
Ligand-Receptor Interactions and Machine Learning in GCGR and GLP-1R Drug Discovery


Abstract
1. Introduction
2. Results
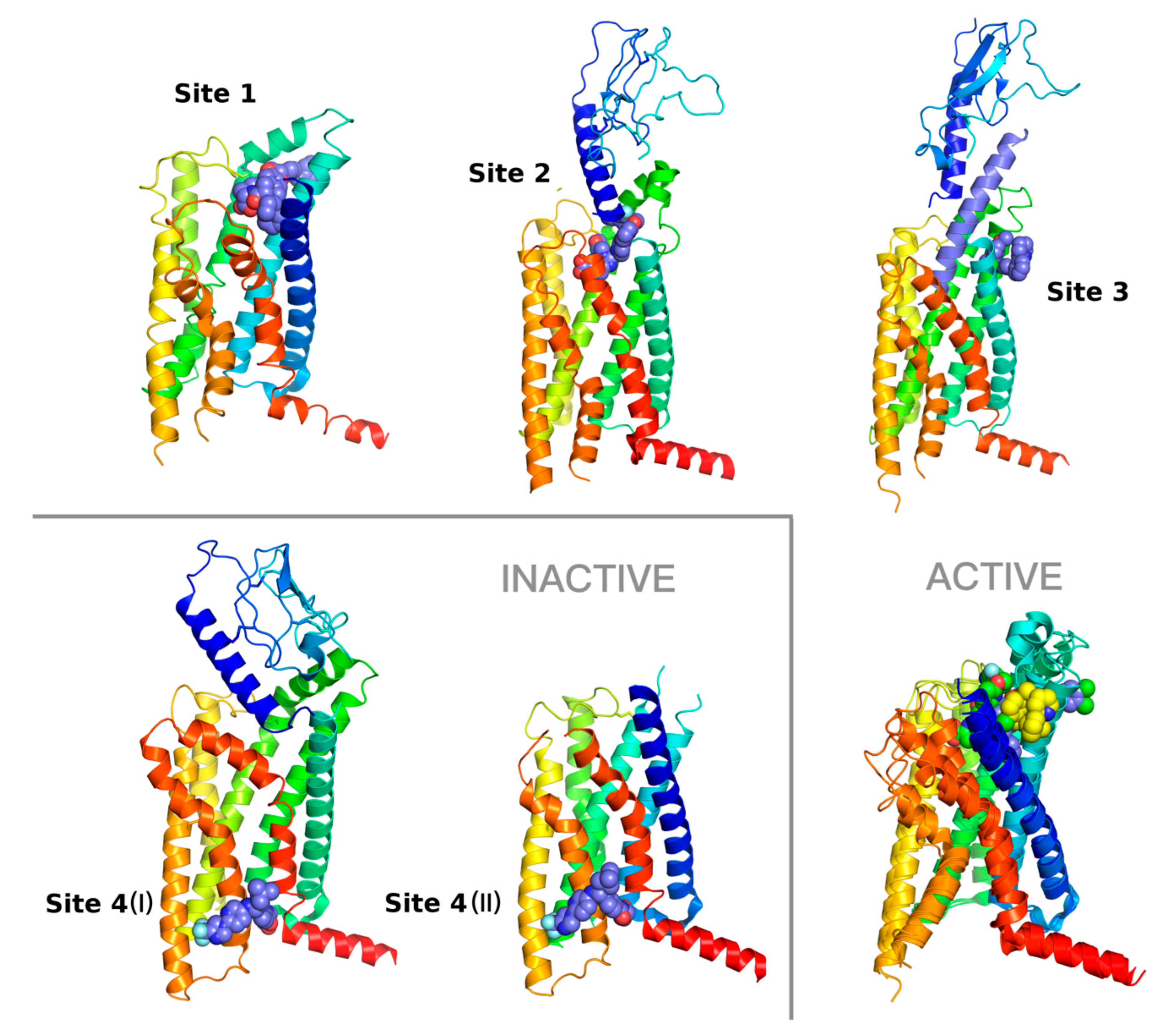
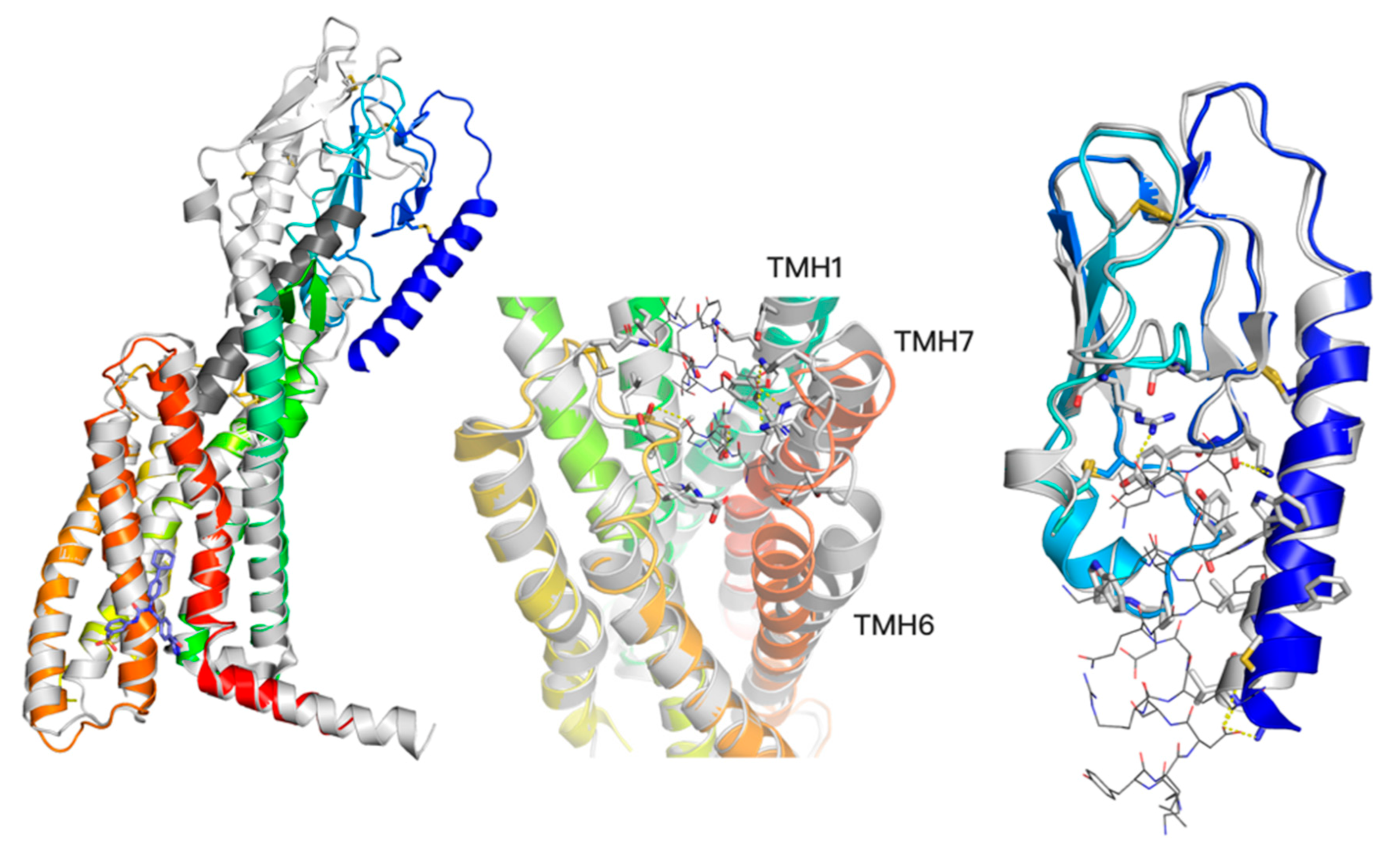
2.1. Recent Advances in Crystallography and Cryo-EM of the Glucagon Receptor Family
2.2. Annotation of Compounds Deposited in ChEMBL
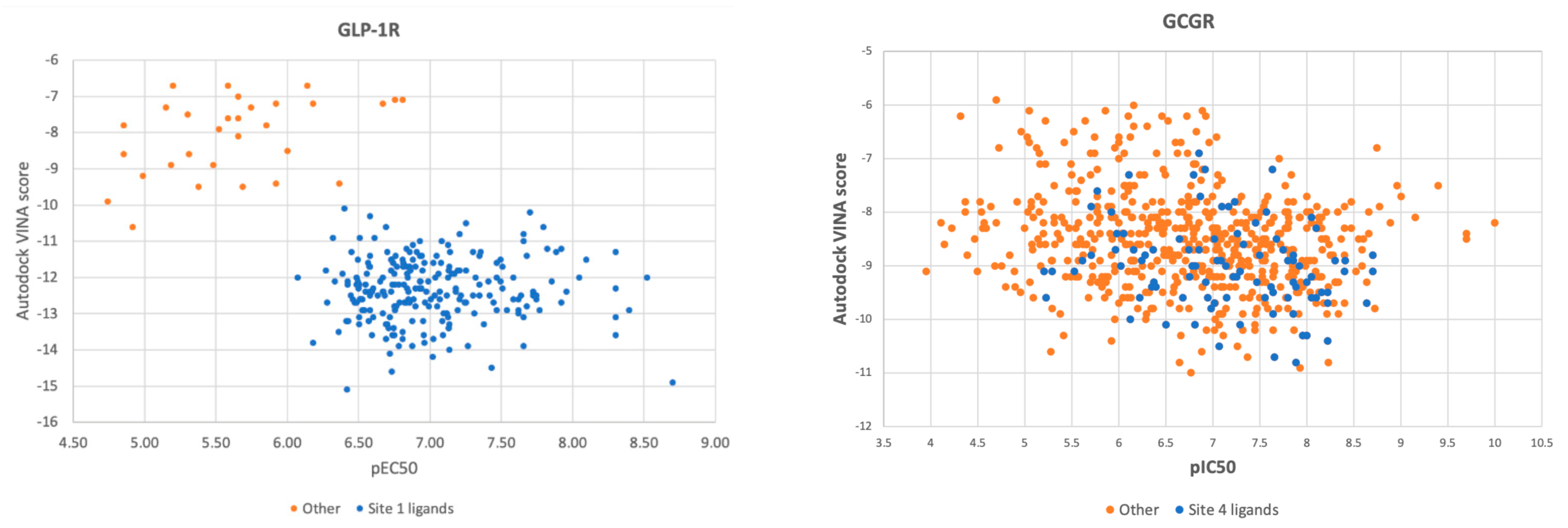
2.3. Response to Drug-Receptor Structure-Based Predictions
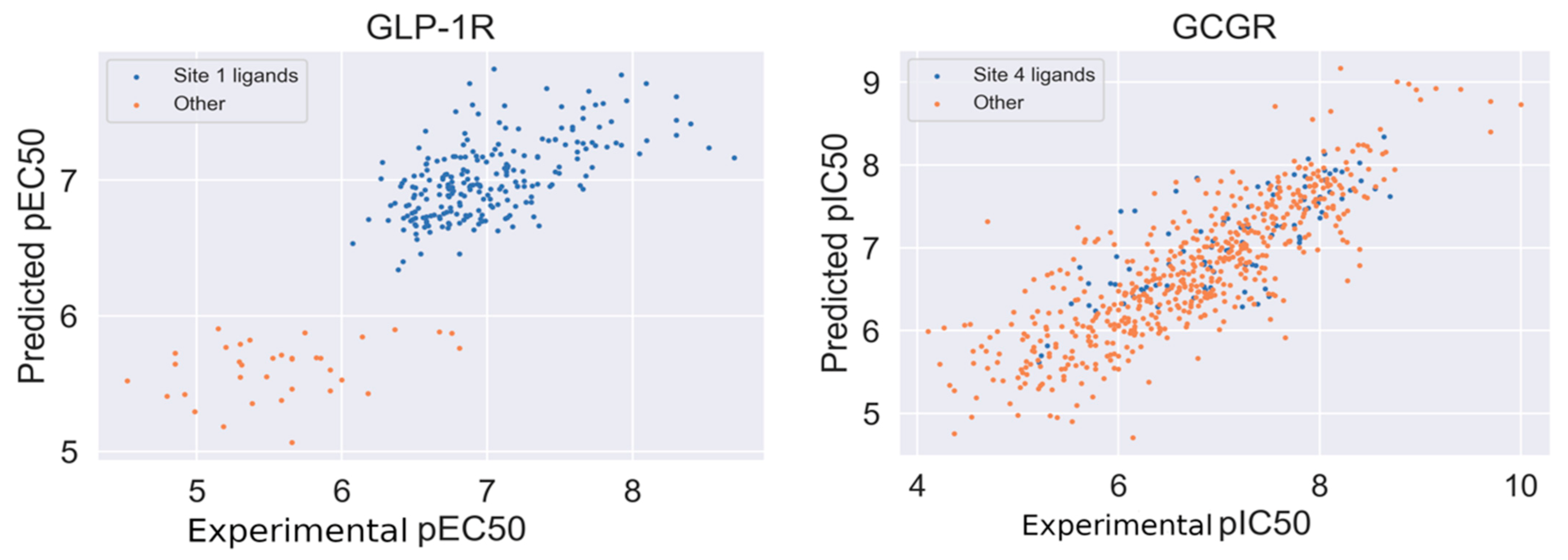
2.4. Response to Drug—Ligand-Based Predictions
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Data Analysis
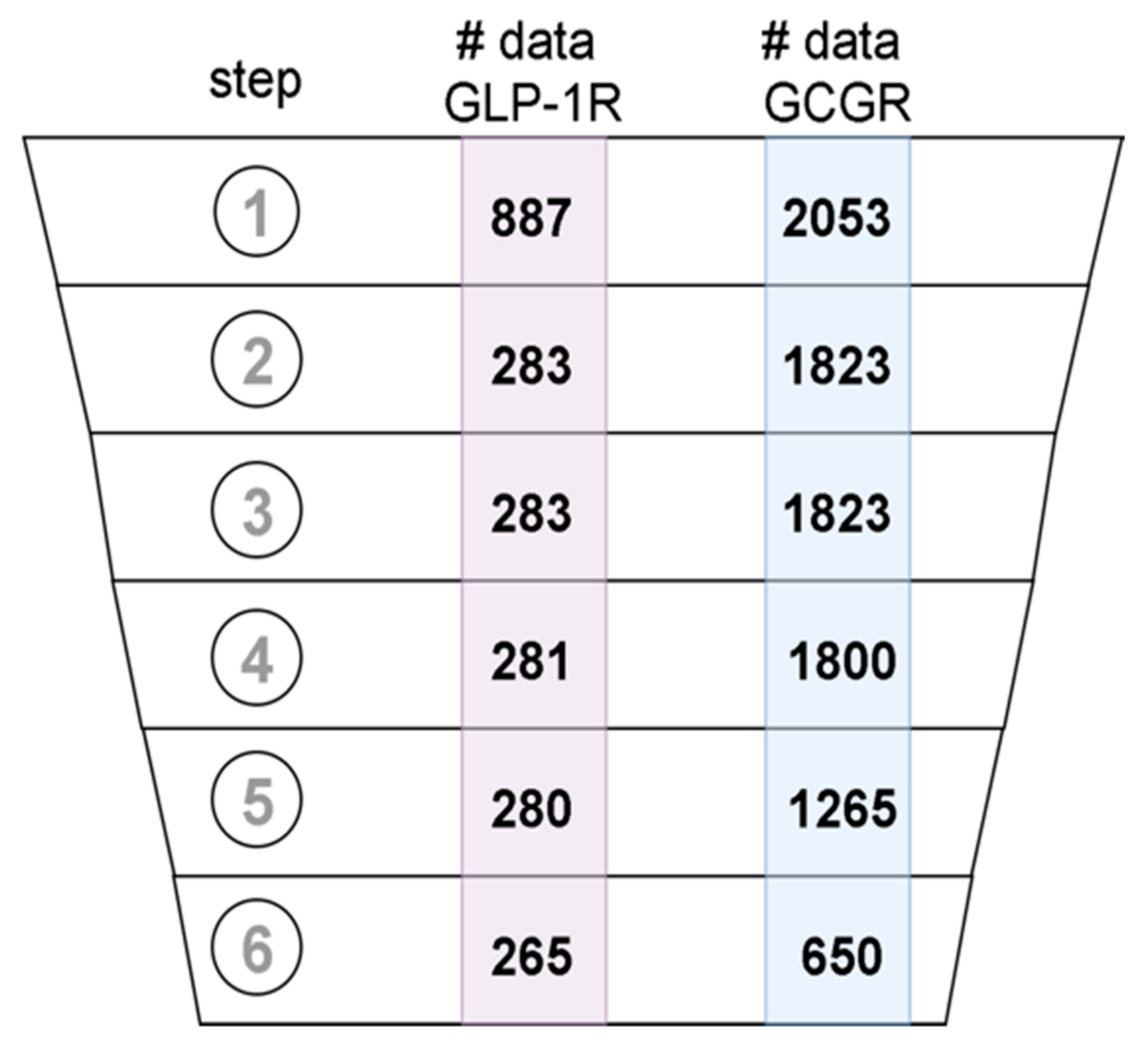
4.3. Data Curation
4.4. Data Storage
4.5. Data Usage
4.5.1. QSAR Modeling and Validation
4.5.2. Molecular Docking
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gribble, F.M.; Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol. 2019, 15, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.; DiMarchi, R.D.; Tschöp, M.H.; Finan, B.; Campbell, J. Targeting the Incretin/Glucagon System With Triagonists to Treat Diabetes. Endocr. Rev. 2018, 39, 719–738. [Google Scholar] [CrossRef]
- Latek, D.; Rutkowska, E.; Niewieczerzal, S.; Cielecka-Piontek, J. Drug-induced diabetes type 2: In silico study involving class B GPCRs. PLoS ONE 2019, 14, e0208892. [Google Scholar] [CrossRef]
- Scheen, A.J.; Paquot, N.; Lefèbvre, P.J. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin. Investig. Drugs 2017, 26, 1373–1389. [Google Scholar] [CrossRef]
- Song, G.; Yang, D.; Wang, Y.; De Graaf, C.; Zhou, Q.; Jiang, S.; Liu, K.; Cai, X.; Dai, A.; Lin, G.; et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nat. Cell Biol. 2017, 546, 312–315. [Google Scholar] [CrossRef]
- Zhang, H.; Qiao, A.; Yang, D.; Yang, L.; Dai, A.; De Graaf, C.; Reedtz-Runge, S.; Dharmarajan, V.; Zhang, H.; Han, G.W.; et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nat. Cell Biol. 2017, 546, 259–264. [Google Scholar] [CrossRef]
- Wu, F.; Yang, L.; Hang, K.; Laursen, M.; Wu, L.; Han, G.W.; Ren, Q.; Roed, N.K.; Lin, G.; Hanson, M.A.; et al. Full-length human GLP-1 receptor structure without orthosteric ligands. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liao, J.; Li, N.; Zhou, C.; Liu, Q.; Wang, G.; Zhang, R.; Zhang, S.; Lin, L.; Chen, K.; et al. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc. Natl. Acad. Sci. USA 2007, 104, 943–948. [Google Scholar] [CrossRef]
- Donnelly, D. The structure and function of the glucagon-like peptide-1 receptor and its ligands. Br. J. Pharmacol. 2012, 166, 27–41. [Google Scholar] [CrossRef]
- Hollenstein, K.; de Graaf, C.; Bortolato, A.; Wang, M.-W.; Marshall, F.H.; Stevens, R.C. Insights into the structure of class B GPCRs. Trends Pharmacol. Sci. 2014, 35, 12–22. [Google Scholar] [CrossRef]
- Bortolato, A.; Doré, A.S.; Hollenstein, K.; Tehan, B.G.; Mason, J.S.; Marshall, F.H. Structure of Class B GPCRs: New horizons for drug discovery. Br. J. Pharmacol. 2014, 171, 3132–3145. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, K.; Kean, J.; Bortolato, A.; Cheng, R.K.Y.; Doré, A.S.; Jazayeri, A.; Cooke, R.M.; Weir, M.; Marshall, F.H. Structure of class B GPCR corticotropin-releasing factor receptor Nat. Cell Biol. 2013, 499, 438–443. [Google Scholar] [CrossRef]
- Zhao, P.; Liang, Y.-L.; Belousoff, M.J.; Deganutti, G.; Fletcher, M.M.; Willard, F.S.; Bell, M.G.; Christe, M.E.; Sloop, K.W.; Inoue, A.; et al. Activation of the GLP-1 receptor by a non-peptidic agonist. Nat. Cell Biol. 2020, 577, 432–436. [Google Scholar] [CrossRef]
- Ma, H.; Huang, W.; Wang, X.; Zhao, L.; Jiang, Y.; Liu, F.; Guo, W.; Sun, X.; Zhong, W.; Yuan, D.; et al. Structural insights into the activation of GLP-1R by a small molecule agonist. Cell Res. 2020, 30, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Bueno, A.B.; Sun, B.; Willard, F.S.; Feng, D.; Ho, J.D.; Wainscott, D.B.; Showalter, A.D.; Vieth, M.; Chen, Q.; Stutsman, C.; et al. Structural insights into probe-dependent positive allosterism of the GLP-1 receptor. Nat. Chem. Biol. 2020, 16, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sun, B.; Yoshino, H.; Feng, D.; Suzuki, Y.; Fukazawa, M.; Nagao, S.; Wainscott, D.B.; Showalter, A.D.; Droz, B.A.; et al. Structural basis for GLP-1 receptor activation by LY3502970, an orally active nonpeptide agonist. Proc. Natl. Acad. Sci. USA 2020, 117, 29959–29967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Belousoff, M.J.; Zhao, P.; Kooistra, A.J.; Truong, T.T.; Ang, S.Y.; Underwood, C.R.; Egebjerg, T.; Šenel, P.; Stewart, G.D.; et al. Differential GLP-1R Binding and Activation by Peptide and Non-peptide Agonists. Mol. Cell 2020, 80, 485–500.e7. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Burggraaff, L.; Van Veen, A.; Lam, C.C.; Van Vlijmen, H.W.T.; Ijzerman, A.P.; Van Westen, G.J.P. Annotation of Allosteric Compounds to Enhance Bioactivity Modeling for Class A GPCRs. J. Chem. Inf. Model. 2020, 60, 4664–4672. [Google Scholar] [CrossRef]
- Fourches, D.; Ash, J. 4D-quantitative structure–activity relationship modeling: Making a comeback. Expert Opin. Drug Discov. 2019, 14, 1227–1235. [Google Scholar] [CrossRef]
- Venugopal, P.P.; Das, B.K.; Soorya, E.; Chakraborty, D.; Pushyaraga, P.V. Effect of hydrophobic and hydrogen bonding interactions on the potency of ß-alanine analogs of G-protein coupled glucagon receptor inhibitors. Proteins Struct. Funct. Bioinform. 2020, 88, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Al-Zamel, N.; Al-Sabah, S.; Luqmani, Y.; Adi, L.; Chacko, S.; Schneider, T.D.; Krasel, C. A Dual GLP-1/GIP Receptor Agonist Does Not Antagonize Glucagon at Its Receptor but May Act as a Biased Agonist at the GLP-1 Receptor. Int. J. Mol. Sci. 2019, 20, 3532. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, D.S.; Bagger, J.I.; Bergmann, N.C.; Lund, A.; Christensen, M.B.; Vilsbøll, T.; Knop, F.K. The Effects of Dual GLP-1/GIP Receptor Agonism on Glucagon Secretion—A Review. Int. J. Mol. Sci. 2019, 20, 4092. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Zhang, X.; Qiao, A.; Dai, A.; Belousoff, M.J.; Tan, Q.; Shao, L.; Zhong, L.; Lin, G.; Liang, Y.-L.; et al. Cryo-electron microscopy structure of the glucagon receptor with a dual-agonist peptide. J. Biol. Chem. 2020, 295, 9313–9325. [Google Scholar] [CrossRef]
- Pan, C.Q.; Buxton, J.M.; Yung, S.L.; Tom, I.; Yang, L.; Chen, H.; MacDougall, M.; Bell, A.; Claus, T.H.; Clairmont, K.B.; et al. Design of a Long Acting Peptide Functioning as Both a Glucagon-like Peptide-1 Receptor Agonist and a Glucagon Receptor Antagonist. J. Biol. Chem. 2006, 281, 12506–12515. [Google Scholar] [CrossRef] [PubMed]
- Claus, T.H.; Pan, C.Q.; Buxton, J.M.; Yang, L.; Reynolds, J.C.; Barucci, N.; Burns, M.; Ortiz, A.A.; Roczniak, S.; Livingston, J.N.; et al. Dual-acting peptide with prolonged glucagon-like petide-1 receptor agonist and glucagon receptor antagonist activity for the treatment of type 2 diabetes. J. Endocrinol. 2007, 192, 371–380. [Google Scholar] [CrossRef]
- Mizera, M.; Latek, D.; Cielecka-Piontek, J. Virtual Screening of C. Sativa Constituents for the Identification of Selective Ligands for Cannabinoid Receptor. Int. J. Mol. Sci. 2020, 21, 5308. [Google Scholar] [CrossRef]
- Siu, F.Y.; He, M.; De Graaf, C.; Han, G.W.; Yang, D.; Zhang, Z.; Zhou, C.; Xu, Q.; Wacker, D.; Joseph, J.S.; et al. Structure of the human glucagon class B G-protein-coupled receptor. Nat. Cell Biol. 2013, 499, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Pasznik, P.; Rutkowska, E.; Niewieczerzal, S.; Cielecka-Piontek, J.; Latek, D. Potential off-target effects of beta-blockers on gut hormone receptors: In silico study including GUT-DOCK—A web service for small-molecule docking. PLoS ONE 2019, 14, e0210705. [Google Scholar] [CrossRef] [PubMed]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem. 2019, 62, 88–127. [Google Scholar] [CrossRef]
- Jazayeri, A.; Doré, A.S.; Lamb, D.; Krishnamurthy, H.; Southall, S.M.; Baig, A.H.; Bortolato, A.; Koglin, M.; Robertson, N.J.; Errey, J.C.; et al. Extra-helical binding site of a glucagon receptor antagonist. Nat. Cell Biol. 2016, 533, 274–277. [Google Scholar] [CrossRef]
- Jazayeri, A.; Rappas, M.; Brown, A.J.H.; Kean, J.; Errey, J.C.; Robertson, N.J.; Fiez-Vandal, C.; Andrews, S.P.; Congreve, M.; Bortolato, A.; et al. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nat. Cell Biol. 2017, 546, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiao, A.; Yang, L.; Van Eps, N.; Frederiksen, K.S.; Yang, D.; Dai, A.; Cai, X.; Zhang, H.; Yi, C.; et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nat. Cell Biol. 2018, 553, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.N.H.; Huy, N.T.; Van Bay, M.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model. 2019, 60, 204–211. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, P.; Wang, Y.; Qin, C.; Chen, S.; He, W.; Tao, L.; Tan, Y.; Gao, D.; Wang, B.; et al. CMAUP: A database of collective molecular activities of useful plants. Nucleic Acids Res. 2019, 47, D1118–D1127. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar]
- Daylight Chemical Information Systems, Inc., Laguna Niguel, CA, USA. Available online: https://www.daylight.com/ (accessed on 17 February 2021).
- Rogers, D.J.; Tanimoto, T.T. A Computer Program for Classifying Plants. Science 1960, 132, 1115–1118. [Google Scholar] [CrossRef]
- Schrödinger, Inc. [Computer Software]. New York, NY, USA. Available online: https://www.schrodinger.com/ (accessed on 17 February 2021).
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.Y. LightGBM: A highly efficient gradient boosting decision tree. In Advances in Neural Information Processing Systems; NIPS: San Diego, CA, USA, 2017. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Chemin. 2011, 3, 33. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | PDB Id 1 | Ligand | Ligand Type | Binding Site |
|---|---|---|---|---|
| GCGR | 5XEZ, 5XF1 [6] |  | NAM 2 | Binding site 4, allosteric, lipids-facing, outside of TMD 3 |
| GLP-1R | 5VEX [5] |  | NAM | Binding site 4, allosteric, lipids-facing, outside of TMD |
| 5VEW [5,7] |  | NAM | Binding site 4, allosteric, lipids-facing, outside of TMD | |
| 6ORV [13] |  | PAM 4 | Binding site 1, allosteric, close to orthosteric 5 | |
| 6VCB [15] |  | PAM | Binding site 3, allosteric, close to orthosteric | |
| 7C2E [14] |  | Full agonist | Binding site 2, orthosteric | |
| 6X1A [17] |  | Full agonist | Binding site 2, orthosteric | |
| 6XOX [16] |  | Full agonist | Binding site 2, orthosteric | |
| 6X19 [17] |  | Full agonist | Binding site 2, orthosteric |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizera, M.; Latek, D. Ligand-Receptor Interactions and Machine Learning in GCGR and GLP-1R Drug Discovery. Int. J. Mol. Sci. 2021, 22, 4060. https://doi.org/10.3390/ijms22084060
Mizera M, Latek D. Ligand-Receptor Interactions and Machine Learning in GCGR and GLP-1R Drug Discovery. International Journal of Molecular Sciences. 2021; 22(8):4060. https://doi.org/10.3390/ijms22084060
Chicago/Turabian StyleMizera, Mikołaj, and Dorota Latek. 2021. "Ligand-Receptor Interactions and Machine Learning in GCGR and GLP-1R Drug Discovery" International Journal of Molecular Sciences 22, no. 8: 4060. https://doi.org/10.3390/ijms22084060
APA StyleMizera, M., & Latek, D. (2021). Ligand-Receptor Interactions and Machine Learning in GCGR and GLP-1R Drug Discovery. International Journal of Molecular Sciences, 22(8), 4060. https://doi.org/10.3390/ijms22084060