
Genetic Neonatal-Onset Epilepsies and Developmental/Epileptic Encephalopathies with Movement Disorders: A Systematic Review

 , ,
, ,  ,
,
Abstract
1. Introduction
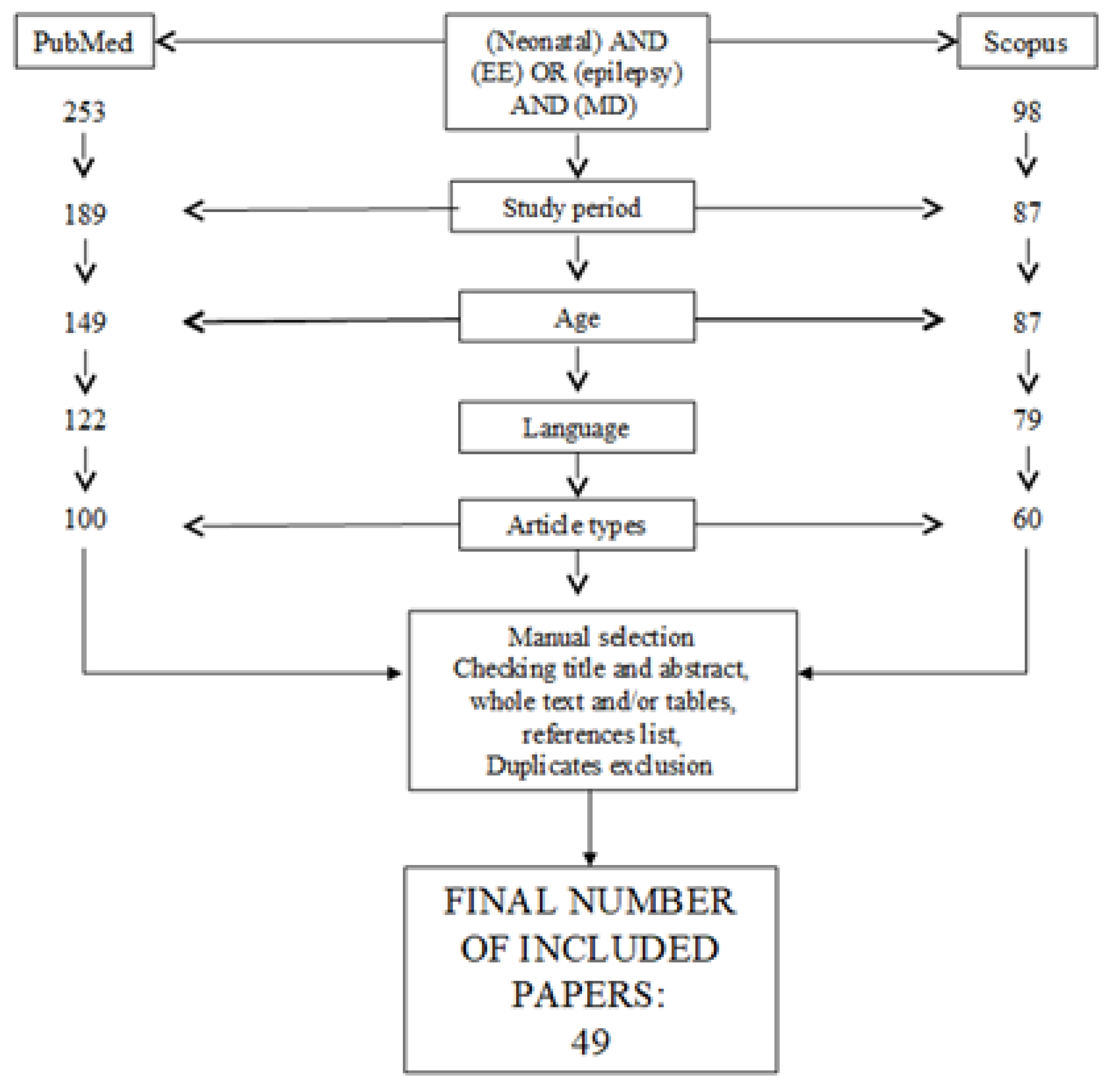
2. Methods
2.1. Protocol
2.1.1. Eligibility Criteria
2.1.2. Information Sources
2.1.3. Search
2.1.4. Study Selection
2.1.5. Data Items
3. Results
3.1. Genetic Results
3.2. Clinical Findings
3.2.1. Epilepsy
3.2.2. Neurological Examination
3.2.3. Neurodevelopmental Aspects
3.2.4. Movement Disorder
3.3. Neuroimaging Findings
3.4. Presentation and Outcome According to Genetic Diagnosis
3.4.1. Enzymes
3.4.2. Synaptopathies
3.4.3. ATPASEs
3.4.4. Channelopathies
3.4.5. Transcription Regulators
3.4.6. Transcription Factors
3.4.7. Growth Factors
3.4.8. Receptors
3.4.9. G-PROTEIN Transduction
3.4.10. Neuronal Connectivity/Signal Transduction
3.4.11. Ubiquitination
3.4.12. Transporters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pisani, F.; Spagnoli, C.; Fusco, C. EEG monitoring of the epileptic newborn. Curr. Neurol. Neurosci. Rep. 2020, 20, 6. [Google Scholar] [CrossRef]
- Pisani, F.; Spagnoli, C.; Falsaperla, R.; Nagarajan, L.; Ramantani, G. Seizures in the neonate: A review of etiologies and outcomes. Seizure 2021, 85, 48–56. [Google Scholar] [CrossRef]
- Shellhaas, R.A.; Wusthoff, C.J.; Tsuchida, T.N.; Glass, H.C.; Chu, C.J.; Massey, S.L.; Soul, J.S.; Wiwattanadittakun, N.; Abend, N.S.; Cilio, M.R.; et al. Profile of neonatal epilepsies: Characteristics of a prospective US cohort. Neurology 2017, 89, 893–899. [Google Scholar] [CrossRef]
- Miceli, F.; Soldovieri, M.V.; Joshi, N.; Weckhuysen, S.; Cooper, E.; Taglialatela, M. KCNQ2-Related Disorders. In GeneReviews®[Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; Seattle (WA): Seattle, DC, USA, 1993–2020. [Google Scholar]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.B.S.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef]
- Gardella, E.; Marini, C.; Trivisano, M.; Fitzgerald, M.P.; Alber, M.; Howell, K.B.; Darra, F.; Siliquini, S.; Bölsterli, B.K.; Masnada, S.; et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 2018, 91, e1112–e1124. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Y.; Pearl, P.L. Metabolic causes of epileptic encephalopathy. Epilepsy Res. Treat. 2013, 2013, 124934. [Google Scholar]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. J. Clin. Epidemiol. 2009, 62, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Jankovic, J.; Hallett, M. Principles and Practice of MovementDisorders, 2nd ed.; Elsevier: Philadelphia, PA, USA, 2011. [Google Scholar]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E. Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef]
- Oxford Centre for Evidence-based Medicine. Available online: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009 (accessed on 16 April 2021).
- Paciorkowski, A.R.; McDaniel, S.S.; Jansen, L.A.; Tully, H.; Tuttle, E.; Ghoneim, D.H.; Tupal, S.; Gunter, S.A.; Vasta, V.; Zhang, Q. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 2015, 56, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Inagaki, H.; Miyake, M.; Kawamura, Y.; Yoshikawa, T.; Kurahashi, H. A case of early onset life-threatening epilepsy associated with a novel ATP1A3 gene variant. Brain Dev. 2019, 41, 285–291. [Google Scholar] [CrossRef]
- Kelly, M.; Park, M.; Mihalek, I.; Rochtus, A.; Gramm, M.; Pérez-Palma, E.; Axeen, E.T.; Hung, C.Y.; Olson, H.; Swanson, L.; et al. Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate-binding region. Epilepsia 2019, 60, 406–418. [Google Scholar] [CrossRef]
- Saitsu, H.; Fukai, R.; Ben-Zeev, B.; Sakai, Y.; Mimaki, M.; Okamoto, N.; Suzuki, Y.; Monden, Y.; Saito, H.; Tziperman, B. Phenotypic spectrum of GNAO1 variants: Epileptic encephalopathy to involuntary movements with severe developmental delay. Eur. J. Hum. Genet. 2016, 24, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kodera, H.; Akita, T.; Shiina, M.; Kato, M.; Hoshino, H.; Terashima, H.; Osaka, H.; Nakamura, S.; Tohyama, J. De Novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am. J. Hum. Genet. 2013, 93, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.C.; Dietel, T.; Evers, C.; Hinderhofer, K.; Korinthenberg, R.; Ezzo, D.; Bönnemann, C.G.; Kirschner, J. Expanding Phenotype of De Novo Mutations in GNAO1: Four New Cases and Review of Literature. Neuropediatrics 2017, 48, 371–377. [Google Scholar] [PubMed]
- Marcé-Grau, A.; Dalton, J.; López-Pisón, J.; García-Jiménez, M.C.; Monge-Galindo, L.; Cuenca-León, E.; Giraldo, J.; Macaya, A. GNAO1 encephalopathy: Further delineation of a severe neurodevelopmental syndrome affecting females. Orphanet. J. Rare Dis. 2016, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Ivanovic, V.; Hendrickx, R.; Van Coster, R.; Hjalgrim, H.; Møller, R.S.; Grønborg, S.; Schoonjans, A.S.; Ceulemans, B.; Heavin, S.B.; et al. Extending the KCNQ2 encephalopathy spectrum: Clinical and neuroimaging findings in 17 patients. Neurology 2013, 81, 1697–1703. [Google Scholar] [CrossRef]
- Blumkin, L.; Suls, A.; Deconinck, T.; De Jonghe, P.; Linder, I.; Kivity, S.; Dabby, R.; Leshinsky-Silver, E.; Lev, D.; Lerman-Sagie, T. Neonatal seizures associated with a severe neonatal myoclonus like dyskinesia due to a familial KCNQ2 gene mutation. Eur. J. Paediatr. Neurol. 2012, 16, 356–360. [Google Scholar] [CrossRef]
- Spagnoli, C.; Salerno, G.G.; Iodice, A.; Frattini, D.; Pisani, F.; Fusco, C. KCNQ2 encephalopathy: A case due to a de novo deletion. Brain Dev. 2018, 40, 65–68. [Google Scholar] [CrossRef]
- Dhamija, R.; Goodkin, H.P.; Bailey, R.; Chambers, C.; Brenton, J.N. A case of KCNQ2-associated movement disorder triggered by fever. J. Child. Neurol. 2017, 32, 1123–1124. [Google Scholar] [CrossRef]
- Denis, J.; Villeneuve, N.; Cacciagli, P.; Mignon-Ravix, C.; Lacoste, C.; Lefranc, J.; Napuri, S.; Damaj, L.; Villega, F.; Pedespan, J.M.; et al. Clinical study of 19 patients with SCN8A-related epilepsy: Two modes of onset regarding EEG and seizures. Epilepsia 2019, 60, 845–856. [Google Scholar] [CrossRef]
- Schreiber, J.M.; Tochen, L.; Brown, M.; Evans, S.; Ball, L.J.; Bumbut, A.; Thewamit, R.; Whitehead, M.T.; Black, C.; Boutzoukas, E.; et al. A multi-disciplinary clinic for SCN8A-related epilepsy. Epilepsy Res. 2020, 159, 106261. [Google Scholar] [CrossRef]
- Vaher, U.; Nõukas, M.; Nikopensius, T.; Kals, M.; Annilo, T.; Nelis, M.; Ounap, K.; Reimand, T.; Talvik, I.; Ilves, P.; et al. De novo SCN8A mutation identified by whole-exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J. Child. Neurol. 2014, 29, NP202-6. [Google Scholar] [CrossRef]
- Singh, R.; Jayapal, S.; Goyal, S.; Jungbluth, H.; Lascelles, K. Early-onset movement disorder and epileptic encephalopathy due to de novo dominant SCN8A mutation. Seizure 2015, 26, 69–71. [Google Scholar] [CrossRef]
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Jugé, C.; Roubertie, A.; Héron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef]
- Hardies, K.; de Kovel, C.G.; Weckhuysen, S.; Asselbergh, B.; Geuens, T.; Deconinck, T.; Azmi, A.; May, P.; Brilstra, E.; Becker, F.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 2015, 138 Pt 11, 3238–3250. [Google Scholar] [CrossRef]
- Milh, M.; Villeneuve, N.; Chouchane, M.; Kaminska, A.; Laroche, C.; Barthez, M.A.; Gitiaux, C.; Bartoli, C.; Borges-Correia, A.; Cacciagli, P.; et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011, 52, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.M.; Conroy, J.; Shahwan, A.; Lynch, B.; Correa, R.G.; Pena, S.D.; McCreary, D.; Magalhães, T.R.; Ennis, S.; Lynch, S.A.; et al. Unexplained early onset epileptic encephalopathy: Exome screening and phenotype expansion. Epilepsia 2016, 57, e12–e17. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, A.; Uddin, M.; Snead, O.C., 3rd; Lira., V.; Silberberg., A.; Weiss, S.; Donner, E.J.; Zak, M.; Bradbury, L.; Scherer, S.W.; et al. STXBP1 encephalopathy is associated with awake bruxism. Epilepsy Behav. 2019, 92, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.J.; Daud, D.; Pyle, A.; Duff, J.; Griffin, H.; He, L.; Alston, C.L.; Steele, H.; Taggart, S.; Basu, A.P.; et al. A novel de novo STXBP1 mutation is associated with mitochondrial complex I deficiency and late- onset juvenile-onset parkinsonism. Neurogenetics 2015, 16, 65–67. [Google Scholar] [CrossRef]
- Howell, K.B.; McMahon, J.M.; Carvill, G.L.; Tambunan, D.; Mackay, M.T.; Rodriguez-Casero, V.; Webster, R.; Clark, D.; Freeman, J.L.; Calvert, S.; et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015, 85, 958–966. [Google Scholar] [CrossRef]
- Schwarz, N.; Hahn, A.; Bast, T.; Müller, S.; Löffler, H.; Maljevic, S.; Gaily, E.; Prehl, I.; Biskup, S.; Joensuu, T.; et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J. Neurol. 2016, 263, 334–343. [Google Scholar] [CrossRef]
- Schwarz, N.; Bast, T.; Gaily, E.; Golla, G.; Gorman, K.M.; Griffiths, L.R.; Hahn, A.; Hukin, J.; King, M.; Korff, C.; et al. Clinical and genetic spectrum of SCN2A-associated episodic ataxia. Eur. J. Paediatr. Neurol. 2019, 23, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Gorman, K.M.; King, M.D. SCN2A p.Ala263Val variant a phenotype of neonatal seizures followed by paroxysmal ataxia in toddlers. Pediatr. Neurol. 2017, 67, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, K.M.; Miranda, M.J.; Lerche, H.; Møller, R.S. Letter to the editor: Confirming neonatal seizure and late onset ataxia in SCN2A Ala263Val. J. Neurol. 2016, 263, 1459–1460. [Google Scholar] [CrossRef] [PubMed]
- Hardies, K.; Cai, Y.; Jardel, C.; Jansen, A.C.; Cao, M.; May, P.; Djémié, T.; Hachon Le Camus, C.; Keymolen, K. Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain 2016, 139, 2420–2430. [Google Scholar] [CrossRef]
- Samanta, D.; Arya, K. Electroclinical findings of SYNJ1 epileptic encephalopathy. J. Pediatr. Neurosci. 2020, 15, 29–33. [Google Scholar]
- Helbig, K.L.; Lauerer, R.J.; Bahr, J.C.; Souza, I.A.; Myers, C.T.; Uysal, B.; Schwarz, N.; Gandini, M.A.; Huang, S.; Keren, B.; et al. De Novo Pathogenic Variants in CACNA1E Cause Developmental and Epileptic Encephalopathy with Contractures, Macrocephaly, and Dyskinesias. Am. J. Hum. Genet. 2018, 103, 666–678. [Google Scholar] [CrossRef]
- Siekierska, A.; Isrie, M.; Liu, Y.; Scheldeman, C.; Vanthillo, N.; Lagae, L.; de Witte, P.A.; Van Esch, H.; Goldfarb, M.; Buyse, G.M. Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology 2016, 86, 2162–2170. [Google Scholar] [CrossRef]
- Assoum, M.; Philippe, C.; Isidor, B.; Perrin, L.; Makrythanasis, P.; Sondheimer, N.; Paris, C.; Douglas, J.; Lesca, G.; Antonarakis, S.; et al. Autosomal-Recessive Mutations in AP3B2, Adaptor-Related Protein Complex 3 Beta 2 Subunit, Cause an Early-Onset Epileptic Encephalopathy with Optic Atrophy. Am. J. Hum. Genet. 2016, 99, 1368–1376. [Google Scholar] [CrossRef]
- Aran, A.; Rosenfeld, N.; Jaron, R.; Renbaum, P.; Zuckerman, S.; Fridman, H.; Zeligson, S.; Segel, R.; Kohn, Y.; Kamal, L.; et al. Loss of function of PCDH12 underlies recessive microcephaly mimicking intrauterine infection. Neurology 2016, 86, 2016–2024. [Google Scholar] [CrossRef]
- Piard, J.; Hawkes, L.; Milh, M.; Villard, L.; Borgatti, R.; Romaniello, R.; Fradin, M.; Capri, Y.; Héron, D.; Nougues, M.C.; et al. The phenotypic spectrum of WWOX-related disorders: 20 additional cases of WOREE syndrome and review of the literature. Genet. Med. 2019, 21, 1308–1318. [Google Scholar] [CrossRef]
- Russo, S.; Marchi, M.; Cogliati, F.; Bonati, M.T.; Pintaudi, M.; Veneselli, E.; Saletti, V.; Balestrini, M.; Ben-Zeev, B.; Larizza, L. Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 2009, 10, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, N.; Lebon, S.; Jeannet, P.Y.; Jacquemont, S.; Billuart, P.; Bienvenu, T. Early-onset encephalopathy with epilepsy associated with a novel splice site mutation in SMC1A. Am. J. Med. Genet. Part. A 2015, 167, 3076–3081. [Google Scholar] [CrossRef]
- von Spiczak, S.; Helbig, K.L.; Shinde, D.N.; Huether, R.; Pendziwiat, M.; Lourenço, C.; Nunes, M.E.; Sarco, D.P.; Kaplan, R.A.; Dlugos, D.J.; et al. DNM1 encephalopathy: A new disease of vesicle fission. Neurology 2017, 89, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Vegas, N.; Cavallin, M.; Maillard, C.; Boddaert, N.; Toulouse, J.; Schaefer, E.; Lerman-Sagie, T.; Lev, D.; Magalie, B.; Moutton, S.; et al. Delineating FOXG1 syndrome: From congenital microcephaly to hyperkinetic encephalopathy. Neurol. Genet. 2018, 4, e281. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H.; Ohba, C.; Kato, M.; Maeda, T.; Araki, K.; Tajima, D.; Matsuo, M.; Hino-Fukuyo, N.; Kohashi, K.; Ishiyama, A.; et al. De novo GABRA1 mutations in Ohtahara and West syndromes. Epilepsia 2016, 57, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef]
- Møller, R.S.; Wuttke, T.V.; Helbig, I.; Marini, C.; Johannesen, K.M.; Brilstra, E.H.; Vaher, U.; Borggraefe, I.; Talvik, I.; Talvik, T.; et al. Mutations in GABRB3: From febrile seizures to epileptic encephalopathies. Neurology 2017, 88, 483–492. [Google Scholar] [CrossRef]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Janowski, R.; Alvi, M.; Self, J.E.; van Essen, T.J.; Vreeburg, M.; Rouhl, R.P.W.; Stevens, S.J.C.; Stegmann, A.P.A.; Schieving, J.; et al. PURA syndrome: Clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J. Med. Genet. 2018, 55, 104–113. [Google Scholar] [CrossRef]
- Salpietro, V.; Malintan, N.T.; Llano-Rivas, I.; Spaeth, C.G.; Efthymiou, S.; Striano, P.; Vandrovcova, J.; Cutrupi, M.C.; Chimenz, R.; David, E.; et al. Mutations in the Neuronal Vesicular SNARE VAMP2 Affect Synaptic Membrane Fusion and Impair Human Neurodevelopment. Am. J. Hum. Genet. 2019, 104, 721–730. [Google Scholar] [CrossRef]
- Straub, J.; Konrad, E.D.H.; Grüner, J.; Toutain, A.; Bok, L.A.; Cho, M.T.; Crawford, H.P.; Dubbs, H.; Douglas, G.; Jobling, R.; et al. Missense Variants in RHOBTB2 Cause a Developmental and Epileptic Encephalopathy in Humans, and Altered Levels Cause Neurological Defects in Drosophila. Am. J. Hum. Genet. 2018, 102, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. GRIN2B encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.B.; Tian, M.Q.; Gao, K.; Jiang, Y.W.; Wu, Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov. Disord. 2015, 30, 1290–1292. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; McWalter, K.; Schmidt, L.; Decker, A.; Picker, J.D.; Lincoln, S.; Sweetser, D.A.; Briere, L.C.; Harini, C.; Members of the Undiagnosed Diseases Network; et al. Expanding the phenotypic spectrum of GABRG2 variants: A recurrent GABRG2 missense variant associated with a severe phenotype. J. Neurogenet. 2017, 31, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, A.; Danti, F.R.; Spaull, R.; Leuzzi, V.; Mctague, A.; Kurian, M.A. The expanding spectrum of movement disorders in genetic epilepsies. Dev. Med. Child. Neurol. 2020, 62, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Cellini, E.; Vignoli, A.; Pisano, T.; Falchi, M.; Molinaro, A.; Accorsi, P.; Bontacchio, A.; Pinelli, L.; Giordano, L.; Guerrini, R.; et al. The Hyperkinetic Movement Disorder of FOXG1-related Epileptic-Dyskinetic Encephalopathy. Dev. Med. Child. Neurol. 2016, 58, 93–97. [Google Scholar] [CrossRef]
- Spagnoli, C.; Soliani, L.; Caraffi, S.G.; Baga, M.; Rizzi, S.; Salerno, G.G.; Frattini, D.; Garavelli, L.; Koskenvuo, J.; Pisani, F.; et al. Paroxysmal movement disorder with response to carbamazepine in a patient with RHOBTB2 developmental and epileptic encephalopathy. Parkinsonism Relat. Disord. 2020, 76, 54–55. [Google Scholar] [CrossRef]
- Wassenberg, T.; Molero-Luis, M.; Jeltsch, K.; Hoffmann, G.F.; Assmann, B.; Blau, N.; Garcia-Cazorla, A.; Artuch, R.; Pons, R.; Pearson, T.S.; et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet. J. Rare Dis. 2017, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Graziano, C.; Wischmeijer, A.; Pippucci, T.; Fusco, C.; Diquigiovanni, C.; Nõukas, M.; Sauk, M.; Kurg, A.; Rivieri, F.; Blau, N.; et al. Syndromic intellectual disability: A new phenotype caused by an aromatic amino acid decarboxylase gene (DDC) variant. Gene 2015, 559, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S. Mitochondrial disease and epilepsy. Dev. Med. Child. Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.L., Jr.; Agadi, S.; Reese, W.; Rehder, C.; Quach, M.M. Infantile spasms and hyperekplexia associated with isolated sulfite oxidase deficiency. JAMA Neurol. 2014, 71, 782–784. [Google Scholar] [CrossRef][Green Version]
- Carecchio, M.; Mencacci, N.E. Emerging monogenic complex hyperkinetic disorders. Curr. Neurol. Neurosci. Rep. 2017, 17, 97. [Google Scholar] [CrossRef]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2017, 15, 304–316. [Google Scholar] [CrossRef]
- Della Sala, G.; Pizzorusso, T. Synaptic plasticity and signaling in Rett syndrome. Dev. Neurobiol. 2014, 74, 178–196. [Google Scholar] [CrossRef]
- Nunes, M.L.; Yozawitz, E.G.; Zuberi, S.; Mizrahi, E.M.; Cilio, M.R.; Moshé, S.L.; Plouin, P.; Vanhatalo, S.; Pressler, R.M. Task Force on Neonatal Seizures, ILAE Commission on Classification & Terminology. Neonatal seizures: Is there a relationship between ictal electroclinical features and etiology? A critical appraisal based on a systematic literature review. Epilepsia Open 2019, 4, 10–29. [Google Scholar]
- He, N.; Lin, Z.J.; Wang, J.; Wei, F.; Meng, H.; Liu, X.R.; Chen, Q.; Su, T.; Shi, Y.W.; Yi, Y.H.; et al. Evaluating the pathogenic potential of genes with de novo variants in epileptic encephalopathies. Genet. Med. 2019, 21, 17–27. [Google Scholar] [CrossRef]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar]
- Kryukov, G.V.; Pennacchio, L.A.; Sunyaev, S.R. Most rare missense alleles are deleterious in humans: Implications for complex disease and association studies. Am. J. Hum. Genet. 2007, 80, 727–739. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Functional Role | Gene Name |
|---|---|
| Channelopathies | CACNA1E, KCNQ2, SCN2A, SCN8A |
| ATPase | ATP1A3 |
| Synaptopathies | AP3B2, DNM1, STXBP1, VAMP2, SYNJ1 |
| G protein transduction | GNAO1 |
| Transcription factors | FOXG1 |
| Transcription regulators | CDKL5, SMC1A, PURA |
| Neuronal connectivity/signal transduction | PCDH12 |
| Ubiquitination | RHOBTB2 |
| Receptors | GABRA1, GABRB2, GABRB3, GABRG2, GRIA2, GRIN2B |
| Enzymes | WWOX |
| Growth factors | FHF1 |
| Transporters | SLC13A5 |
| Electroclinical Phenotype | Gene Name |
|---|---|
| EOEE (22/27; 81.5%) | AP3B2, ATP1A3, CACNA1E, CDKL5, DNM1, FOXG1, FXF1, GABRA1, GABRB2, GABRB3, GABRG2, GNAO1, KCNQ2, PCDH12, RHOBTB2, SCN2A, SCN8A, SLC13A5, SMC1A, STXBP1, SYNJ1,WWOX |
| BFIS (1/27; 3.7%) | SCN2A |
| KCNQ2-RELATED ENCEPHALOPATHY (1/27; 3.7%) | KCNQ2 |
| NOT CATHEGORIZED AS EE/DE (4/27; 14.8%) | GRIA2, GRIN2B, KCNMA1, PURA, VAMP2 |
| Type of MD | Gene Name |
|---|---|
| HYPERKINETIC MD | |
| Ataxia | FHF1 KCNQ2 SCN8A STXBP1 SLC13A5 |
| Dystonia | AP3B2 CACNA1E DNM1 GABRB2 GNAO1 KCNQ2 PCDH12 RHOBTB2 SCN2A SCN8A STXBP1 WWOX SLC13A5 SYNJ1 |
| Status dystonicus | GNAO1 |
| Stereotypies | AP3B2 CDKL5 FOXG1 KCNQ2 GABRG2 SCN2A SMC1A STXBP1 |
| Tremor | KCNQ2 SCN8A STXBP1 |
| Chorea | CACNA1E GNAO1 KCNQ2 (with fever) RHOBTB2 SCN2A STXBP1 VAMP2 SLC13A5 |
| Choreo-athethosis | GABRA1 STXBP1 SLC13A5 |
| Athethosis | ATP1A3 PCDH12 |
| Dyskinesia | AP3B2 CACNA1E FOXG1 GABRB3 GNAO1 SCN8A STXBP1 SLC13A5 KCNQ2 (myoclonus-like) |
| Akathisia | GNAO1 |
| Myoclonus | CACNA1E GABRA1 GRIA2 KCNQ2 SCN8A STXBP1 WWOX |
| Oculogyric crises | GRIA2 SCN2A |
| Paroxysmal dyskinesia | KCNMA1 RHOBTB2 |
| Episodic ataxia | SCN2A |
| Paroxysmal non-epileptic polymorphous events | ATP1A3 SCN8A |
| Paroxysmal involuntary movements | WWOX |
| Startle/hyperekplexia | GNAO1 SCN8A STXBP1 WWOX |
| HYPOKINETIC MD | |
| Bradykinesia | |
| Hypokinesia | GRIA2 WWOX |
| Hypokinetic-rigid syndrome | STXBP1 |
| UNSPECIFIED | |
| PURA | |
| Brain MRI Findings | Genes |
|---|---|
| NORMAL | CACNA1E, CDKL5, DNM1, FXF1, GABRA1, GABRG2, GNAO1, GRIN2B, KCNMA1, KCNQ2, PURA, SCN8A, SLC13A5, STXBP1, VAMP2, WWOX |
| Cerebral atrophy | ATP1A3, CACNA1E, FOXG, GABRA1, GNAO1, KCNQ2, SCN8A, STXBP1 SYNJ1 |
| White matter abnormalities (including hypomyelination) | CACNA1E, GABRB2, GABRB3, GRIA2, KCNQ2, SCN2A, SLC13A5 SYNJ1 (periventricular WM gliosis) |
| Cerebellar atrophy | FHF1, GABRA1, GRIA2 |
| Corpus callosum hypoplasia | FOXG1,GABRA1, WWOX |
| Thin CC | AP3B2, CACNA1E, GNAO1, KCNQ2, SMC1A, SYNJ1, WWOX |
| Myelination delay | ATP1A3, CACN1E, FOXG1, GNAO1, RHOBTB2, SCN8A |
| Basal Ganglia hyperintensities | ATP1A3 (GP), CACNA1E, GNAO1 (GP), KCNQ2 (BG and THALAMI, neonatal age), SCN2A |
| Cortical malformations | FOXG1, KCNQ2 (simplified gyral pattern) |
| Cerebellar hypoplasia | GRIA2 (vermis) |
| Enlarged extra-axial space | AP3B2, WWOX |
| Brainstem hyperintensities | SCN2A |
| Midbrain, hypothalamus and optic trait dysplasia | PCDH12 |
| Small frontal lobes | SMC1A |
| Small thalami | KCNQ2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spagnoli, C.; Fusco, C.; Percesepe, A.; Leuzzi, V.; Pisani, F. Genetic Neonatal-Onset Epilepsies and Developmental/Epileptic Encephalopathies with Movement Disorders: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 4202. https://doi.org/10.3390/ijms22084202
Spagnoli C, Fusco C, Percesepe A, Leuzzi V, Pisani F. Genetic Neonatal-Onset Epilepsies and Developmental/Epileptic Encephalopathies with Movement Disorders: A Systematic Review. International Journal of Molecular Sciences. 2021; 22(8):4202. https://doi.org/10.3390/ijms22084202
Chicago/Turabian StyleSpagnoli, Carlotta, Carlo Fusco, Antonio Percesepe, Vincenzo Leuzzi, and Francesco Pisani. 2021. "Genetic Neonatal-Onset Epilepsies and Developmental/Epileptic Encephalopathies with Movement Disorders: A Systematic Review" International Journal of Molecular Sciences 22, no. 8: 4202. https://doi.org/10.3390/ijms22084202
APA StyleSpagnoli, C., Fusco, C., Percesepe, A., Leuzzi, V., & Pisani, F. (2021). Genetic Neonatal-Onset Epilepsies and Developmental/Epileptic Encephalopathies with Movement Disorders: A Systematic Review. International Journal of Molecular Sciences, 22(8), 4202. https://doi.org/10.3390/ijms22084202