
AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery



Abstract
1. Introduction
2. Results and Discussion
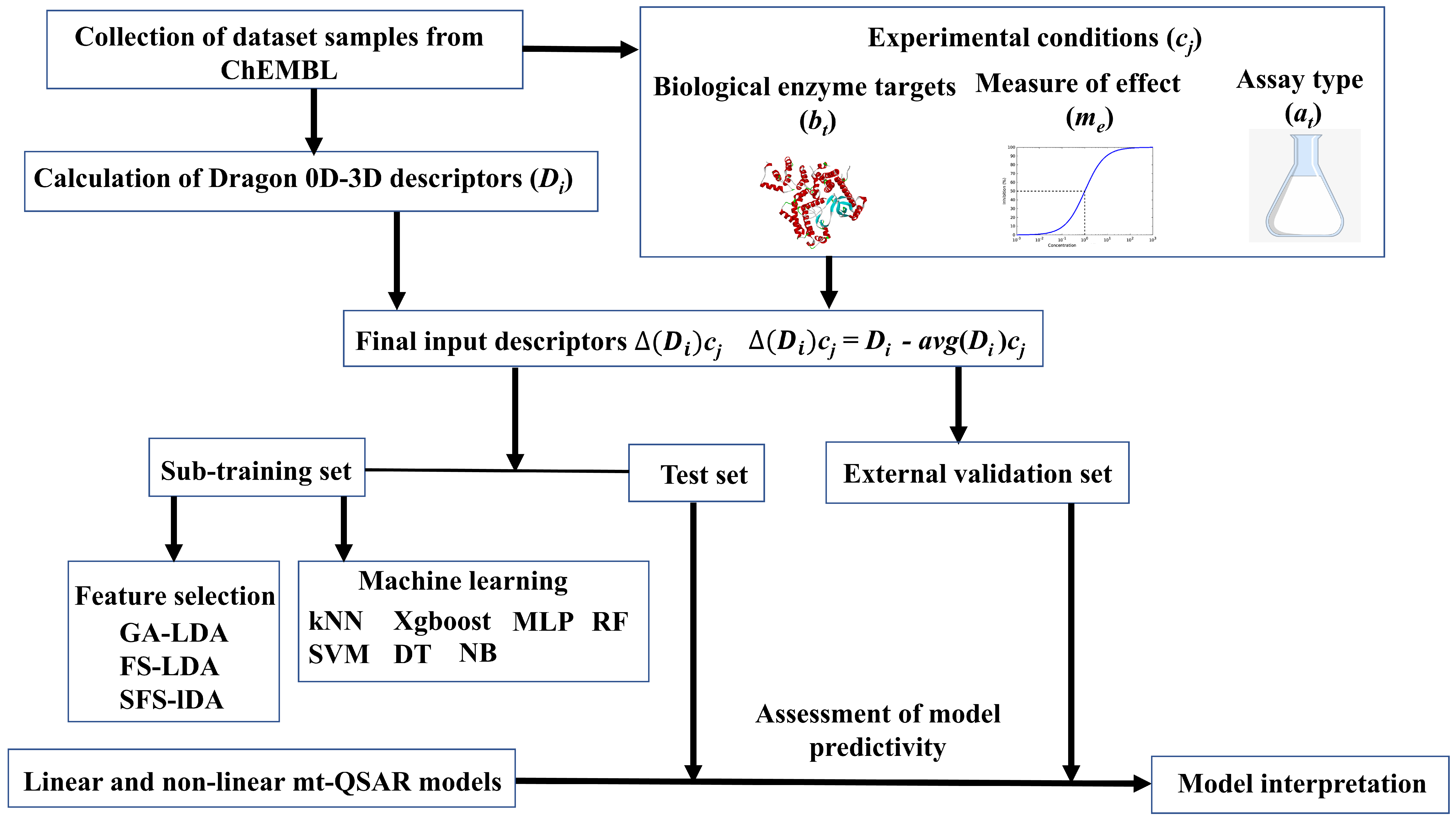
2.1. Dataset Collection and Preparation
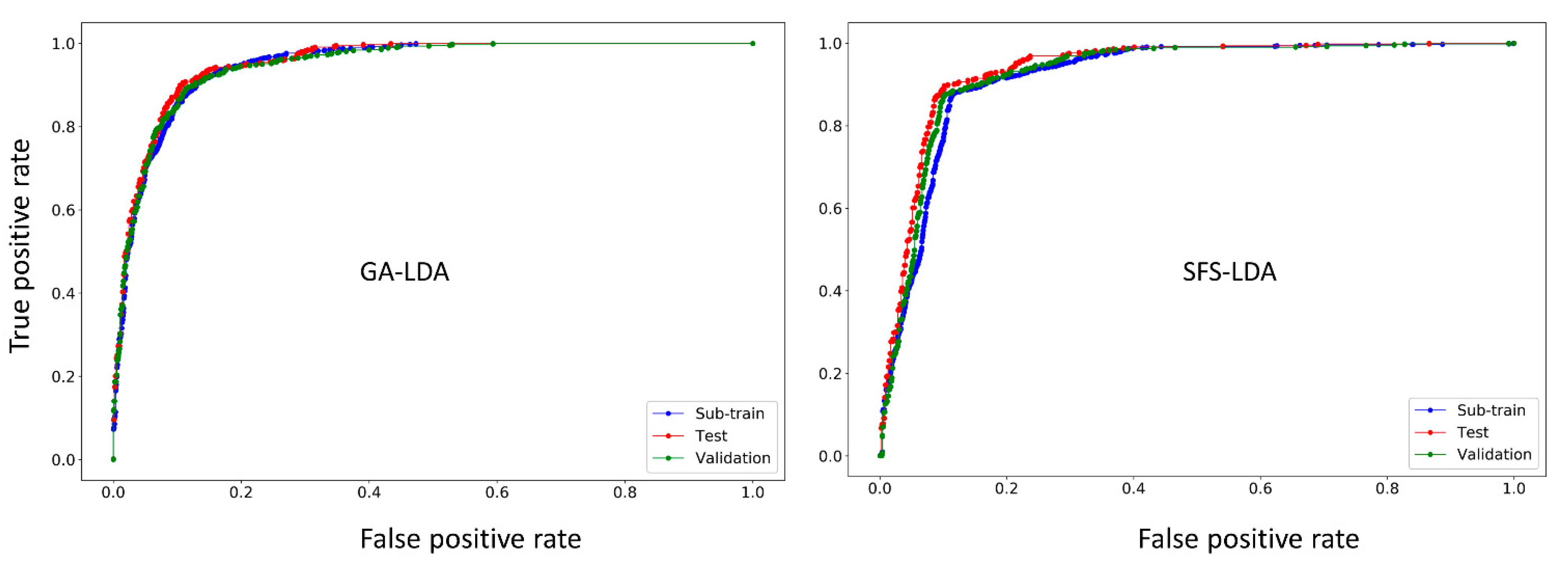
2.2. Linear Interpretable Mt-QSAR Models
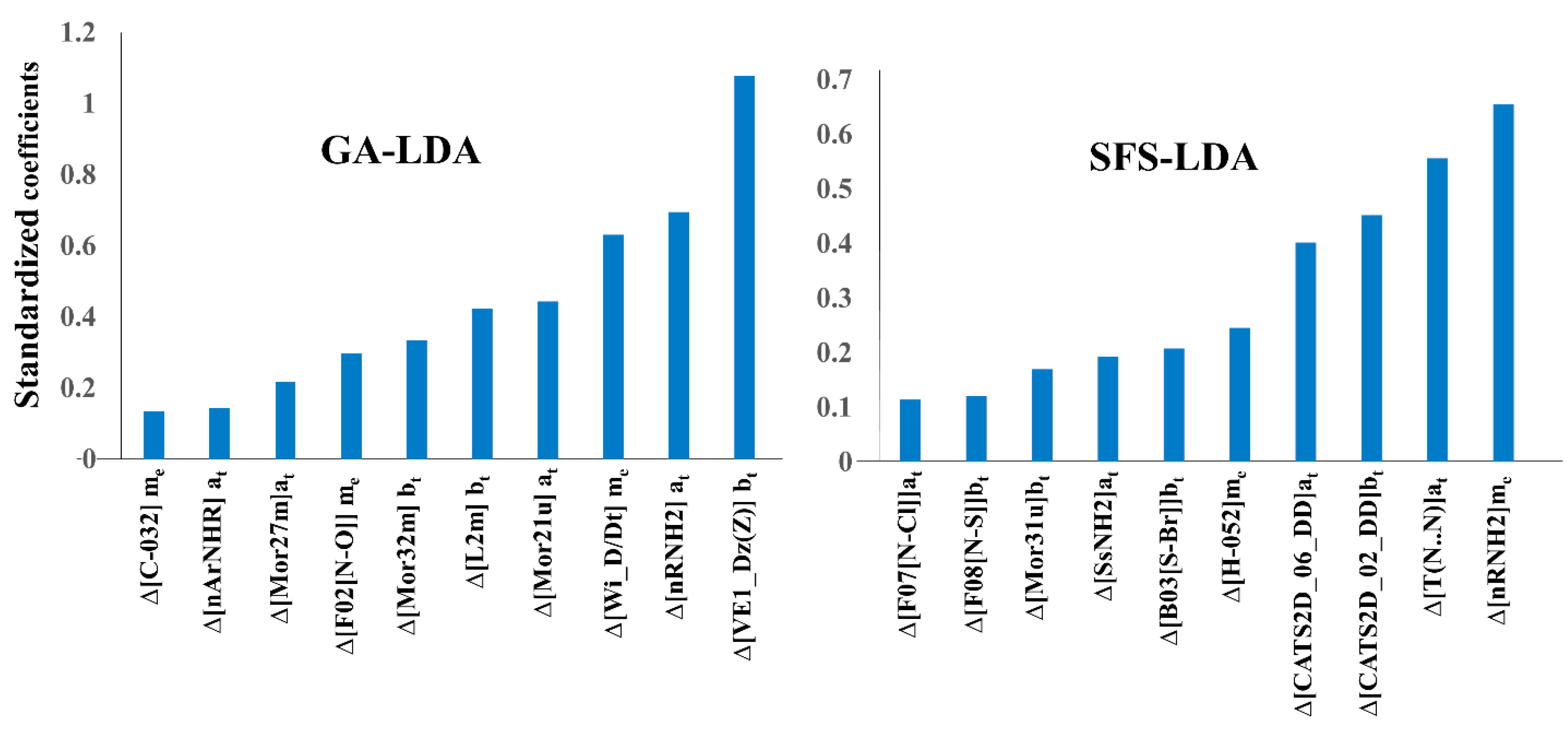
2.3. Interpretation of Molecular Descriptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Deviation Descriptors | cj | Core Descriptor | Description | Descriptor Type a |
|---|---|---|---|---|---|
| GA-LDA | biological target | VE1_Dz(Z) | Coefficient sum of the last eigenvector (absolute values) from Barysz matrix weighted by atomic number | 2D matrix-based | |
| biological target | Mor32m | Signal 32/weighted by mass | 3D-MoRSE | ||
| biological target | L2m | 2nd component size/weighted by mass | WHIM directional | ||
| measure of effect | C−032 | X--CX--X | Atom-centered fragments | ||
| measure of effect | F02[N−O] | Frequency of N−O at topological distance 2 | 2D Atom Pairs | ||
| measure of effect | Wi_D/Dt | Wiener-like index from distance/detour matrix | 2D matrix-based | ||
| assay type | nRNH2 | Number of primary amines (aliphatic) | Functional group counts | ||
| assay type | nArNHR | Number of secondary amines (aromatic) | Functional group counts | ||
| assay type | Mor27m | Signal 27/weighted by mass | 3D-MoRSE | ||
| assay type | Mor21u | Signal 21/unweighted | 3D-MoRSE | ||
| FS-LDA | biological target | D/Dtr05 | Distance/detour ring index of order 5 | Ring | |
| biological target | C−030 | X--CH--X | Atom-centered fragments | ||
| biological target | nCt | Number of total tertiary C(sp3) | Functional group counts | ||
| biological target | L2m | 2nd component size/weighted by mass | WHIM directional | ||
| biological target | CATS3D_18_DL | Donor-Lipophilic BIN 18 (18–19 Å) | 3D-CATS | ||
| biological target | CATS3D_10_PL | Positive-Lipophilic BIN 10 (10–11 Å) | 3D-CATS | ||
| measure of effect | nPyridines | Number of Pyridines | Functional group counts | ||
| measure of effect | T(N..O) | Sum of topological distances between N..O | 2D Atom Pairs | ||
| measure of effect | CATS3D_07_DA | Donor-Acceptor BIN 7 (7–8 Å) | 3D-CATS | ||
| assay type | nRNH2 | Number of primary amines (aliphatic) | Functional group counts | ||
| SFS-LDA | biological target | F08[N−S] | Frequency of N−S at topological distance 8 | 2D Atom Pairs | |
| biological target | B03[S−Br] | Presence/absence of S−Br at topological distance 3 | 2D Atom Pairs | ||
| biological target | Mor31u | Signal 31/unweighted | 3D-MoRSE | ||
| biological target | CATS2D_02_DD | Donor-Donor at lag 2 | 2D-CATS | ||
| measure of effect | H−052 | H attached to C0(sp3) with 1X attached to next C | Atom-centered fragments | ||
| measure of effect | nRNH2 | Number of primary amines (aliphatic) | Functional group counts | ||
| assay type | T(N..N) | Sum of topological distances between N..N | 2D Atom Pairs | ||
| assay type | F07[N−Cl] | Frequency of N-Cl at topological distance 7 | 2D Atom Pairs | ||
| assay type | SsNH2 | Sum of sNH2 E-states | Atom-type E-state indices | ||
| assay type | CATS2D_06_DD | Donor-Donor at lag 6 | 2D-CATS |
2.4. Non-Linear Predictive Mt-QSAR Models
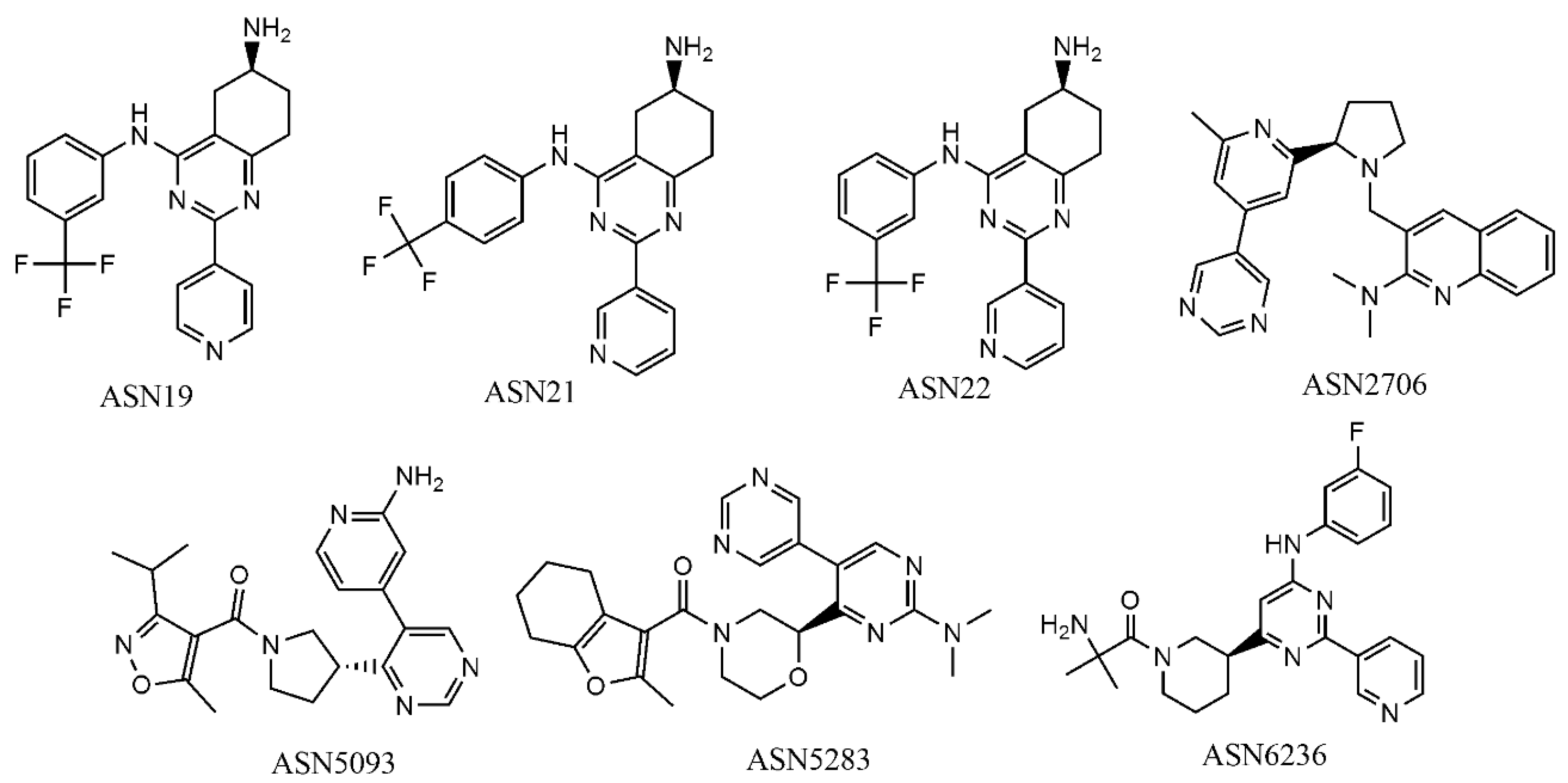
2.5. Virtual Screening
2.6. Pharmacophore Based Biological Target Identification
2.7. Structure-Based Prediction of the Virtual Hits
3. Materials and Methods
3.1. Descriptor Calculation
3.2. Development of Linear Interpretable Models
3.3. Non-Linear Model Development
3.4. PharmMapper Based Prediction of Biological Targets
3.5. Homology Modeling
3.6. Molecular Docking
3.7. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Santi, S.A.; Douglas, A.C.; Lee, H. The AKT isoforms, their unique functions and potential as anticancer therapeutic targets. Biomol. Concepts 2010, 1, 389–401. [Google Scholar] [CrossRef]
- Hinz, N.; Jucker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef]
- Barile, E.; De, S.K.; Carlson, C.B.; Chen, V.; Knutzen, C.; Riel-Mehan, M.; Yang, L.; Dahl, R.; Chiang, G.; Pellecchia, M. Design, Synthesis, and structure-activity relationships of 3-Ethynyl-1H-indazoles as Inhibitors of the phosphatidylintositol 3-kinase signaling pathway. J. Med. Chem. 2010, 53, 8368–8375. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Gradinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The AKT pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. AKT kinase pathway: A leading target in cancer research. Sci. World J. 2013, 2013, 756134. [Google Scholar] [CrossRef]
- Dumble, M.; Crouthamel, M.C.; Zhang, S.Y.; Schaber, M.; Levy, D.; Robell, K.; Liu, Q.; Figueroa, D.J.; Minthorn, E.A.; Seefeld, M.A.; et al. Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor. PLoS ONE 2014, 9, e100880. [Google Scholar] [CrossRef]
- Mundi, P.S.; Sachdev, J.; McCourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956. [Google Scholar] [CrossRef]
- Hers, I.; Vincent, E.E.; Tavare, J.M. AKT signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. AKT inhibitors in cancer treatment: The long journey from drug discovery to clinical use. Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef]
- Song, M.Q.; Bode, A.M.; Dong, Z.G.; Lee, M.H. AKT as a therapeutic target for cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef]
- Narayan, R.S.; Fedrigo, C.A.; Brands, E.; Dik, R.; Stalpers, L.J.A.; Baumert, B.G.; Slotman, B.J.; Westerman, B.A.; Peters, G.J.; Sminia, P. The allosteric AKT inhibitor MK2206 shows a synergistic interaction with chemotherapy and radiotherapy in glioblastoma spheroid cultures. BMC Cancer 2017, 17, 204. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M.N.D.S. Development of multi-target chemometric models for the inhibition of class i PI3K enzyme isoforms: A case study using qsar-co tool. Int. J. Mol. Sci. 2019, 20, 4191. [Google Scholar] [CrossRef]
- Lima, A.N.; Philot, E.A.; Trossini, G.H.G.; Scott, L.P.B.; Maltarollo, V.G.; Honorio, K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 225–239. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Kausar, S.; Falcao, A.O. An automated framework for QSAR model building. J. Cheminform. 2018, 10, 1. [Google Scholar] [CrossRef]
- Halder, A.K.; Giri, A.K.; Cordeiro, M.N.D.S. Multi-target chemometric modelling, fragment analysis and virtual screening with erk inhibitors as potential anticancer agents. Molecules 2019, 24, 3909. [Google Scholar] [CrossRef]
- Lewis, R.A.; Wood, D. Modern 2D QSAR for drug discovery. Wires Comput. Mol. Sci. 2014, 4, 505–522. [Google Scholar] [CrossRef]
- Ambure, P.; Halder, A.K.; Gonzalez Diaz, H.; Cordeiro, M. QSAR-Co: An open source software for developing robust multitasking or multitarget classification-based QSAR models. J. Chem. Inf. Model. 2019, 59, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A. Multi-Scale modeling in drug discovery against infectious diseases. Mini Rev. Med. Chem. 2019, 19, 1560–1563. [Google Scholar] [CrossRef]
- Speck-Planche, A. Multiple perspectives in anti-cancer drug discovery: From old targets and natural products to innovative computational approaches. Anticancer Agents Med. Chem. 2019, 19, 146–147. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Scotti, M.T. BET bromodomain inhibitors: Fragment-based in silico design using multi-target QSAR models. Mol. Divers. 2019, 23, 555–572. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3716. [Google Scholar] [CrossRef]
- Vyas, V.K.; Ghate, M.; Gupta, N. 3D QSAR and HQSAR analysis of protein kinase B (PKB/AKT) inhibitors using various alignment methods. Arab. J. Chem. 2017, 10, S2182–S2195. [Google Scholar] [CrossRef]
- Dong, X.W.; Jiang, C.Y.; Hu, H.Y.; Yan, J.Y.; Chen, J.; Hu, Y.Z. QSAR study of AKT/protein kinase B (PKB) inhibitors using support vector machine. Eur. J. Med. Chem. 2009, 44, 4090–4097. [Google Scholar] [CrossRef]
- Fei, J.; Zhou, L.; Liu, T.; Tang, X.Y. Pharmacophore modeling, virtual screening, and molecular docking studies for discovery of novel AKT2 inhibitors. Int. J. Med. Sci. 2013, 10, 265–275. [Google Scholar] [CrossRef]
- Akhtar, N.; Jabeen, I. A 2D-QSAR and grid-independent molecular descriptor (grind) analysis of quinoline-type inhibitors of AKT2: Exploration of the binding mode in the pleckstrin homology (ph) domain. PLoS ONE 2016, 11, e0168806. [Google Scholar] [CrossRef] [PubMed]
- Al-Sha’er, M.A.; Taha, M.O. Ligand-based modeling of AKT3 lead to potent dual AKT1/AKT3 inhibitor. J. Mol. Graph. Model. 2018, 83, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Ajmani, S.; Agrawal, A.; Kulkarni, S.A. A comprehensive structure-activity analysis of protein kinase B-alpha (AKT1) inhibitors. J. Mol. Graph. Model. 2010, 28, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Muddassar, M.; Pasha, F.A.; Neaz, M.M.; Saleem, Y.; Cho, S.J. Elucidation of binding mode and three dimensional quantitative structure-activity relationship studies of a novel series of protein kinase B/AKT inhibitors. J. Mol. Model. 2009, 15, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M.N.D.S. Probing the environmental toxicity of deep eutectic solvents and their components: An in silico modeling approach. ACS Sustain. Chem. Eng. 2019, 7, 10649–10660. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Ruso, J.M.; Speck-Planche, A.; Cordeiro, M.N.D.S. Enabling the discovery and virtual screening of potent and safe antimicrobial peptides. Simultaneous prediction of antibacterial activity and cytotoxicity. ACS Comb. Sci. 2016, 18, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Fragment-based in silico modeling of multi-target inhibitors against breast cancer-related proteins. Mol. Divers. 2017, 21, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A. alvaDesc: A tool to calculate and analyze molecular descriptors and fingerprints. In Ecotoxicological QSARs; Roy, K., Ed.; Springer: New York, NY, USA, 2020; pp. 801–820. [Google Scholar]
- Rucker, C.; Rucker, G.; Meringer, M. Y-Randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Ojha, P.K.; Roy, K. Comparative QSARs for antimalarial endochins: Importance of descriptor-thinning and noise reduction prior to feature selection. Chemom. Intellig. Lab. Syst. 2011, 109, 146–161. [Google Scholar] [CrossRef]
- Brown, M.T.; Wicker, L.R. 8—Discriminant analysis. In Handbook of Applied Multivariate Statistics and Mathematical Modeling; Tinsley, H.E.A., Brown, S.D., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 209–235. [Google Scholar]
- Boughorbel, S.; Jarray, F.; El-Anbari, M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS ONE 2017, 12, e0177678. [Google Scholar] [CrossRef]
- Hanczar, B.; Hua, J.; Sima, C.; Weinstein, J.; Bittner, M.; Dougherty, E.R. Small-sample precision of ROC-related estimates. Bioinformatics 2010, 26, 822–830. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intellig. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Speeding up early drug discovery in antiviral research: A fragment-based in silico approach for the design of virtual anti-hepatitis C leads. ACS Comb. Sci. 2017, 19, 501–512. [Google Scholar] [CrossRef]
- Gramatica, P. WHIM descriptors of shape. QSAR Comb. Sci. 2006, 25, 327–332. [Google Scholar] [CrossRef]
- Reutlinger, M.; Koch, C.P.; Reker, D.; Todoroff, N.; Schneider, P.; Rodrigues, T.; Schneider, G. Chemically Advanced Template Search (CATS) for scaffold-hopping and prospective target prediction for "Orphan’ molecules. Mol. Inform. 2013, 32, 133–138. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V.; Todeschini, R. Molecular Descriptors for Chemoinformatics, 2nd ed.; John Wiley Distributor: Weinheim, Germany; Chichester, UK, 2009; p. 1. [Google Scholar]
- Devinyak, O.; Havrylyuk, D.; Lesyk, R. 3D-MoRSE descriptors explained. J. Mol. Graph. Model. 2014, 54, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cover, T.M.; Hart, P.E. Nearest neighbor pattern classification. IEEE T. Inform. Theory 1967, 13, 21–29. [Google Scholar] [CrossRef]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. A training algorithm for optimal margin classifiers. In Proceedings of the 5th annual workshop on Computational learning theory, Pittsburgh, PA, USA, 27–29 July 1992; pp. 144–152. [Google Scholar]
- Huang, G.B.; Babri, H.A. Upper bounds on the number of hidden neurons in feedforward networks with arbitrary bounded nonlinear activation functions. IEEE Trans. Neural Netw. 1998, 9, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Hammann, F.; Drewe, J. Decision tree models for data mining in hit discovery. Expert Opin. Drug Discov. 2012, 7, 341–352. [Google Scholar] [CrossRef]
- McCallum, A.; Nigam, K. A comparison of event models for naive bayes text classification. Work Learn. Text Categ. 2001, 752, 41–48. [Google Scholar]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.L.; Huan, J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Cheminform. 2017, 9, 42. [Google Scholar] [CrossRef]
- Stalring, J.C.; Carlsson, L.A.; Almeida, P.; Boyer, S. AZOrange—High performance open source machine learning for QSAR modeling in a graphical programming environment. J. Cheminform. 2011, 3, 28. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.H.; Wang, S.W.; Li, S.L.; Zhang, W.L.; Liu, X.F.; Lai, L.H.; Pei, J.F.; Li, H.L. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Ouyang, S.S.; Yu, B.A.; Liu, Y.B.; Huang, K.; Gong, J.Y.; Zheng, S.Y.; Li, Z.H.; Li, H.L.; Jiang, H.L. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Haddad, Y.; Adam, V.; Heger, Z. Ten quick tips for homology modeling of high-resolution protein 3D structures. PLoS Comput. Biol. 2020, 16, e1007449. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- ChemAxon. Standardizer, Version 15.9.14.0 Software; ChemAxon: Budapest, Hungary, 2010. [Google Scholar]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef]
- Sadowski, J.; Gasteiger, J.; Klebe, G. Comparison of automatic three-dimensional model builders using 639 x-ray structures. J. Chem. Inf. Comput. Sci. 1994, 34, 1000–1008. [Google Scholar] [CrossRef]
- Ambure, P.; Aher, R.B.; Gajewicz, A.; Puzyn, T.; Roy, K. “NanoBRIDGES” software: Open access tools to perform QSAR and nano-QSAR modeling. Chemom. Intellig. Lab. Syst. 2015, 147, 1–13. [Google Scholar] [CrossRef]
- Menzies, T.; Kocagüneli, E.; Minku, L.; Peters, F.; Turhan, B. Chapter 22—Complexity: Using assemblies of multiple models. In Sharing Data and Models in Software Engineering; Menzies, T., Kocagüneli, E., Minku, L., Peters, F., Turhan, B., Eds.; Morgan Kaufmann: Boston, MA, USA, 2015; pp. 291–304. [Google Scholar]
- Wilks, S.S. Certain generalizations in the analysis of variance. Biometrika 1932, 24, 471–494. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIRES Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2005, 26, 114. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Wang, L.; Chen, L.; Yu, M.; Xu, L.H.; Cheng, B.; Lin, Y.S.; Gu, Q.; He, X.H.; Xu, J. Discovering new mTOR inhibitors for cancer treatment through virtual screening methods and in vitro assays. Sci. Rep. 2016, 6, 18987. [Google Scholar] [CrossRef]
- Berishvili, V.P.; Kuimov, A.N.; Voronkov, A.E.; Radchenko, E.V.; Kumar, P.; Choonara, Y.E.; Pillay, V.; Kamal, A.; Palyulin, V.A. Discovery of novel tankyrase inhibitors through molecular docking-based virtual screening and molecular dynamics simulation studies. Molecules 2020, 25, 3171. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Schoning, V.; Hammann, F. How far have decision tree models come for data mining in drug discovery? Expert Opin. Drug Discov. 2018, 13, 1067–1069. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.F.; Byrne, R.; Schneider, G.; Yang, S.Y. Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef]
- Zhao, L.L.; Ciallella, H.L.; Aleksunes, L.M.; Zhu, H. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov. Today 2020, 25, 1624–1638. [Google Scholar] [CrossRef]





| Method | Model | λ | χ2 | D2 | p | F (10,2696) |
|---|---|---|---|---|---|---|
| GA-LDA | 0.414 | 2381.34 | 5.89 | <10−16 | 374.53 | |
| FS-LDA | 0.408 | 2420.04 | 6.156 | <10−16 | 391.07 | |
| SFS-LDA | 0.507 | 1831.98 | 4.120 | <10−16 | 261.77 |
| Classification a | GA-LDA | FS-LDA | SFS-LDA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sub-Training | Test | Validation | Sub-Training | Test | Validation | Sub-Training | Test | Validation | |
| NDTotal b | 2707 | 1160 | 1656 | 2707 | 1160 | 1656 | 2707 | 1160 | 1656 |
| NDactive b | 1027 | 459 | 620 | 1027 | 459 | 620 | 1027 | 459 | 620 |
| CCDactive c | 916 | 413 | 553 | 901 | 399 | 541 | 905 | 409 | 546 |
| Sensitivity (%) | 89.2 | 90.0 | 88.6 | 87.4 | 88.6 | 88.5 | 88.2 | 89.9 | 88.7 |
| NDinactive b | 1680 | 701 | 1036 | 1680 | 701 | 1036 | 1680 | 1680 | 1036 |
| CCDinactive c | 1472 | 626 | 918 | 1468 | 621 | 917 | 1481 | 630 | 919 |
| Specificity (%) | 87.6 | 89.3 | 89.2 | 87.7 | 86.9 | 87.3 | 88.1 | 89.1 | 88.1 |
| F-measure | 0.852 | 0.872 | 0.857 | 0.842 | 0.851 | 0.845 | 0.849 | 0.871 | 0.851 |
| Accuracy (%) | 88.2 | 89.6 | 88.8 | 87.5 | 87.9 | 88.0 | 88.1 | 89.6 | 88.5 |
| MCC d | 0.756 | 0.785 | 0.767 | 0.741 | 0.75 | 0.749 | 0.753 | 0.784 | 0.758 |
| Classification a | GA-LDA Model | Consensus LDA Model | ||||
|---|---|---|---|---|---|---|
| Sub-Training | Test | Validation | Sub-Training | Test | Validation | |
| NDTotal | 2707 | 1160 | 1656 | 2707 | 1160 | 1656 |
| NDactive | 1027 | 459 | 620 | 1027 | 459 | 620 |
| CCDactive | 916 | 413 | 553 | 902 | 406 | 541 |
| Sensitivity (%) | 89.19 | 89.98 | 88.61 | 87.83 | 88.45 | 87.26 |
| NDinactive | 1680 | 701 | 1036 | 1680 | 701 | 1036 |
| CCDinactive | 1472 | 626 | 918 | 1486 | 633 | 927 |
| Specificity (%) | 87.62 | 89.30 | 89.19 | 88.45 | 90.30 | 89.48 |
| F-measure | 0.852 | 0.872 | 0.857 | 0.850 | 0.870 | 0.852 |
| Accuracy (%) | 88.21 | 89.57 | 88.83 | 88.22 | 89.57 | 88.65 |
| MCC | 0.756 | 0.785 | 0.767 | 0.754 | 0.783 | 0.760 |
| Method | Parameters Tuned | Parameters Selected | 10-Fold CV Accuracy (%) a |
|---|---|---|---|
| RF | Bootstrap: True/False | False | |
| Criterion: Gini, Entropy, | Gini | ||
| Maximum depth: 10, 30, 50, 70, 90, 100, None | 90 | ||
| Maximum features: Auto, Sqrt | Sqrt | 91.02 | |
| Minimum samples leaf: 1, 2, 4 | 1 | ||
| Minimum samples split: 2, 5, 10 | 5 | ||
| Number of estimators: 50, 100, 200,500 | 200 | ||
| kNN | Number of neighbors: 1–31 | 20 | |
| Weight options: Uniform, Distance | Distance | 79.20 | |
| Algorithms: Auto, Ball tree, kd_tree, brute | Auto | ||
| Xgboost | Minimum child weight: 1,5,10 | 1 | |
| Gamma: 0, 0.5, 1, 1.5, 2, 5 | 0 | ||
| Sum sample: 0.6, 0.8, 1.0 | 0.8 | 91.54 | |
| Number of estimators: 50, 100, 200,300 | 100 | ||
| Maximum depth: 3, 4, 5 | 5 | ||
| RBF-SVC | C: 0.1, 1, 10, 100, 1000 | 1 | 62.30 |
| Gamma: 1, 0.1, 0.01, 0.001 | 1 | ||
| MLP | Hidden layer sizes:(50,50,50), (50,100,50), (100,) | (100,) | |
| Activation: Identity, Logistic, Tanh, Relu | Relu | ||
| Solver: SGD, Adam | Adam | 82.97 | |
| Alpha: 0.0001, 0.001, 0.01,1 | 0.0001 | ||
| Learning rate: Constant, Adaptive, Inverse scaling | Adaptive | ||
| DT | Criterion: Gini, EntropyMaximum depth: 10,30,50,70,90,100, NoneMaximum features: Auto, SqrtMinimum samples leaf: 1,2,4Minimum samples split: 2–50 | Entropy100Sqrt113 | 84.33 |
| NB | Alpha: 1,0.5,0.1Fit prior: True, False | 0.1True | 69.40 |
| Classification a | RF | Xgboost | ||||
|---|---|---|---|---|---|---|
| Sub-Training (10-CV) | Test | Validation | Sub-Training (10-CV) | Test | Validation | |
| NDTotal | 2707 | 1160 | 1656 | 2707 | 1160 | 1656 |
| NDactive | 1027 | 459 | 620 | 1027 | 459 | 620 |
| CCDactive | 919 | 417 | 573 | 932 | 422 | 578 |
| Sensitivity (%) | 89.48 | 92.58 | 93.53 | 90.75 | 91.87 | 93.24 |
| NDinactive | 1680 | 701 | 1036 | 1680 | 701 | 1036 |
| CCDinactive | 1545 | 649 | 969 | 1546 | 644 | 966 |
| Specificity (%) | 91.96 | 90.85 | 92.42 | 92.02 | 91.94 | 93.23 |
| F-measure | 0.883 | 0.899 | 0.909 | 0.891 | 0.900 | 0.912 |
| Accuracy (%) | 91.02 | 91.90 | 93.11 | 91.54 | 91.90 | 93.24 |
| MCC | 0.810 | 0.831 | 0.854 | 0.822 | 0.832 | 0.857 |
| Test Set | External Validation Set | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NSamplea | Xgboost | GA-LDA | NSamplea | Xgboost | GA-LDA | ||||||||
| # Incorrect b | % Accuracy | # Incorrect b | % Accuracy | # Incorrect b | % Accuracy | # Incorrect b | % Accuracy | ||||||
| 1 | IC50 | B | AKT | 509 | 80 | 84.28 | 95 | 81.34 | 707 | 95 | 86.56 | 142 | 79.92 |
| 9 | Ki | F | AKT2 | 163 | 2 | 98.77 | 2 | 98.77 | 238 | 0 | 100.00 | 5 | 97.90 |
| 10 | Ki | F | AKT3 | 148 | 1 | 99.32 | 3 | 97.97 | 236 | 2 | 99.15 | 2 | 99.15 |
| 8 | Ki | F | AKT | 156 | 3 | 98.08 | 2 | 98.72 | 203 | 1 | 99.51 | 2 | 99.01 |
| 2 | IC50 | B | AKT2 | 121 | 2 | 98.35 | 14 | 88.43 | 167 | 8 | 95.21 | 23 | 86.23 |
| 3 | IC50 | B | AKT3 | 35 | 4 | 88.57 | 4 | 88.57 | 53 | 2 | 96.23 | 3 | 94.34 |
| 5 | Ki | B | AKT | 17 | 2 | 88.24 | 1 | 94.12 | 27 | 2 | 92.59 | 3 | 88.89 |
| 6 | Ki | B | AKT2 | 4 | 0 | 100.00 | 0 | 100.00 | 12 | 1 | 91.67 | 1 | 91.67 |
| 4 | IC50 | F | AKT | 4 | 0 | 100.00 | 0 | 100.00 | 9 | 1 | 88.89 | 3 | 66.67 |
| 7 | Ki | B | AKT3 | 3 | 0 | 100.00 | 0 | 100.00 | 4 | 0 | 100.00 | 1 | 75.00 |
| Compound | No of Experimental Conditions (GA-LDA) | No of Experimental Conditions (Xgboost) |
|---|---|---|
| Asn0019 | 10 | 6 |
| Asn0021 | 10 | 6 |
| Asn0022 | 10 | 6 |
| Asn2706 | 7 | 6 |
| Asn5093 | 8 | 6 |
| Asn5283 | 7 | 6 |
| Asn6236 | 7 | 6 |
| Compound | PDB ID | Target Name | Feature Type a | No of Features | Fit Score |
|---|---|---|---|---|---|
| Asn0019 | 3CQU | AKT1 | 2H,A,D | 4 | 2.925 |
| Asn0019 | 2UW9 | AKT2 | 3H,P,A,D | 6 | 3.078 |
| Asn0021 | 3CQU | AKT1 | 2H,A,D | 4 | 2.274 |
| Asn0021 | 2UW9 | AKT2 | 3H,P,A,D | 6 | 3.385 |
| Asn0022 | 3CQU | AKT1 | 2H,A,D | 4 | 2.724 |
| Asn0022 | 2UW9 | AKT2 | 3H,P,A,D | 6 | 3.000 |
| Asn5093 | 3CQU | AKT1 | 2H,A,D | 4 | 2.637 |
| Asn5093 | 2UW9 | AKT2 | 3H,P,A,D | 6 | 3.135 |
| Asn6236 | 3CQU | AKT1 | 2H,A,D | 4 | 3.126 |
| Asn6236 | 2WU9 | AKT2 | 3H,P,A,D | 6 | 2.955 |
| Compound | AKT1 | AKT2 | AKT3 |
|---|---|---|---|
| Asn0019 | −32.54 | −27.61 | −43.61 |
| Asn0021 | −25.81 | −29.03 | −39.04 |
| Asn0022 | −27.75 | −23.56 | −37.29 |
| Asn5093 | −36.34 | −29.44 | −22.67 |
| Asn6236 | −18.08 | −19.72 | −26.82 |
| GSK690693 | −46.88 | −29.78 | −43.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halder, A.K.; Cordeiro, M.N.D.S. AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery. Int. J. Mol. Sci. 2021, 22, 3944. https://doi.org/10.3390/ijms22083944
Halder AK, Cordeiro MNDS. AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery. International Journal of Molecular Sciences. 2021; 22(8):3944. https://doi.org/10.3390/ijms22083944
Chicago/Turabian StyleHalder, Amit Kumar, and M. Natália D. S. Cordeiro. 2021. "AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery" International Journal of Molecular Sciences 22, no. 8: 3944. https://doi.org/10.3390/ijms22083944
APA StyleHalder, A. K., & Cordeiro, M. N. D. S. (2021). AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery. International Journal of Molecular Sciences, 22(8), 3944. https://doi.org/10.3390/ijms22083944