1. Introduction
The Epstein–Barr virus-induced gene 2 (EBI2, GPR183) is highly expressed in the immune tissue and cells where it regulates innate and adaptive immune responses [
1,
2]. Accordingly, it is highly expressed in B and T lymphocytes and its signaling is implicated in a range of autoimmune diseases including type 1 diabetes, inflammatory bowel disease, rheumatoid arthritis and multiple sclerosis (MS) [
3,
4,
5,
6,
7,
8]. Specifically in MS, EBI2 was abundantly present in the human brain in infiltrating macrophages and various subsets of lymphocytes in the brains of MS patients [
7]. EBI2 expression and function in non-immune tissue and cells were also demonstrated in bone osteoclasts, hepatocytes and astrocytes [
9,
10,
11]. Human and mouse astrocytes are the only brain-specific cells in which EBI2 protein and function has so far been shown [
11,
12].
The receptor expression and function seem particularly important under neuroinflammatory conditions. For instance, treatment with bacterial lipopolysaccharide (LPS) induces sharp downregulation of EBI2 in primary mouse astrocytes and release of various oxysterols including the EBI2 ligand 7α,25-dihydroxycholesterol (7α,25OHC) [
13]. In primary human macrophages, LPS treatment leads to temporary upregulation of the receptor and release of oxysterols [
14]. Other factors such as the pro-inflammatory cytokines IL1β and IL23 upregulate and sustain EBI2 expression, while TGFβ/IL6 suppress EBI2 expression in differentiating T helper cells [
7]. Importantly, EBI2 expression increases during B cell maturation and after interaction with T helper cells [
15,
16,
17]. In the experimental autoimmune encephalomyelitis (EAE) model of MS, EBI2 expression increased in pathogenic Th17 cells and was substantially lower in naïve cells in comparison to memory T cells [
7,
8]. In untreated relapsing-remitting MS (RRMS), a similar level of EBI2 expression was found in patients and the healthy controls; however, there was significant variability in EBI2 receptor levels found in CD4+ and CD1 T cells in the MS patients’ samples [
18]. Moreover, treatment with an anti-α4-integrin humanized antibody (natalizumab) induces a 3-fold increase in EBI2 expression only in CD4+ T cells, an effect not observed after another MS treatment—dimethyl fumarate [
8].
The endogenous EBI2 ligand 7α,25OHC is enzymatically synthesized from cholesterol with cholesterol 25-hydroxylase (CH25H) and 25-hydroxycholesterol 7-alpha-hydroxylase (CYP7B1) [
19,
20]. Upon activation with 7α,25OHC, EBI2 exhibits chemotactic properties and induces chemotaxis of various EBI2 positive cells. Primary B and T lymphocytes from healthy and MS patients, primary human macrophages and mouse and human astrocytes were all shown to migrate specifically in response to EBI2 activation with 7α,25OHC [
7,
8,
11,
14,
18]. In addition to the studies investigating the effects of exogenously added 7α,25OHC on cellular chemotaxis in vitro, we also demonstrated that macrophages migrate towards astrocyte-released 7α,25OHC [
13]. In the EAE model, the concentration of 7α,25OHC in the central nervous system (CNS) was sharply elevated as a result of increased release of CH25H and CYP7B1 enzymes by microglia and CNS infiltrating immune cells, respectively [
7]. Wanke and colleagues also showed that EBI2 enhances CNS infiltration of EBI2 expressing pathogenic Th17 cells in the passive EAE. Furthermore, in the EAE model, Chalmin and colleagues found that deficiency of the CH25H enzyme limits trafficking of CD4+ T cells to the inflamed CNS [
18]. Similarly, primary human lymphocytes from MS patients exhibit strong migratory response towards 7α,25OHC in vitro. This migratory response was potentiated in lymphocytes from natalizumab treated patients [
8].
The roles of EBI2 in the EAE model, MS and astrocytes have been investigated; however, the key question about its expression and function in oligodendrocytes, the cells most affected in MS, remains to be answered. We showed before that EBI2 is involved in myelination under normal and demyelinating conditions. In our studies, normal myelin development was delayed in mice deficient in EBI2 [
21]. In organotypic cerebellar slices challenged with lysophosphatidylcholine (LPC), simultaneous treatment with the EBI2 agonist 7α,25OHC prevented demyelination. This protective effect was absent in EBI2-deficient slices and in the EBI2 antagonist NIBR189 treated slices, indicating a direct involvement of EBI2 signaling in protection from demyelination. Importantly, long-term inhibition of EBI2 signals with the antagonist NIBR189 leads to the inhibition of normal myelin development in organotypic cerebellar slices.
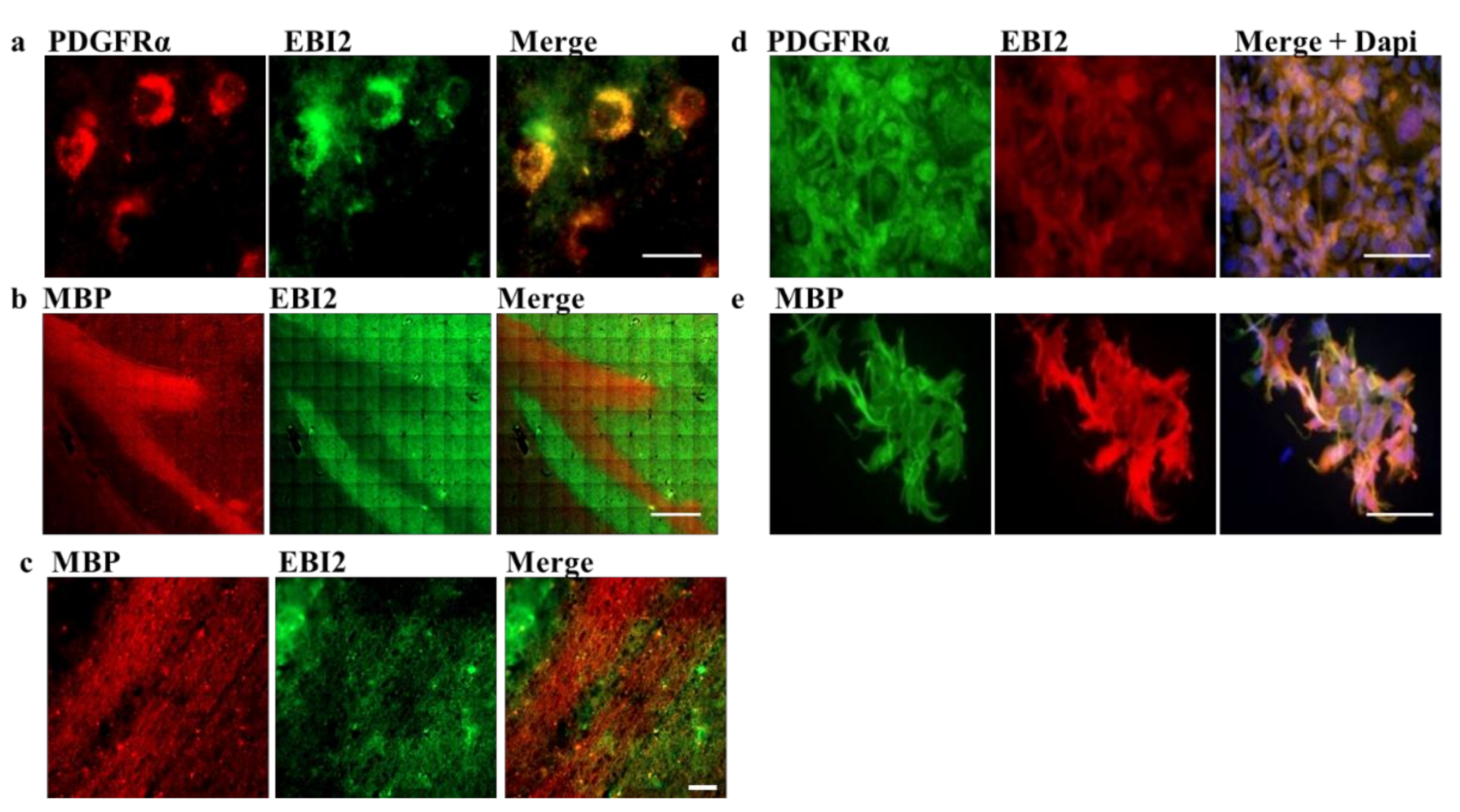
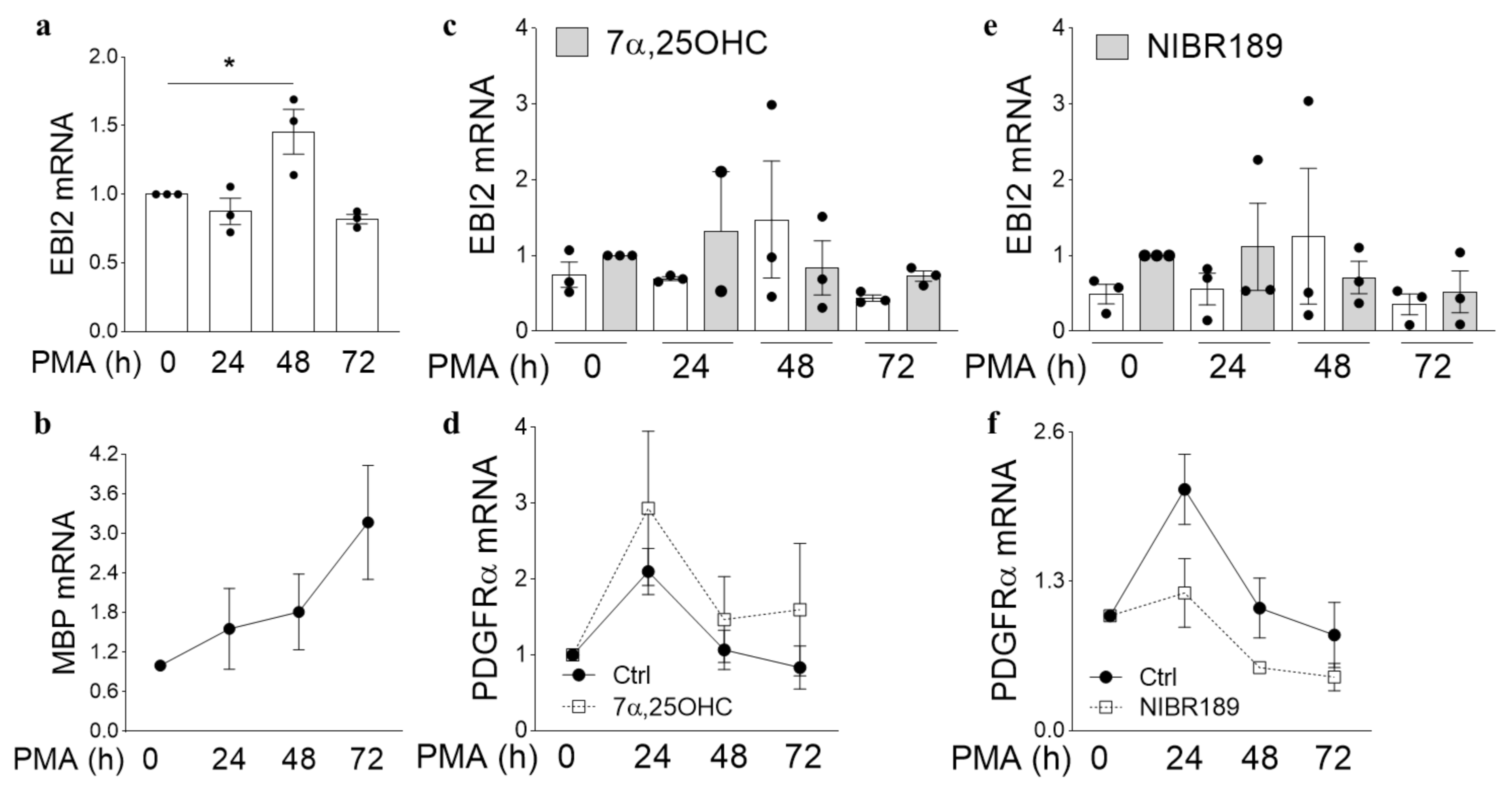
Here, to investigate the function of EBI2 in oligodendrocytes and remyelination, we first examined its expression in oligodendrocyte progenitor cells (OPCs, PDGFRα+ cells) and myelinating oligodendrocytes (myelin basic protein (MBP)+ cells) in the human brain. We then inspected the changes in EBI2 expression during oligodendrocyte maturation and its chemotactic properties in MO3.13 oligodendrocytes. Finally, the receptor’s involvement in protection from demyelination in CH25H-deficient mice and remyelination in the wild-type (WT) mice was also explored in the cerebellar organotypic slice model.
3. Discussion
The EBI2 receptor’s role in MS pathophysiology and myelination were studied in human and animal models before [
7,
8,
18,
21]. However, its expression and function in oligodendrocytes have never been investigated. Here, we studied EBI2 expression and function in oligodendrocytes in the human brain and MO3.13 cells and its involvement in myelination in organotypic slices of the cerebellum. The data, for the first time, showed EBI2 in OPCs and microglia and confirmed the receptor’s presence in astrocytes in the human brain. Our studies showed that oligodendrocytes upregulate EBI2 expression during maturation but that EBI2-signaling is non-essential for oligodendrocyte maturation to proceed normally.
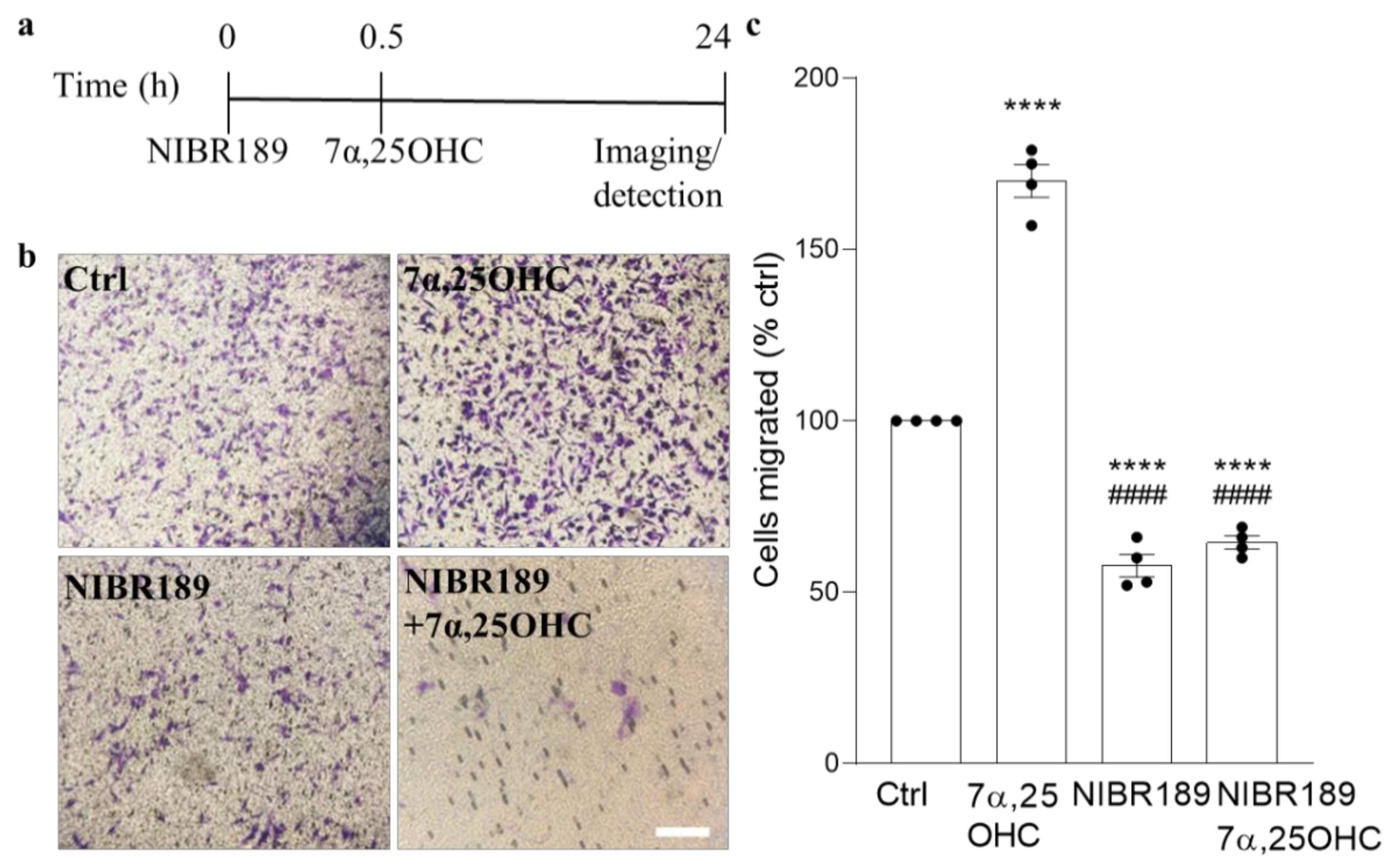
Importantly, to our knowledge, this is the first report of EBI2 upregulation in a non-inflammatory or non-immune setting suggesting that EBI2 may have other roles to play in the CNS beside immune regulation. The data reported here also showed that EBI2 in MO3.13 oligodendrocytes is functional and that activation of EBI2 induces migration of MO3.13 oligodendrocytes towards higher concentrations of 7α,25OHC. The ability to induce migration in immature oligodendrocytes may prove very useful in treating demyelinating diseases where OPCs need to migrate towards the demyelinated area. Perhaps the temporary upregulation of EBI2 in maturing oligodendrocytes is linked to cellular migration. By upregulating EBI2, the cells may migrate to and better position themselves in demyelinated areas where oxysterol concertation is increased as a result of cholesterol release from damaged myelin sheets and synthesis by astrocytes and other infiltrating immune cells.
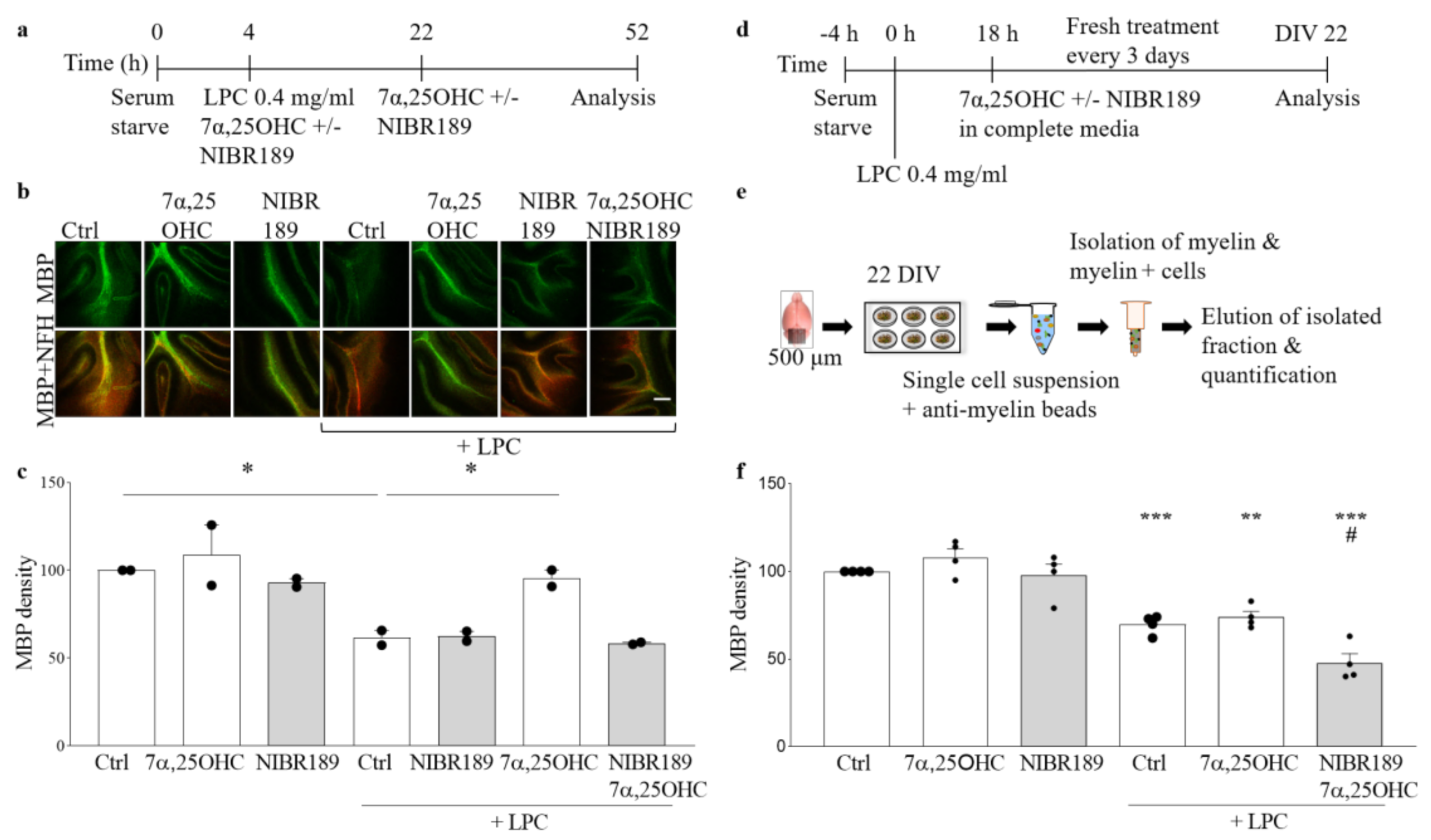
In the organotypic cerebellar slice model, induction of EBI2 signaling with its agonist 7α,25OHC did not have any additional effect on normally occurring remyelination post-LPC-induced demyelination. However, when EBI2 signaling was blocked with the antagonist, remyelination was inhibited. These data show that even though increased activation of EBI2 does not enhance normally occurring remyelination it is essential to proceed normally. Oxysterols, including EBI2 agonist 7α,25OHC, are present in the serum in slice media and are released by the injured tissue post-LPC-treatment. These endogenous oxysterols induced EBI2 signaling in the control slices and thus no additional effect was observed after further addition of exogenous 7α,25OHC. Remyelination was not enhanced above the control levels in 7α,25OHC-treated slices most likely as a result of the limits in the inherent capacity to synthesize myelin. However, long-term antagonism of EBI2 revealed that its signaling is essential for remyelination in this model as the antagonist blocked EBI2 signaling induced by the endogenous as well as the exogenously added oxysterols and inhibited remyelination. Furthermore, persistent antagonism of EBI2 most likely inhibited EBI2-mediated OPC migration towards the demyelinated tissue, where oxysterol levels are increased.
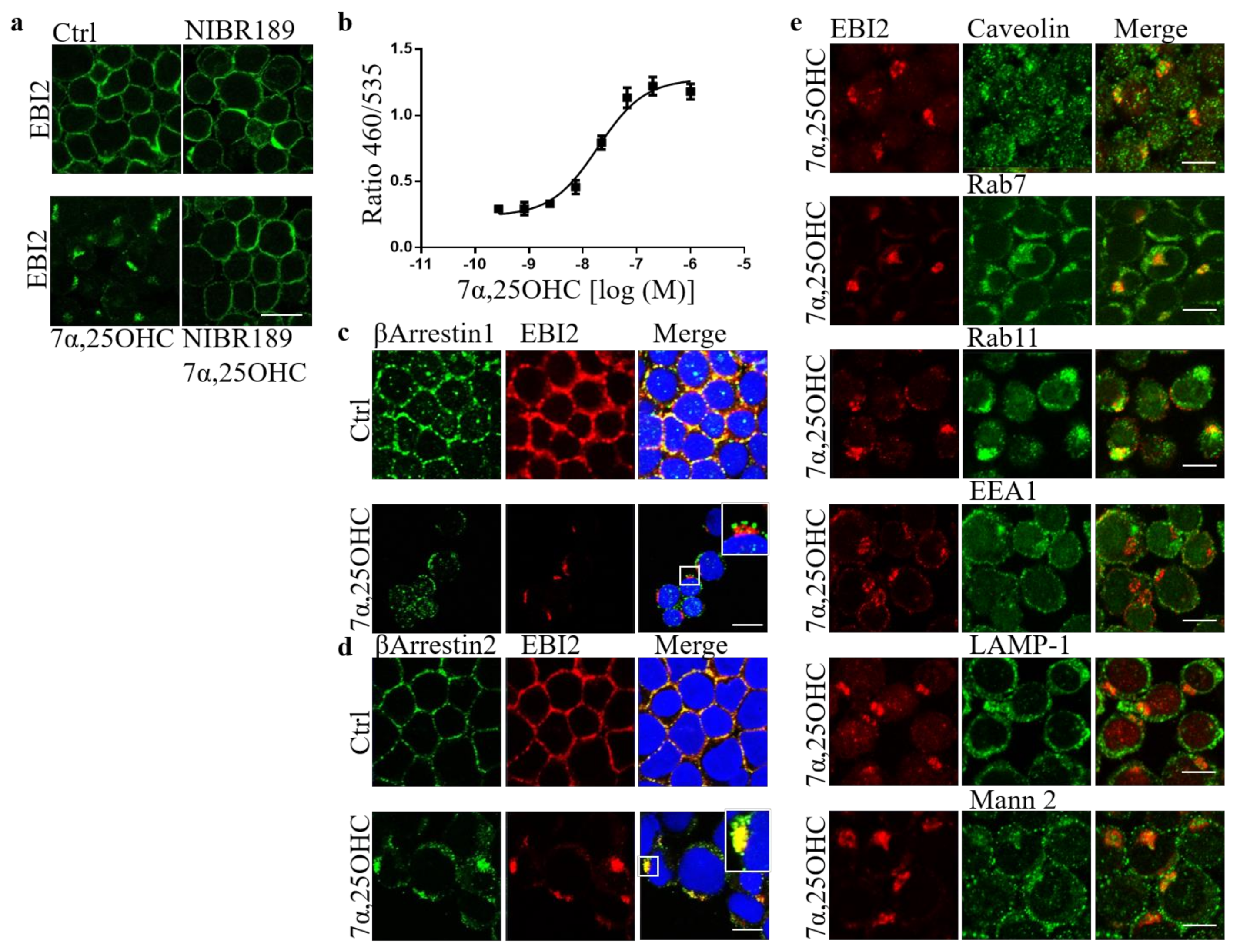
Our previous work showed that in EBI2-deficient mice, normal myelination is delayed in the post-natal period [
21]. We also discovered before that long-term and persistent antagonism of EBI2 inhibits normal myelin development in cerebellar slices from P10 mice [
21]. We confirmed here that EBI2 is indeed internalized after 7α,25OHC treatment and showed that its internalization is βArrestin2 dependent. The trafficking and intracellular organelle data suggested that after short-term stimulation, EBI2 is recycled back to the cell membrane. We could not show with the current model that long-term EBI2 antagonism or stimulation leads to receptor downregulation and degradation and needs to be clarified. Altogether, the results reported here demonstrate a direct role of EBI2 signaling in remyelination. The EBI2 agonist 7α,25OHC is an oxysterol and oxysterols are derivatives of cholesterol, which is the main component of myelin. EBI2 signaling might therefore be necessary for correct cholesterol/myelin synthesis under normal and demyelinating conditions.
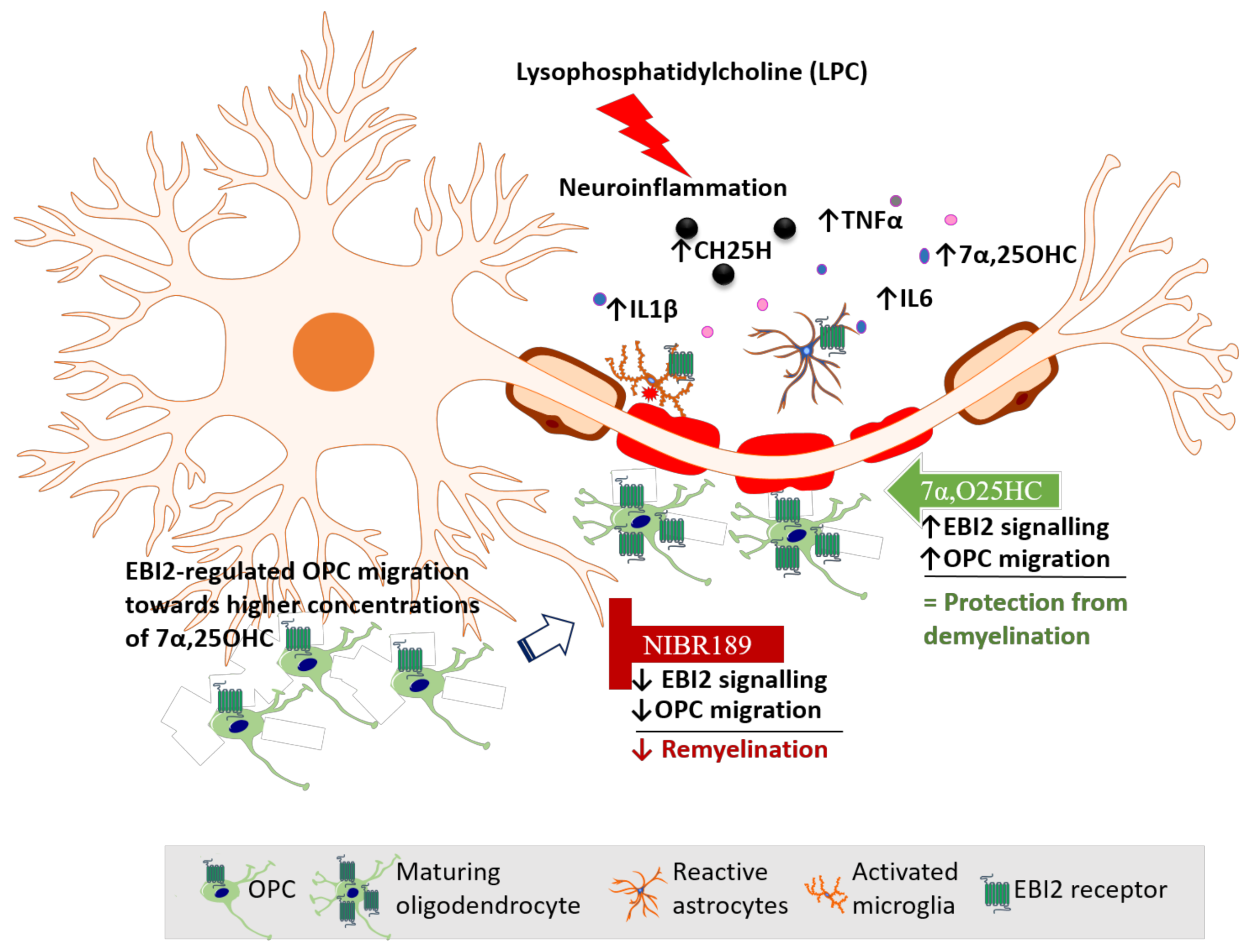
Based on our data reported here and elsewhere, we propose a model of EBI2 function in the CNS (
Figure 6). In this model, LPC challenge induces inflammation and demyelination in cerebellar slices. Activated EBI2+ astrocytes and microglia respond to myelin damage and release pro-inflammatory cytokines and oxysterols. EBI2-expressing OPCs upregulate EBI2 and begin to migrate towards the affected areas in EBI2-dependent manner in response to increased concentration of 7α,25OHC and EBI2-independent manner in response to other chemotactic molecules. Once at the site of injury, OPCs mature, start to synthesize myelin and downregulate EBI2. Exogenous addition of 7α,25OHC increases migration of OPCs while inhibition of EBI2 signaling with NIBR189 leads to downregulation of EBI2 signaling and inhibition of OPC migration, which negatively affects remyelination.
4. Materials and Methods
4.1. Mice
The Ch25h(−/−) knock-out (KO) mice (C57BL/6) were provided by Dr. David W. Russell (University of Texas Southwestern, TX, USA) [
25]. All animals were kept in filter-top cages under specific pathogen-free conditions with ad libitum access to standard diet and water. All animal tissue was harvested according to the national and institutional ethical guidelines.
4.2. Immunohistochemistry of Human Brain Sections
Post-mortem human brain sections from MS donors preserved in formalin were received from the Rocky Mountain MS Center Tissue Bank (Englewood, CO, USA) after approval by the Medical University of Gdansk (Poland) bioethics committee (NKBBN/253/2018). Portions of 1 cm × 1 cm containing white and grey matter from periventricular region were removed and soaked in 15% followed by 30% sucrose solution to dehydrate. The Tissue-Tek OCT Compound (4583, Sakura, The Netherlands)-embedded brain portions were cut into 30-micron thick slices on a cryostat. The non-specific binding was reduced by incubation in 10% normal goat serum (NGS) in 0.3% triton-x in PBS (blocking solution) for two hours at RT. Then, the sections were washed 3 × 15 min in PBS and incubated in a primary antibody cocktail o/n in the blocking solution. After further washes, the tissue was incubated o/n in secondary antibody cocktail (Cy3 goat anti-rabbit (Abcam, ab6939, RRID:AB_955021) or/and Alexa 488 goat anti-mouse (ThermoFisher, A-11001, RRID:AB_2534069)) after which it was washed, inserted onto glass cover slides and imaged using an automated Z1 system (Zeiss, Oberkochen, Germany) using the Zeiss Zen 3.1 software (Zeiss, Oberkochen, Germany). Primary antibodies used were: Rabbit PDGFRα (ThermoFisher, PA5-16571, RRID: AB_10981626), rabbit MBP (CellSignaling, 78896, RRID: AB_2799920), chicken NFH (Millipore, AB5539, RRID:AB_11212161), rabbit GFAP (CellSignaling, 12389, RRID:AB_2631098, mouse EBI2 (clone 57C9B51C9, Novartis, Basel, Switzerland [
7]).
4.3. Organotypic Slice Culture, Treatments and Immunohostochemistry
Organotypic cerebellar slices were made from 10-day-old (P10) CH25H−/− KO or wild-type (WT) C57BL/6 mice according to previously published protocol [
21]. Briefly, pups were decapitated with scissors, the skull was opened, and the cerebellum removed and placed in cold Opti-MEM medium (11058021, ThermoFisher, Warsaw, Poland). Parasagittal 400 μm thick slices of the cerebellum were cut using the McIlwain tissue chopper WPI, Friedberg, Germany). Several (4–6) slices were cultured on a single insert (Merck, Millicell, PICMORG50) in a humidified incubator at a reduced temperature of 35.5 °C and standard 5% CO
2 concentration. The growth medium consisted of 50% Opti-MEM, 25% Hanks’ buffered salt solution (HBSS), 25% heat-inactivated horse serum supplemented with Glutamax (2 mM), D-glucose (28 mM), 1% pen/strep, HEPES (10 mM) for 12 days in vitro. After 12 days in vitro, slices were placed in serum free media for 4 h and then again in fresh serum-free media supplemented with LPC (0.4 mg/mL) (Sigma, L-4129) with or without 7α,25OHC (1 μM) and/or NIBR189 (NIBR189, 1 μM) for 18 h. Following the 18 h LPC, 7α,25OHC and/or NIBR189 treatment, the slices were cultured in medium containing 7α,25OHC (1 μM) and/or NIBR189 (1 μM) for further 30 h (a total of 48 h) (
Figure 4a,b). When the treatments were finished, the slices were washed twice with PBS and fixed in 4% paraformaldehyde (PFA) for 5 min at RT and permeabilized with 20% ice cold methanol for 5 min. Subsequently, the slices were washed 2 × 5 min in PBS and incubated overnight in the blocking buffer that consisted of Triton-X100 (0.5%), bovine serum albumin (10%, BSA; Sigma Aldrich), NGS (1%). The slices were then incubated overnight at 4 °C with the primary antibodies (rabbit anti-myelin basic protein (MBP, Abcam ab40390, RRID:AB_1141521) and chicken anti-neurofilament heavy chain (NFH), Millipore, AB5539, RRID:AB_11212161) in PBS supplemented with BSA (0.5%) and Tween-20 (0.05%), washed twice in the above solution and incubated with secondary antibodies overnight at 4 °C. Finally, the slices were washed twice with wash solution as was described earlier and mounted on glass cover slides with Tissue-Tek OCT Compound. Confocal images were taken from the central part of the slices and relative fluorescence intensity was analyzed with ImageJ software (Bethesda, MD, USA).
4.4. Organotypic Slice Culture, Treatments and Myelin Content Analysis with Myelin-Coated Beads
Cerebella from the WT mice were placed in a stainless-steel brain matrix (AgnTho’s, Lidingö, Sweden) for cutting of 500 μm thick mouse sagittal slices with a sterile blade. The cut slices were moved into a petri dish containing Opti-MEM medium and slices were separated from each other using needles under a microscope. Four slices per condition were cultured on one organotypic insert at 35 °C, 5% CO
2. Media change was done on day in vitro (DIV) 1, 4 and 7 with media consisting of 50% Opti-MEM, 25% HBSS, 25% horse serum, 1% glucose, 1% HEPES buffer, 1% GlutaMax and 1% pen/strep. At DIV 10, slices were serum-starved with media containing 75% HBSS, 25% Opti-MEM, 1% GlutaMax, 1% glucose, 1% pen/strep and 1% HEPES for 4h before adding LPC 0.4 mg/mL for 18h. After 18h incubation with LPC, media was removed and slices were transferred to serum-containing media with 0.25 µM 7α,25OHC or 100 µM NIBR189 for another 12 days, changing to fresh media with compounds every 3 days (
Figure 4d,e). At DIV 22, slices were collected, and brain cells were dissociated using the MACS Neural Tissue Dissociation Kit P (Miltenyi, Bergisch Gladbach, Germany, 130-092-628) according to the accompanying product protocol with the MACS separator. The dissociated cells were incubated with myelin beads (Miltenyi, Bergisch Gladbach, Germany, 130-104-257) and separated using LS columns (Miltenyi, Bergisch Gladbach, Germany, 130-042-401). The amount of myelin was determined using a BCA assay (Thermofisher, Waltham, MA, USA, 23225) as a ratio of myelin/total.
4.5. Cell Culture and Differentiation
MO3.13 human oligodendrocyte cell line (RRID:CVCL_D357) was purchased from Tebu-Bio (2018, CLU301, batch 131117 P25) and grown in high glucose DMEM, 10% FBS and 1% pen/strep. Cells were differentiated with 100 nM PMA (Sigma-Aldrich, P1585) with or without the EBI2 agonist oxysterol 7a,25-dihydroxycholesterol (7α,25-OHC) (1 μM) or EBI2 antagonist NIBR189 (1 μM) in T25 flasks for 0–72 h.
U937 cells were cultured in RPMI 1640 medium containing Glutamax, 10% FBS, 1% MEM non-essential amino acids (NEA, Invitrogen, Carlsbad, CA, USA, 11140), 1% sodium pyruvate (Invitrogen, 11360-070), 1% pen/strep and 0.1% 2-Mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA, M7154) in T75 culture flasks.
4.6. Migration Assay
MO3.13 undifferentiated oligodendrocytes were starved for 3 h before treatment in serum-free media. Serum-free DMEM with or without 0.1 µM 7α,25OHC or 1 µM NIBR189 was pipetted in a 24 well-plate. Then, 150 µl cell suspension of 2 million cells per ml were plated in the top chamber of the transwell assay insert (ThermoFisher, 141006, 8 µm pores). After 30 min of incubation with the antagonist, the insert with cells was moved into wells containing the oxysterol with or without NIBR189 for 6 h. After 6 h, the media inside the transwell was discarded, and the cells that did not migrate were removed with a cotton swab. The transwells were put in 400 µL of crystal violet staining solution (Cell Biolabs, San Diego, CA, USA, CBA-100) for 10 min at RT. Then, they were dipped carefully in a beaker with water and left to air dry. When dried, images were taken with a light microscope and transwells were incubated in 200 µL of extraction solution (Methanol, Cell Biolabs, CBA-100) for 10 min on a shaker. Then, the solution was transferred to a 96-wellplate in order to read absorbance at 590 nm.
4.7. Immunocytochemistry
MO3.13 oligodendrocytes in 8-well imaging plates (Milicell, Millipore) were washed with cold PBS, fixed for 10 min on ice in 4% PFA and then permeabilized for 60 s in coldmethanol (100%). The non-specific binding was blocked in the incubation in 0.5% NGS, 1% BSA, 0.1% tween-20 in PBS for 60 min and then incubated o/n at 4 °C with primary antibodies in PBS containing BSA (0.5%), tween-20 (0.05%). The following primary antibodies were used: Rabbit PDGFRα (ThermoFisher, PA5-16571, RRID: AB_10981626), rabbit MBP (CellSignaling, 78896, RRID: AB_2799920), mouse EBI2 (clone 607B, Novartis). The cells were then washed with PBS 3 × 5 min and incubated for 60 min with the following secondary antibodies: Anti-rabbit Alexa 488 and anti-mouse 546. Then they were washed again, incubated with Hoechst (ThermoFisher, H1399) for 10 min, washed and imaged with Nikon Eclipse Ti fluorescent microscope (Tokyo, Japan).
The U927 monocytes were washed with PBS and treated for 1 h at 37 °C with 7α,25OHC (10 μM) or NIBR189 (10 μM). After the treatment, the cells were spun in a Cytospin 4 Cytocentrifuge (Thermo Scientific, A78300003) and adhered to glass slides. Subsequently, the U937s on glass slides were washed with PBS and fixed with fixation/permeabilisation kit (BD Cytofix/Cytoperm, 554714) according to the manufacturer’s protocol (20 min on ice). The monocytes were then incubated for 1 h in a blocking solution consisting of PBS, 0.1% tween-20 and 1% BSA. The cells were then incubated overnight at 4 °C with the primary antibodies in 0.05% tween-20 and 0.5% BSA in PBS, washed twice and incubated with secondary antibodies for 1 h in the same solution. The primary antibodies used were mouse EBI2 (clone 57C, Novartis), rabbit Rab11 (Invitrogen, 71-5300, RRID:AB_87868), rabbit EEA1 (Abcam, ab2900, RRID:AB_2262056), rabbit caveolin (BD, 610060, RRID:AB_397472), rabbit LAMP-1 (Abcam, ab24170, RRID:AB_775978), rabbit Rab7 (CellSignaling, D95F2, RRID:AB_1904103), rabbit mannosidase2 (Abcam, ab136497, RRID:AB_722896), rabbit βArrestin 1 (Abcam, ab32099, RRID:AB_722896) and goat βArrestin 2 (Abcam, ab31294, RRID:AB_2060265). The secondary antibodies used were anti-rabbit Alexa 488 (Invitrogen, A11008, RRID:AB_143165), anti-goat Alexa 488 (Invitrogen), anti-mouse Alexa 633 (Invitrogen, A21052, RRID:AB_2535719). After two subsequent washes, the U973s were incubated with Hoechst for 10 min, washed twice again, covered in PBS and kept covered in aluminum foil at 4 °C until imaged. The Zeiss LSM 700 confocal microscope was used to take images of the cells.
4.8. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Human OPC cDNA extracted from normal primary cells was purchased from ScienCell (1604, Carslbad, CA, USA). Human microglia cell total RNA extracted from primary cells was purchased from Celprogen (37089RNA, Torrance, CA, USA). Treated MO3.13 oligodendrocytes were washed in PBS, scraped, spun for 10 min at maximum speed and resuspended in 150 μL of lysis buffer (Sigma-Aldrich, RTN70-1KT). Total RNA was extracted using the GenElute total RNA kit (Sigma-Aldrich, RTN70-1KT). Isolated total RNA was frozen at −20 °C until used. Total RNA was reverse-transcribed with cDNA transcription kit (ThermoFisher, 4368814) according to the manufacturer’s instructions. RT-qPCR was performed with TaqMan master mix (ThermoFisher, 4444556) using the LightCycler 480 (Roche, Basel, Switzerland) according to the standard protocol. FAM dye-labelled TaqMan (Applied Biosystems, Foster City, CA, USA) probes were used in all experiments. The relative mRNA expression using the ΔΔCt method was calculated from absolute quantification after normalization to the reference gene.
4.9. βArrestin Assay
The Tango assay (Invitrogen, Carlsbad, CA, USA) is a cell-based system for detection of GPCR activation and βArrestin interaction. The assay was run on EBI2-bla U2OS cells (Invitrogen, K1828A), which express the human EBI2 receptor associated with a TEV protease site and a Gal4-VP16 transcription factor stably integrated into the Tango GPCR-bla U2OS parental cell line. The parental cells stably express a βArrestin/TEV protease fusion protein and the β-lactamase reporter gene under the control of a UAS response element. The EBI2-bla U2OS cells were grown in McCoy’s 5A media (Invitrogen, 36600-088) supplemented with 10% dialyzed FBS (Invitrogen, 26400-036), 0.1 mM NEAA, 25 mM Hepes, 200 µg/mL Zeocin (Invitrogen, R250-01), pen/strep. The experiments were conducted according to manufacturer’s protocol, in brief: EBI2-blaU2OS cells in Freestyle Expression media (Invitrogen, 12338-018) were placed in 384 clear bottom/black walls plates and incubated for 48 h at 37 °C. On the day of the experiment, the cells were treated with 7α,25OHC for 16 h in the incubator. Subsequently, a substrate mixture was loaded in the absence of direct strong light and incubated for another 2 h at RT in the dark. The fluorescence was read with Envision (PerkinElmer, Waltham, MA, USA) plate reader.
4.10. Statistical Analysis
Statistical analysis was performed on GraphPad Prism 8 using one-way analysis of variance (ANOVA) followed by Sidak’s multiple comparisons post hoc tests for comparisons of differences between pre-selected groups or Dunnett’s post hoc test for comparisons of the control with the mean of every other column. Data are shown as means ± standard error of the mean (SEM). Where appropriate, p values are written in the figure legends, and group comparisons derived from post hoc analysis are given in the figures with significant effects indicated by asterisks: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






