physiol. 2011, 2, 83. [Google Scholar] [CrossRef] [PubMed]
- Ait Mou, Y.; Toth, A.; Cassan, C.; Czuriga, D.; de Tombe, P.P.; Papp, Z.; Lacampagne, A.; Cazorla, O. Beneficial effects of SR33805 in failing myocardium. Cardiovasc. Res. 2011, 91, 412–419. [Google Scholar] [CrossRef]
- Scruggs, S.B.; Hinken, A.C.; Thawornkaiwong, A.; Robbins, J.; Walker, L.A.; de Tombe, P.P.; Geenen, D.L.; Buttrick, P.M.; Solaro, R.J. Ablation of Ventricular Myosin Regulatory Light Chain Phosphorylation in Mice Causes Cardiac Dysfunction in Situ and Affects Neighboring Myofilament Protein Phosphorylation. J. Biol. Chem. 2009, 284, 5097–5106. [Google Scholar] [CrossRef]
- Chang, A.N.; Battiprolu, P.K.; Cowley, P.M.; Chen, G.; Gerard, R.D.; Pinto, J.R.; Hill, J.A.; Baker, A.J.; Kamm, K.E.; Stull, J.T. Constitutive Phosphorylation of Cardiac Myosin Regulatory Light Chain in Vivo. J. Biol. Chem. 2015, 290, 10703–10716. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.C.; Patel, J.R.; Fitzsimons, D.P.; Walker, J.W.; Moss, R.L. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2712–H2718. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.K.; Zhi, G.; Stull, J.T.; Trewhella, J. Neutron-Scattering Studies Reveal Further Details of the Ca2+/Calmodulin-Dependent Activation Mechanism of Myosin Light Chain Kinase†. Biochemistry 1998, 37, 13997–14004. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Dedicated Myosin Light Chain Kinases with Diverse Cellular Functions. J. Biol. Chem. 2001, 276, 4527–4530. [Google Scholar] [CrossRef] [PubMed]
- Sevrieva, I.R.; Brandmeier, B.; Ponnam, S.; Gautel, M.; Irving, M.; Campbell, K.S.; Sun, Y.-B.; Kampourakis, T. Cardiac myosin regulatory light chain kinase modulates cardiac contractility by phosphorylating both myosin regulatory light chain and troponin I. J. Biol. Chem. 2020, 295, 4398–4410. [Google Scholar] [CrossRef]
- Chang, A.N.; Chen, G.; Gerard, R.D.; Kamm, K.E.; Stull, J.T. Cardiac Myosin Is a Substrate for Zipper-interacting Protein Kinase (ZIPK). J. Biol. Chem. 2010, 285, 5122–5126. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Surks, H.K.; Mochizuki, S.; Mochizuki, N.; Mendelsohn, M.E. Identification and Characterization of Zipper-interacting Protein Kinase as the Unique Vascular Smooth Muscle Myosin Phosphatase-associated Kinase. J. Biol. Chem. 2004, 279, 42055–42061. [Google Scholar] [CrossRef]
- Haystead, T.A. ZIP kinase, a key regulator of myosin protein phosphatase 1. Cell. Signal. 2005, 17, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Hagerty, L.; Weitzel, D.H.; Chambers, J.; Fortner, C.N.; Brush, M.H.; Loiselle, D.; Hosoya, H.; Haystead, T.A.J. ROCK1 Phosphorylates and Activates Zipper-interacting Protein Kinase. J. Biol. Chem. 2007, 282, 4884–4893. [Google Scholar] [CrossRef] [PubMed]
- Rajashree, R.; Blunt, B.C.; Hofmann, P.A. Modulation of myosin phosphatase targeting subunit and protein phosphatase 1 in the heart. Am. J. Physiol. Circ. Physiol. 2005, 289, H1736–H1743. [Google Scholar] [CrossRef]
- Venema, R.C.; Raynor, R.L.; A Noland, T.; Kuo, J.F. Role of protein kinase C in the phosphorylation of cardiac myosin light chain 2. Biochem. J. 1993, 294, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Eikemo, H.; Moltzau, L.R.; Hussain, R.I.; Nguyen, C.H.; Qvigstad, E.; Levy, F.O.; Skomedal, T.; Osnes, J.-B. CaMKII in addition to MLCK contributes to phosphorylation of regulatory light chain in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 471, 219–225. [Google Scholar] [CrossRef]
- Goshe, M.B. Characterizing phosphoproteins and phosphoproteomes using mass spectrometry. Briefings Funct. Genom. Proteom. 2006, 4, 363–376. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kinoshita-Kikuta, E.; Kinoshita, E.; Koike, T. Phos-tag beads as an immunoblotting enhancer for selective detection of phosphoproteins in cell lysates. Anal. Biochem. 2009, 389, 83–85. [Google Scholar] [CrossRef]
- Tsunehiro, M.; Meki, Y.; Matsuoka, K.; Kinoshita-Kikuta, E.; Kinoshita, E.; Koike, T. A Phos-tag-based magnetic-bead method for rapid and selective separation of phosphorylated biomolecules. J. Chromatogr. B 2013, 925, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Inagaki, M. Production of a site- and phosphorylation state-specific antibody. Nat. Protoc. 2007, 2, 2574–2581. [Google Scholar] [CrossRef]
- Taylor, K.A.; Rahmani, H.; Edwards, R.J.; Reedy, M.K. Insights into Actin-Myosin Interactions within Muscle from 3D Electron Microscopy. Int. J. Mol. Sci. 2019, 20, 1703. [Google Scholar] [CrossRef]
- Nealon, J.O.; Philomina, L.S.; McGuffin, L.J. Predictive and Experimental Approaches for Elucidating Protein–Protein Interactions and Quaternary Structures. Int. J. Mol. Sci. 2017, 18, 2623. [Google Scholar] [CrossRef]
- Stadtländer, C. Scanning electron microscopy and transmission electron microscopy of mollicutes: Challenges and opportunities. Mod. Res. Educ. Top. Microsc. 2007, 1, 122–131. [Google Scholar]
- Ross, F.M. Opportunities and challenges in liquid cell electron microscopy. Science 2015, 350, aaa9886. [Google Scholar] [CrossRef]
- Kampourakis, T.; Sun, Y.-B.; Irving, M. Orientation of the N- and C-terminal lobes of the myosin regulatory light chain in cardiac muscle. Biophys. J. 2015, 108, 304–314. [Google Scholar] [CrossRef]
- Nayak, A.; Wang, T.; Franz, P.; Steffen, W.; Chizhov, I.; Tsiavaliaris, G.; Amrute-Nayak, M. Single molecule analysis reveals the role of regulatory light chains in fine-tuning skeletal myosin-II function. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Huber, I.; Itzhaki, I.; Caspi, O.; Arbel, G.; Tzukerman, M.; Gepstein, A.; Habib, M.; Yankelson, L.; Kehat, I.; Gepstein, L. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. FASEB J. 2007, 21, 2551–2563. [Google Scholar] [CrossRef] [PubMed]
- Bizy, A.; Guerrero-Serna, G.; Hu, B.; Ponce-Balbuena, D.; Willis, B.C.; Zarzoso, M.; Ramirez, R.J.; Sener, M.F.; Mundada, L.V.; Klos, M.; et al. Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res. 2013, 11, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Veevers, J.; Farah, E.N.; Corselli, M.; Witty, A.D.; Palomares, K.; Vidal, J.G.; Emre, N.; Carson, C.T.; Ouyang, K.; Liu, C.; et al. Cell-Surface Marker Signature for Enrichment of Ventricular Cardiomyocytes Derived from Human Embryonic Stem Cells. Stem Cell Rep. 2018, 11, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Chirikian, O.; Goodyer, W.R.; Dzilic, E.; Serpooshan, V.; Buikema, J.W.; McKeithan, W.; Wu, H.; Li, G.; Lee, S.; Merk, M.; et al. CRISPR/Cas9-based targeting of fluorescent reporters to human iPSCs to isolate atrial and ventricular-specific cardiomyocytes. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.-D.J.; Osteil, P.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598.e8. [Google Scholar] [CrossRef]
- Hernández, D.; Millard, R.; Sivakumaran, P.; Wong, R.C.B.; Crombie, D.E.; Hewitt, A.W.; Liang, H.; Hung, S.S.C.; Pébay, A.; Shepherd, R.K.; et al. Electrical Stimulation Promotes Cardiac Differentiation of Human Induced Pluripotent Stem Cells. Stem Cells Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Bos, J.M.; Ye, D.; Tester, D.J.; Hrstka, S.; Maleszewski, J.J.; Ommen, S.R.; Nishimura, R.A.; Schaff, H.V.; Kim, C.S.; et al. Induced Pluripotent Stem Cell–Derived Cardiomyocytes from a Patient with MYL2-R58Q-Mediated Apical Hypertrophic Cardiomyopathy Show Hypertrophy, Myofibrillar Disarray, and Calcium Perturbations. J. Cardiovasc. Transl. Res. 2019, 12, 394–403. [Google Scholar] [CrossRef]
- Perrie, W.T.; Perry, S.V. An electrophoretic study of the low-molecular-weight components of myosin. Biochem. J. 1970, 119, 31–38. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Species | Normal Ventricles (mol Pi/mol RLC) | Diseased Ventricles (mol Pi/mol RLC) | References |
|---|---|---|---|
| Human | 0.39–0.40 | 0–0.6 (End-stage heart failure) | [1,72] |
| Pigs | 0.39 | Decrease (Ischaemic failing heart model) | [72,89] |
| Rat | 0.39 | 0.76 (Ischaemic failing heart model) Decrease (Ischaemic failing heart model) | [1,72,90] |
| Rabbit | 0.36 | – | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markandran, K.; Poh, J.W.; Ferenczi, M.A.; Cheung, C. Regulatory Light Chains in Cardiac Development and Disease. Int. J. Mol. Sci. 2021, 22, 4351. https://doi.org/10.3390/ijms22094351
Markandran K, Poh JW, Ferenczi MA, Cheung C. Regulatory Light Chains in Cardiac Development and Disease. International Journal of Molecular Sciences. 2021; 22(9):4351. https://doi.org/10.3390/ijms22094351
Chicago/Turabian StyleMarkandran, Kasturi, Jane Wenjin Poh, Michael A. Ferenczi, and Christine Cheung. 2021. "Regulatory Light Chains in Cardiac Development and Disease" International Journal of Molecular Sciences 22, no. 9: 4351. https://doi.org/10.3390/ijms22094351
APA StyleMarkandran, K., Poh, J. W., Ferenczi, M. A., & Cheung, C. (2021). Regulatory Light Chains in Cardiac Development and Disease. International Journal of Molecular Sciences, 22(9), 4351. https://doi.org/10.3390/ijms22094351