
Fish-Ing for Enhancers in the Heart



Abstract
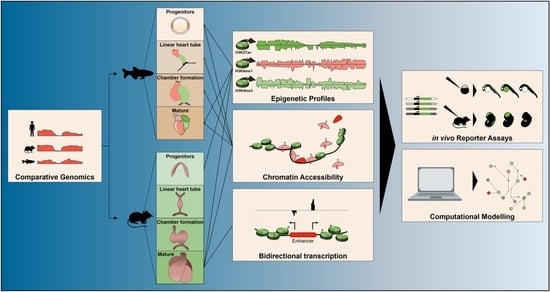
1. Introduction
2. The Quest for Enhancers Involved in Heart Development and Function
2.1. Targeted Analysis of Gene Promoter Regions Pinpoints Regulatory Elements Driving Tissue-Specific Expression
2.2. Large-Scale Enhancer Discovery by Enhancer Trapping Generates Live Markers for Developmental Studies
2.3. Comparative Genomics Identify Highly Conserved Developmental Enhancers Regulating Heart Development
2.4. Genome-Wide Enhancers Discovery Generates Valuable Resource on Gene Regulation in Heart Development and Function
2.5. Dynamics of Chromatin Landscape during Cardiogenesis Reveals Enhancers Implicated in Heart Development
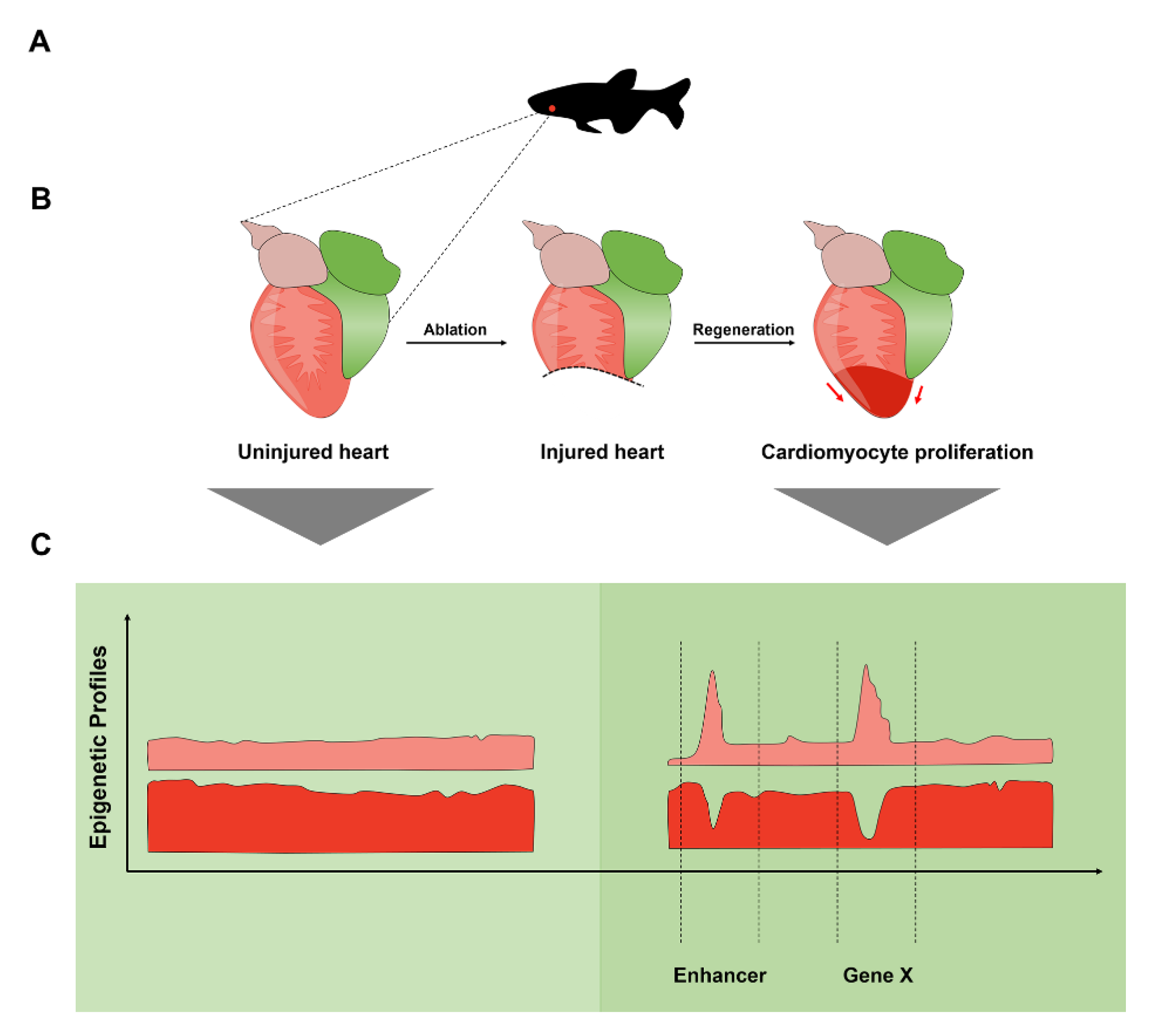
2.6. Enhancers Direct Gene Expression in the Regenerating Zebrafish Heart
3. Technological Advances in Enhancer Discovery Provides Future Opportunities for Identification of Cardiac Enhancers
3.1. Transcription of Enhancer Regions Pinpoints the Presence of Active Enhancers
3.2. Computational Modeling Approaches Allows Integrative Analyses of Genomic Data for Large Scale Enhancers Discovery
4. Conclusions and Future Perspectives for the Role of Enhancers in Human Health and Diseases
Author Contributions
Funding
Conflicts of Interest
References
- Olson, E.N. Gene Regulatory Networks in the Evolution and Development of the Heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef]
- Ounzain, S.; Pezzuto, I.; Micheletti, R.; Burdet, F.; Sheta, R.; Nemir, M.; Gonzales, C.; Sarre, A.; Alexanian, M.; Blow, M.J.; et al. Functional Importance of Cardiac Enhancer-Associated Noncoding RNAs in Heart Development and Disease. J. Mol. Cell. Cardiol. 2014, 76, 55–70. [Google Scholar] [CrossRef]
- Doane, A.S.; Elemento, O. Regulatory Elements in Molecular Networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1374. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Nir, R.; Lubliner, S.; Sharon, E.; Kalma, Y.; Weinberger, A.; Segal, E. Systematic Interrogation of Human Promoters. Genome Res. 2019, 29, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, N.D.; Jajodia, A.; Mishra, A.; Hawkins, R.D. Candidate Silencer Elements for the Human and Mouse Genomes. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- West, A.G. Insulators: Many Functions, Many Mechanisms. Genes Dev. 2002, 16, 271–288. [Google Scholar] [CrossRef]
- Wittkopp, P.J. Genomic Sources of Regulatory Variation in Cis and in Trans. Cell. Mol. Life Sci. CMLS 2005, 62, 1779–1783. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, S.; Bhagat, S.; Matsuki, Y.; Takegami, Y.; Uehata, T.; Kanemaru, A.; Itoh, M.; Shirakawa, K.; Takaori-Kondo, A.; Takeuchi, O. NET-CAGE Characterizes the Dynamics and Topology of Human Transcribed Cis-Regulatory Elements. Nat. Genet. 2019, 51, 1369–1379. [Google Scholar] [CrossRef]
- Gasperini, M.; Tome, J.M.; Shendure, J. Towards a Comprehensive Catalogue of Validated and Target-Linked Human Enhancers. Nat. Rev. Genet. 2020, 21, 292–310. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, J.; Olson, L.; Tyndall, C.; Schaffner, W. Transcriptional ‘Enhancers’ from SV40 and Polyoma Virus Show a Cell Type Preference. Nucleic Acids Res. 1982, 10, 7965–7976. [Google Scholar] [CrossRef]
- Spandidos, D.A.; Wilkie, N.M. Host-Specificities of Papillomavirus, Moloney Murine Sarcoma Virus and Simian Virus 40 Enhancer Sequences. EMBO J. 1983, 2, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Hansen, U.; Sharp, P.A. Sequences Controlling in Vitro Transcription of SV40 Promoters. EMBO J. 1983, 2, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Schirm, S.; Weber, F.; Schaffner, W.; Fleckenstein, B. A Transcription Enhancer in the Herpesvirus Saimiri Genome. EMBO J. 1985, 4, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Banerji, J.; Olson, L.; Schaffner, W. A Lymphocyte-Specific Cellular Enhancer Is Located Downstream of the Joining Region in Immunoglobulin Heavy Chain Genes. Cell 1983, 33, 729–740. [Google Scholar] [CrossRef]
- Gillies, S.D.; Morrison, S.L.; Oi, V.T.; Tonegawa, S. A Tissue-Specific Transcription Enhancer Element Is Located in the Major Intron of a Rearranged Immunoglobulin Heavy Chain Gene. Cell 1983, 33, 717–728. [Google Scholar] [CrossRef]
- Mercola, M.; Wang, X.; Olsen, J.; Calame, K. Transcriptional Enhancer Elements in the Mouse Immunoglobulin Heavy Chain Locus. Science 1983, 221, 663–665. [Google Scholar] [CrossRef]
- Zeller, R.W.; Griffith, J.D.; Moore, J.G.; Kirchhamer, C.V.; Britten, R.J.; Davidson, E.H. A Multimerizing Transcription Factor of Sea Urchin Embryos Capable of Looping DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 2989–2993. [Google Scholar] [CrossRef]
- Bulger, M.; Groudine, M. Looping versus Linking: Toward a Model for Long-Distance Gene Activation. Genes Dev. 1999, 13, 2465–2477. [Google Scholar] [CrossRef]
- Ong, C.-T.; Corces, V.G. Enhancer Function: New Insights into the Regulation of Tissue-Specific Gene Expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and Coordinated Epigenetic Regulation of Developmental Transitions in the Cardiac Lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Barozzi, I.; Mietton, F.; Ghisletti, S.; Polletti, S.; Tusi, B.K.; Muller, H.; Ragoussis, J.; Wei, C.-L.; Natoli, G. A Large Fraction of Extragenic RNA Pol II Transcription Sites Overlap Enhancers. PLoS Biol. 2010, 8, e1000384. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Hughes, J.R.; Garrick, D.; Lynch, M.D.; Sharpe, J.A.; Sloane-Stanley, J.A.; McGowan, S.J.; Gobbi, M.D.; Hosseini, M.; Vernimmen, D.; et al. Intragenic Enhancers Act as Alternative Promoters. Mol. Cell 2012, 45, 447–458. [Google Scholar] [CrossRef]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs Associate with Mediator to Enhance Chromatin Architecture and Transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef]
- Lam, M.T.Y.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer RNAs and Regulated Transcriptional Programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Nemer, G.; Nemer, M. Regulation of Heart Development and Function through Combinatorial Interactions of Transcription Factors. Ann. Med. 2001, 33, 604–610. [Google Scholar] [CrossRef]
- Chang, C.-P.; Bruneau, B.G. Epigenetics and Cardiovascular Development. Annu. Rev. Physiol. 2012, 74, 41–68. [Google Scholar] [CrossRef]
- Li, X.; Martinez-Fernandez, A.; Hartjes, K.A.; Kocher, J.-P.A.; Olson, T.M.; Terzic, A.; Nelson, T.J. Transcriptional Atlas of Cardiogenesis Maps Congenital Heart Disease Interactome. Physiol. Genom. 2014, 46, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular Regulation of Cardiomyocyte Differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Langenbacher, A.; Chen, J.-N. Transcriptional Regulation of Heart Development in Zebrafish. J. Cardiovasc. Dev. Dis. 2016, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Niescierowicz, K.; Winata, C.L. Decoding the Heart through Next Generation Sequencing Approaches. Genes 2018, 9, 289. [Google Scholar] [CrossRef]
- Bruneau, B.G. Transcriptional Regulation of Vertebrate Cardiac Morphogenesis. Circ. Res. 2002, 90, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.J.; Olson, E.N. Building the Heart Piece by Piece: Modularity of Cis-Elements Regulating Nkx2-5 Transcription. Development 1999, 126, 4187–4192. [Google Scholar]
- Srivastava, D.; Olson, E.N. A Genetic Blueprint for Cardiac Development. Nature 2000, 407, 221–226. [Google Scholar] [CrossRef]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the Mammalian Heart from Two Sources of Myocardial Cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Fishman, M.C.; Olson, E.N. Parsing the Heart: Genetic Modules for Organ Assembly. Cell 1997, 91, 153–156. [Google Scholar] [CrossRef]
- OOta, S.; Saitou, N. Phylogenetic Relationship of Muscle Tissues Deduced from Superimposition of Gene Trees. Mol. Biol. Evol. 1999, 16, 856–867. [Google Scholar] [CrossRef]
- Arendt, D. The Evolution of Cell Types in Animals: Emerging Principles from Molecular Studies. Nat. Rev. Genet. 2008, 9, 868–882. [Google Scholar] [CrossRef]
- Bishopric, N.H. Evolution of the Heart from Bacteria to Man. Ann. N. Y. Acad. Sci. 2005, 1047, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Kawakoshi, A.; Hyodo, S.; Yasuda, A.; Takei, Y. A Single and Novel Natriuretic Peptide Is Expressed in the Heart and Brain of the Most Primitive Vertebrate, the Hagfish (Eptatretus Burgeri). J. Mol. Endocrinol. 2003, 31, 209–220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Becker, D.L.; Cook, J.E.; Davies, C.S.; Evans, W.H.; Gourdie, R.G. Expression of Major Gap Junction Connexin Types in the Working Myocardium of Eight Chordates. Cell Biol. Int. 1998, 22, 527–543. [Google Scholar] [CrossRef]
- Holland, N.D.; Venkatesh, T.V.; Holland, L.Z.; Jacobs, D.K.; Bodmer, R. Amphink2-Tin, an Amphioxus Homeobox Gene Expressed in Myocardial Progenitors: Insights into Evolution of the Vertebrate Heart. Dev. Biol. 2003, 255, 128–137. [Google Scholar] [CrossRef]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.-L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef]
- Tu, C.-T.; Yang, T.-C.; Tsai, H.-J. Nkx2.7 and Nkx2.5 Function Redundantly and Are Required for Cardiac Morphogenesis of Zebrafish Embryos. PLoS ONE 2009, 4, e4249. [Google Scholar] [CrossRef]
- Bakkers, J. Zebrafish as a Model to Study Cardiac Development and Human Cardiac Disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef]
- Kuo, C.T.; Morrisey, E.E.; Anandappa, R.; Sigrist, K.; Lu, M.M.; Parmacek, M.S.; Soudais, C.; Leiden, J.M. GATA4 Transcription Factor Is Required for Ventral Morphogenesis and Heart Tube Formation. Genes Dev. 1997, 11, 1048–1060. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Lin, Q.; Duncan, S.A.; Olson, E.N. Requirement of the Transcription Factor GATA4 for Heart Tube Formation and Ventral Morphogenesis. Genes Dev. 1997, 11, 1061–1072. [Google Scholar] [CrossRef]
- Patient, R.K.; McGhee, J.D. The GATA Family (Vertebrates and Invertebrates). Curr. Opin. Genet. Dev. 2002, 12, 416–422. [Google Scholar] [CrossRef]
- King, T.; Brown, N.A. Embryonic Asymmetry: The Left Side Gets All the Best Genes. Curr. Biol. 1999, 9, R18–R22. [Google Scholar] [CrossRef]
- Campione, M.; Steinbeisser, H.; Schweickert, A.; Deissler, K.; van Bebber, F.; Lowe, L.A.; Nowotschin, S.; Viebahn, C.; Haffter, P.; Kuehn, M.R. The Homeobox Gene Pitx2: Mediator of Asymmetric Left-Right Signaling in Vertebrate Heart and Gut Looping. Development 1999, 126, 1225–1234. [Google Scholar] [PubMed]
- Noël, E.S.; Verhoeven, M.; Lagendijk, A.K.; Tessadori, F.; Smith, K.; Choorapoikayil, S.; Den Hertog, J.; Bakkers, J. A Nodal-Independent and Tissue-Intrinsic Mechanism Controls Heart-Looping Chirality. Nat. Commun. 2013, 4, 2754. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, N.; Savic, D.; Nobrega, M.A. Transcriptional Enhancers in Development and Disease. Genome Biol. 2012, 13, 238. [Google Scholar] [CrossRef]
- Postma, A.V.; Bezzina, C.R.; Christoffels, V.M. Genetics of Congenital Heart Disease: The Contribution of the Noncoding Regulatory Genome. J. Hum. Genet. 2016, 61, 13–19. [Google Scholar] [CrossRef]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Chahal, G.; Tyagi, S.; Ramialison, M. Navigating the Non-Coding Genome in Heart Development and Congenital Heart Disease. Differentiation 2019, 107, 11–23. [Google Scholar] [CrossRef]
- Blow, M.J.; McCulley, D.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-Seq Identification of Weakly Conserved Heart Enhancers. Nat. Genet. 2010, 42, 806–810. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-Scale Discovery of Enhancers from Human Heart Tissue. Nat. Genet. 2012, 44, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Thomas, S.; Stoick-Cooper, C.L.; Wang, H.; Maves, L.; Sandstrom, R.; Pabon, L.; Reinecke, H.; Pratt, G.; Keller, G.; et al. A Temporal Chromatin Signature in Human Embryonic Stem Cells Identifies Regulators of Cardiac Development. Cell 2012, 151, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Kedzierska, K.Z.; Migdal, M.; Nahia, K.A.; Ramilowski, J.A.; Bugajski, L.; Hashimoto, K.; Marconi, A.; Piwocka, K.; Carninci, P.; et al. Dynamics of Cardiomyocyte Transcriptome and Chromatin Landscape Demarcates Key Events of Heart Development. Genome Res. 2019, 29, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, L.; Sakabe, N.J.; Blanski, A.A.; Arimura, F.E.; Westlund, J.M.; Nobrega, M.A.; Ovcharenko, I. Genome-Wide Discovery of Human Heart Enhancers. Genome Res. 2010, 20, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Dickel, D.E.; Barozzi, I.; Zhu, Y.; Fukuda-Yuzawa, Y.; Osterwalder, M.; Mannion, B.J.; May, D.; Spurrell, C.H.; Plajzer-Frick, I.; Pickle, C.S.; et al. Genome-Wide Compendium and Functional Assessment of in Vivo Heart Enhancers. Nat. Commun. 2016, 7, 12923. [Google Scholar] [CrossRef]
- Poon, K.-L.; Liebling, M.; Kondrychyn, I.; Garcia-Lecea, M.; Korzh, V. Zebrafish Cardiac Enhancer Trap Lines: New Tools for in Vivo Studies of Cardiovascular Development and Disease. Dev. Dyn. 2010, 239, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Q.; Wu, F.; Zhang, Y.; Sun, S.; Wang, X.; Gui, Y.; Li, Q. Identification of a 42-bp Heart-specific Enhancer of the Notch1b Gene in Zebrafish Embryos. Dev. Dyn. 2019, 248, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Van den Boogaard, M.; van Weerd, J.H.; Bawazeer, A.C.; Hooijkaas, I.B.; van de Werken, H.J.G.; Tessadori, F.; de Laat, W.; Barnett, P.; Bakkers, J.; Christoffels, V.M. Identification and Characterization of a Transcribed Distal Enhancer Involved in Cardiac Kcnh2 Regulation. Cell Rep. 2019, 28, 2704–2714.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, F.; Wu, F.; Wang, Y.; Wang, X.; Gui, Y.; Li, Q. Tnni1b-ECR183-D2, an 87 Bp Cardiac Enhancer of Zebrafish. PeerJ 2020, 8, e10289. [Google Scholar] [CrossRef]
- The modENCODE Consortium; Roy, S.; Ernst, J.; Kharchenko, P.V.; Kheradpour, P.; Negre, N.; Eaton, M.L.; Landolin, J.M.; Bristow, C.A.; Ma, L.; et al. Identification of Functional Elements and Regulatory Circuits by Drosophila ModENCODE. Science 2010, 330, 1787–1797. [Google Scholar] [CrossRef]
- Phifer-Rixey, M.; Nachman, M.W. Insights into Mammalian Biology from the Wild House Mouse Mus Musculus. eLife 2015, 4, e05959. [Google Scholar] [CrossRef]
- Han, S.; Yang, A.; Lee, S.; Lee, H.-W.; Park, C.B.; Park, H.-S. Expanding the Genetic Code of Mus Musculus. Nat. Commun. 2017, 8, 14568. [Google Scholar] [CrossRef]
- Woods, I.G.; Kelly, P.D.; Chu, F.; Ngo-Hazelett, P.; Yan, Y.-L.; Huang, H.; Postlethwait, J.H.; Talbot, W.S. A Comparative Map of the Zebrafish Genome. Genome Res. 2000, 10, 1903–1914. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Lee-Liu, D.; Méndez-Olivos, E.E.; Muñoz, R.; Larraín, J. The African Clawed Frog Xenopus Laevis: A Model Organism to Study Regeneration of the Central Nervous System. Neurosci. Lett. 2017, 652, 82–93. [Google Scholar] [CrossRef]
- Borodinsky, L.N. Xenopus Laevis as a Model Organism for the Study of Spinal Cord Formation, Development, Function and Regeneration. Front. Neural Circuits 2017, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of Clones of Homozygous Diploid Zebra Fish (Brachydanio Rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Staudt, D.; Stainier, D. Uncovering the Molecular and Cellular Mechanisms of Heart Development Using the Zebrafish. Annu. Rev. Genet. 2012, 46, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Luan, Y.; Liu, T.; Lee, H.J.; Fang, L.; Wang, Y.; Wang, X.; Zhang, B.; Jin, Q.; Ang, K.C.; et al. A Map of Cis-Regulatory Elements and 3D Genome Structures in Zebrafish. Nature 2020, 588, 337–343. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef]
- Gerstein, M.B.; Lu, Z.J.; Van Nostrand, E.L.; Cheng, C.; Arshinoff, B.I.; Liu, T.; Yip, K.Y.; Robilotto, R.; Rechtsteiner, A.; Ikegami, K.; et al. Integrative Analysis of the Caenorhabditis Elegans Genome by the ModENCODE Project. Science 2010, 330, 1775–1787. [Google Scholar] [CrossRef]
- Tan, H.; Onichtchouk, D.; Winata, C. DANIO-CODE: Toward an Encyclopedia of DNA Elements in Zebrafish. Zebrafish 2016, 13, 54–60. [Google Scholar] [CrossRef]
- Meng, A.; Tang, H.; Yuan, B.; Ong, B.A.; Long, Q.; Lin, S. Positive and Negative Cis-Acting Elements Are Required for Hematopoietic Expression of Zebrafish GATA-1. Blood 1999, 93, 500–508. [Google Scholar] [CrossRef]
- Long, Q.; Meng, A.; Wang, H.; Jessen, J.R.; Farrell, M.J.; Lin, S. GATA-1 Expression Pattern Can Be Recapitulated in Living Transgenic Zebrafish Using GFP Reporter Gene. Development 1997, 124, 4105–4111. [Google Scholar]
- Muller, F.; Chang, B.; Albert, S.; Fischer, N.; Tora, L.; Strahle, U. Intronic Enhancers Control Expression of Zebrafish Sonic Hedgehog in Floor Plate and Notochord. Development 1999, 126, 2103–2116. [Google Scholar]
- Müller, F.; Williams, D.W.; Kobolák, J.; Gauvry, L.; Goldspink, G.; Orbán, L.; Maclean, N. Activator Effect of Coinjected Enhancers on the Muscle-Specific Expression of Promoters in Zebrafish Embryos. Mol. Reprod. Dev. Inc. Gamete Res. 1997, 47, 404–412. [Google Scholar] [CrossRef]
- Chang, B.-E.; Blader, P.; Fischer, N.; Ingham, P.W.; Strähle, U. Axial (HNF3β) and Retinoic Acid Receptors Are Regulators of the Zebrafish Sonic Hedgehog Promoter. EMBO J. 1997, 16, 3955–3964. [Google Scholar] [CrossRef] [PubMed]
- Heicklen-Klein, A.; Evans, T. T-Box Binding Sites Are Required for Activity of a Cardiac GATA-4 Enhancer. Dev. Biol. 2004, 267, 490–504. [Google Scholar] [CrossRef]
- Kawakami, K.; Shima, A.; Kawakami, N. Identification of a Functional Transposase of the Tol2 Element, an Ac-like Element from the Japanese Medaka Fish, and Its Transposition in the Zebrafish Germ Lineage. Proc. Natl. Acad. Sci. USA 2000, 97, 11403–11408. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Takeda, H.; Kawakami, N.; Kobayashi, M.; Matsuda, N.; Mishina, M. A Transposon-Mediated Gene Trap Approach Identifies Developmentally Regulated Genes in Zebrafish. Dev. Cell 2004, 7, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Parinov, S.; Kondrichin, I.; Korzh, V.; Emelyanov, A. Tol2 Transposon-Mediated Enhancer Trap to Identify Developmentally Regulated Zebrafish Genes in Vivo. Dev. Dyn. 2004, 231, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Choo, B.; Kondrichin, I.; Parinov, S.; Emelyanov, A.; Go, W.; Toh, W.; Korzh, V. Zebrafish Transgenic Enhancer TRAP Line Database (ZETRAP). BMC Dev. Biol. 2006, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kondrychyn, I.; Garcia-Lecea, M.; Emelyanov, A.; Parinov, S.; Korzh, V. Genome-Wide Analysis of Tol2 Transposon Reintegration in Zebrafish. BMC Genom. 2009, 10, 418. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-Resolution Mapping and Characterization of Open Chromatin across the Genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac Separates Active from Poised Enhancers and Predicts Developmental State. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and Predictive Chromatin Signatures of Transcriptional Promoters and Enhancers in the Human Genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone Modifications at Human Enhancers Reflect Global Cell-Type-Specific Gene Expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A Unique Chromatin Signature Uncovers Early Developmental Enhancers in Humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T. An Atlas of Active Enhancers across Human Cell Types and Tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Nishikawa, K.; Seno, S.; Matsuda, H.; Takayanagi, H.; Ishii, M. Roles of Enhancer RNAs in RANKL-Induced Osteoclast Differentiation Identified by Genome-Wide Cap-Analysis of Gene Expression Using CRISPR. Sci. Rep. 2018, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Galang, G.; Mandla, R.; Ruan, H.; Jung, C.; Sinha, T.; Stone, N.R.; Wu, R.S.; Mannion, B.J.; Allu, P.K.R.; Chang, K.; et al. ATAC-Seq Reveals an Isl1 Enhancer That Regulates Sinoatrial Node Development and Function. Circ. Res 2020, 127, 1502–1518. [Google Scholar] [CrossRef] [PubMed]
- Balciunas, D.; Davidson, A.E.; Sivasubbu, S.; Hermanson, S.B.; Welle, Z.; Ekker, S.C. Enhancer Trapping in Zebrafish Using the Sleeping Beauty Transposon. BMC Genom. 2004, 5, 62. [Google Scholar] [CrossRef]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Kawakami, Y.; Rodríguez-León, J.; Koth, C.M.; Büscher, D.; Itoh, T.; Raya, Á.; Ng, J.K.; Esteban, C.R.; Takahashi, S.; Henrique, D.; et al. MKP3 Mediates the Cellular Response to FGF8 Signalling in the Vertebrate Limb. Nat. Cell Biol. 2003, 5, 513–519. [Google Scholar] [CrossRef]
- Chen, L.; Fish, A.E.; Capra, J.A. Prediction of Gene Regulatory Enhancers across Species Reveals Evolutionarily Conserved Sequence Properties. PLoS Comput Biol. 2018, 14, e1006484. [Google Scholar] [CrossRef]
- Yuan, X.; Song, M.; Devine, P.; Bruneau, B.G.; Scott, I.C.; Wilson, M.D. Heart Enhancers with Deeply Conserved Regulatory Activity Are Established Early in Zebrafish Development. Nat. Commun. 2018, 9, 4977. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.S.; Zheng, D.; Tan, S.Z.; Bower, N.I.; Garside, V.; Vanwalleghem, G.; Gaiti, F.; Scott, E.; Hogan, B.M.; Kikuchi, K.; et al. Deep Conservation of the Enhancer Regulatory Code in Animals. Science 2020, 370, eaax8137. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Morrison, A.; Gould, A.; Gilthorpe, J.; Chaudhuri, C.; Rigby, P.; Krumlauf, R.; Brenner, S. Detecting Conserved Regulatory Elements with the Model Genome of the Japanese Puffer Fish, Fugu Rubripes. Proc. Natl. Acad. Sci. USA 1995, 92, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Ansari-Lari, M.A.; Oeltjen, J.C.; Schwartz, S.; Zhang, Z.; Muzny, D.M.; Lu, J.; Gorrell, J.H.; Chinault, A.C.; Belmont, J.W.; Miller, W.; et al. Comparative Sequence Analysis of a Gene-Rich Cluster at Human Chromosome 12p13 and Its Syntenic Region in Mouse Chromosome 6. Genome Res. 1998, 8, 29–40. [Google Scholar]
- Ellsworth, R.E.; Jamison, D.C.; Touchman, J.W.; Chissoe, S.L.; Braden Maduro, V.V.; Bouffard, G.G.; Dietrich, N.L.; Beckstrom-Sternberg, S.M.; Lyer, L.M.; Weintraub, L.A.; et al. Comparative Genomic Sequence Analysis of the Human and Mouse Cystic Fibrosis Transmembrane Conductance Regulator Genes. Proc. Natl. Acad. Sci. USA 2000, 97, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, M.A. Scanning Human Gene Deserts for Long-Range Enhancers. Science 2003, 302, 413. [Google Scholar] [CrossRef]
- Sandelin, A.; Bailey, P.; Bruce, S.; Engström, P.G.; Klos, J.M.; Wasserman, W.W.; Ericson, J.; Lenhard, B. Arrays of Ultraconserved Non-Coding Regions Span the Loci of Key Developmental Genes in Vertebrate Genomes. BMC Genom. 2004, 5, 1–9. [Google Scholar] [CrossRef]
- Woolfe, A.; Goodson, M.; Goode, D.K.; Snell, P.; McEwen, G.K.; Vavouri, T.; Smith, S.F.; North, P.; Callaway, H.; Kelly, K.; et al. Highly Conserved Non-Coding Sequences Are Associated with Vertebrate Development. PLoS Biol. 2004, 3, e7. [Google Scholar] [CrossRef]
- Pennacchio, L.A.; Ahituv, N.; Moses, A.M.; Prabhakar, S.; Nobrega, M.A.; Shoukry, M.; Minovitsky, S.; Dubchak, I.; Holt, A.; Lewis, K.D.; et al. In Vivo Enhancer Analysis of Human Conserved Non-Coding Sequences. Nature 2006, 444, 499–502. [Google Scholar] [CrossRef]
- Li, Y.; Hiroi, Y.; Liao, J.K. Notch Signaling as an Important Mediator of Cardiac Repair and Regeneration After Myocardial Infarction. Trends Cardiovasc. Med. 2010, 20, 228–231. [Google Scholar] [CrossRef]
- Dong, X.; Navratilova, P.; Fredman, D.; Drivenes, Ø.; Becker, T.S.; Lenhard, B. Exonic Remnants of Whole-Genome Duplication Reveal Cis-Regulatory Function of Coding Exons. Nucleic Acids Res. 2010, 38, 1071–1085. [Google Scholar] [CrossRef]
- Cai, C.; Sang, C.; Du, J.; Jia, H.; Tu, J.; Wan, Q.; Bao, B.; Xie, S.; Huang, Y.; Li, A.; et al. Knockout of Tnni1b in Zebrafish Causes Defects in Atrioventricular Valve Development via the Inhibition of the Myocardial Wnt Signaling Pathway. FASEB J. 2019, 33, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, Z.; Hwang, P.M. Cardiac Troponin Complex: Cardiac Troponin C (TNNC1), Cardiac Troponin I (TNNI3), and Cardiac Troponin T (TNNT2). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: New York, NY, USA, 2016; pp. 1–10. ISBN 978-1-4614-6438-9. [Google Scholar]
- Hallikas, O.; Palin, K.; Sinjushina, N.; Rautiainen, R.; Partanen, J.; Ukkonen, E.; Taipale, J. Genome-Wide Prediction of Mammalian Enhancers Based on Analysis of Transcription-Factor Binding Affinity. Cell 2006, 124, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Chiang, I.K.-N.; Fritzsche, M.; Pichol-Thievend, C.; Neal, A.; Holmes, K.; Lagendijk, A.; Overman, J.; D’Angelo, D.; Omini, A.; Hermkens, D.; et al. SoxF Factors Induce Notch1 Expression via Direct Transcriptional Regulation during Early Arterial Development. Development 2017, 144, 2629–2639. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.Y.; Everman, D.B.; Murphy, K.K.; Gurrieri, F.; Schwartz, C.E.; Ahituv, N. Functional Characterization of Tissue-Specific Enhancers in the DLX5/6 Locus. Hum. Mol. Genet. 2012, 21, 4930–4938. [Google Scholar] [CrossRef]
- Becker, P.W.; Sacilotto, N.; Nornes, S.; Neal, A.; Thomas, M.O.; Liu, K.; Preece, C.; Ratnayaka, I.; Davies, B.; Bou-Gharios, G.; et al. An Intronic Flk1 Enhancer Directs Arterial-Specific Expression via RBPJ-Mediated Venous Repression. Arter. Thromb. Vasc. Biol. 2016, 36, 1209–1219. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-Generation DNA Sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and Analysis of Functional Elements in 1% of the Human Genome by the ENCODE Pilot Project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Myers, R.M.; Stamatoyannopoulos, J.; Snyder, M.; Dunham, I.; Hardison, R.C.; Bernstein, B.E.; Gingeras, T.R.; Kent, W.J.; Birney, E.; Wold, B.; et al. A User’s Guide to the Encyclopedia of DNA Elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium an Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, 991–995. [Google Scholar] [CrossRef]
- Gorkin, D.U.; Barozzi, I.; Zhao, Y.; Zhang, Y.; Huang, H.; Lee, A.Y.; Li, B.; Chiou, J.; Wildberg, A.; Ding, B.; et al. An Atlas of Dynamic Chromatin Landscapes in Mouse Fetal Development. Nature 2020, 583, 744–751. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–329. [Google Scholar] [CrossRef]
- Krishnan, A.; Samtani, R.; Dhanantwari, P.; Lee, E.; Yamada, S.; Shiota, K.; Donofrio, M.T.; Leatherbury, L.; Lo, C.W. A Detailed Comparison of Mouse and Human Cardiac Development. Pediatr. Res. 2014, 76, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Shulha, H.P.; Lin, J.M.; Vales, T.R.; Fu, Y.; Bodine, D.M.; McKay, R.D.G.; Chenoweth, J.G.; Tesar, P.J.; Furey, T.S.; et al. Identification and Characterization of Cell Type-Specific and Ubiquitous Chromatin Regulatory Structures in the Human Genome. PLoS Genet. 2007, 3, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Crawford, G.E.; Davis, S.; Scacheri, P.C.; Renaud, G.; Halawi, M.J.; Erdos, M.R.; Green, R.; Meltzer, P.S.; Wolfsberg, T.G.; Collins, F.S. DNase-Chip: A High-Resolution Method to Identify DNase I Hypersensitive Sites Using Tiled Microarrays. Nat. Methods 2006, 3, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Jurkiewicz, N.K. Two Components of Cardiac Delayed Rectifier K+ Current. Differential Sensitivity to Block by Class III Antiarrhythmic Agents. J. Gen. Physiol. 1990, 96, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Arking, D.E.; Pulit, S.L.; Crotti, L.; van der Harst, P.; Munroe, P.B.; Koopmann, T.T.; Sotoodehnia, N.; Rossin, E.J.; Morley, M.; Wang, X.; et al. Genetic Association Study of QT Interval Highlights Role for Calcium Signaling Pathways in Myocardial Repolarization. Nat. Genet. 2014, 46, 826–836. [Google Scholar] [CrossRef]
- Méndez-Giráldez, R.; Gogarten, S.M.; Below, J.E.; Yao, J.; Seyerle, A.A.; Highland, H.M.; Kooperberg, C.; Soliman, E.Z.; Rotter, J.I.; Kerr, K.F.; et al. GWAS of the Electrocardiographic QT Interval in Hispanics/Latinos Generalizes Previously Identified Loci and Identifies Population-Specific Signals. Sci. Rep. 2017, 7, 17075. [Google Scholar] [CrossRef]
- Caballero, R.; Utrilla, R.G.; Amorós, I.; Matamoros, M.; Pérez-Hernández, M.; Tinaquero, D.; Alfayate, S.; Nieto-Marín, P.; Guerrero-Serna, G.; Liu, Q.; et al. Tbx20 Controls the Expression of the KCNH2 Gene and of HERG Channels. Proc. Natl. Acad. Sci. USA 2017, 114, E416–E425. [Google Scholar] [CrossRef]
- Boogerd, C.J.; Zhu, X.; Aneas, I.; Sakabe, N.; Zhang, L.; Sobreira, D.R.; Montefiori, L.; Bogomolovas, J.; Joslin, A.C.; Zhou, B.; et al. Tbx20 Is Required in Mid-Gestation Cardiomyocytes and Plays a Central Role in Atrial Development. Circ. Res. 2018, 123, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread Transcription at Neuronal Activity-Regulated Enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef]
- Devine, W.P.; Wythe, J.D.; George, M.; Koshiba-Takeuchi, K.; Bruneau, B.G. Early Patterning and Specification of Cardiac Progenitors in Gastrulating Mesoderm. eLife 2014, 3, e03848. [Google Scholar] [CrossRef]
- Goldman, J.A.; Poss, K.D. Gene Regulatory Programmes of Tissue Regeneration. Nat. Rev. Genet. 2020, 21, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Hu, J.; Karra, R.; Dickson, A.L.; Tornini, V.A.; Nachtrab, G.; Gemberling, M.; Goldman, J.A.; Black, B.L.; Poss, K.D. Modulation of Tissue Repair by Regeneration Enhancer Elements. Nature 2016, 532, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Ou, J.; Lee, N.; Shin, K.; Cigliola, V.; Song, L.; Crawford, G.E.; Kang, J.; Poss, K.D. Identification and Requirements of Enhancers That Direct Gene Expression during Zebrafish Fin Regeneration. Development 2020, 147, dev191262. [Google Scholar] [CrossRef]
- Potts, H.G.; Stockdale, W.T.; Mommersteeg, M.T.M. Unlocking the Secrets of the Regenerating Fish Heart: Comparing Regenerative Models to Shed Light on Successful Regeneration. J. Cardiovasc. Dev. Dis. JCDD 2021, 8, 4. [Google Scholar] [CrossRef]
- Honkoop, H.; de Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; de Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A.; et al. Single-Cell Analysis Uncovers That Metabolic Reprogramming by ErbB2 Signaling Is Essential for Cardiomyocyte Proliferation in the Regenerating Heart. eLife 2019, 8, e50163. [Google Scholar] [CrossRef]
- Begeman, I.J.; Shin, K.; Osorio-Méndez, D.; Kurth, A.; Lee, N.; Chamberlain, T.J.; Pelegri, F.J.; Kang, J. Decoding an Organ Regeneration Switch by Dissecting Cardiac Regeneration Enhancers. Development 2020, 147, dev194019. [Google Scholar] [CrossRef]
- Goldman, J.A.; Kuzu, G.; Lee, N.; Karasik, J.; Gemberling, M.; Foglia, M.J.; Karra, R.; Dickson, A.L.; Sun, F.; Tolstorukov, M.Y.; et al. Resolving Heart Regeneration by Replacement Histone Profiling. Dev. Cell 2017, 40, 392–404.e5. [Google Scholar] [CrossRef]
- Ding, M.; Liu, Y.; Liao, X.; Zhan, H.; Liu, Y.; Huang, W. Enhancer RNAs (ERNAs): New Insights into Gene Transcription and Disease Treatment. J. Cancer 2018, 9, 2334–2340. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M. Enhancer RNAs: A Class of Long Noncoding RNAs Synthesized at Enhancers. Cold Spring Harb. Perspect. Biol. 2015, 7, 3–5. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as Non-Coding RNA Transcription Units: Recent Insights and Future Perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs Are an Important Regulatory Layer of the Epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M.; et al. Cage: Cap Analysis of Gene Expression. Nat. Methods 2006, 3, 211. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap Analysis Gene Expression for High-Throughput Analysis of Transcriptional Starting Point and Identification of Promoter Usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef]
- Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; De Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A Promoter-Level Mammalian Expression Atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An Atlas of Human Long Non-Coding RNAs with Accurate 5′ Ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef]
- Takahashi, H.; Lassmann, T.; Murata, M.; Carninci, P. 5′ End-Centered Expression Profiling Using Cap-Analysis Gene Expression and Next-Generation Sequencing. Nat. Protoc. 2012, 7, 542–561. [Google Scholar] [CrossRef]
- Abugessaisa, I.; Shimoji, H.; Sahin, S.; Kondo, A.; Harshbarger, J.; Lizio, M.; Hayashizaki, Y.; Carninci, P.; Forrest, A.; Kasukawa, T.; et al. FANTOM5 Transcriptome Catalog of Cellular States Based on Semantic MediaWiki. Database 2016, 2016, baw105. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawaji, H.; Kasukawa, T.; Forrest, A.; Carninci, P.; Hayashizaki, Y. The FANTOM5 Collection, a Data Series Underpinning Mammalian Transcriptome Atlases in Diverse Cell Types. Sci. Data 2017, 4, 170113. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Arakawa, T.; Fukuda, S.; Furuno, M.; Hasegawa, A.; Hori, F.; Ishikawa-Kato, S.; Kaida, K.; Kaiho, A.; Kanamori-Katayama, M.; et al. FANTOM5 CAGE Profiles of Human and Mouse Samples. Sci. Data 2017, 4, 170112. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; di Iulio, J.; Maleri, S.; Eser, U.; Vierstra, J.; Reynolds, A.; Sandstrom, R.; Stamatoyannopoulos, J.A.; Churchman, L.S. Native Elongating Transcript Sequencing Reveals Human Transcriptional Activity at Nucleotide Resolution. Cell 2015, 161, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Young, R.S.; Kumar, Y.; Bickmore, W.A.; Taylor, M.S. Bidirectional Transcription Initiation Marks Accessible Chromatin and Is Not Specific to Enhancers. Genome Biol. 2017, 18, 242. [Google Scholar] [CrossRef] [PubMed]
- Galas, D.J.; Schmitz, A. DNAase Footprinting a Simple Method for the Detection of Protein-DNA Binding Specificity. Nucleic Acids Res. 1978, 5, 3157–3170. [Google Scholar] [CrossRef] [PubMed]
- Barozzi, I.; Bora, P.; Morelli, M.J. Comparative Evaluation of DNase-Seq Footprint Identification Strategies. Front. Genet. 2014, 5, 1–6. [Google Scholar] [CrossRef]
- Piper, J.; Elze, M.C.; Cauchy, P.; Cockerill, P.N.; Bonifer, C.; Ott, S. Wellington: A Novel Method for the Accurate Identification of Digital Genomic Footprints from DNase-Seq Data. Nucleic Acids Res. 2013, 41, e201. [Google Scholar] [CrossRef]
- Gusmao, E.G.; Allhoff, M.; Zenke, M.; Costa, I.G. Analysis of Computational Footprinting Methods for DNase Sequencing Experiments. Nat. Methods 2016, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Bentsen, M.; Goymann, P.; Schultheis, H.; Klee, K.; Petrova, A.; Wiegandt, R.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; et al. ATAC-Seq Footprinting Unravels Kinetics of Transcription Factor Binding during Zygotic Genome Activation. Nat. Commun. 2020, 11, 4267. [Google Scholar] [CrossRef]
- Zhu, T.; Liao, K.; Zhou, R.; Xia, C.; Xie, W. ATAC-Seq with Unique Molecular Identifiers Improves Quantification and Footprinting. Commun. Biol. 2020, 3, 675. [Google Scholar] [CrossRef] [PubMed]
- Salehin, N.; Tam, P.P.L.; Osteil, P. Prenet: Predictive Network from ATAC-SEQ Data. J. Bioinform. Comput. Biol. 2020, 18, 2040003. [Google Scholar] [CrossRef]
- Karabulut, M.; Ibrikci, T. Identification of Transcription Factor Binding Sites Using Gaussian Mixture Models. Expert Syst. 2014, 31, 70–80. [Google Scholar] [CrossRef]
- Won, K.J.; Chepelev, I.; Ren, B.; Wang, W. Prediction of Regulatory Elements in Mammalian Genomes Using Chromatin Signatures. BMC Bioinform. 2008, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Won, K.-J.; Ren, B.; Wang, W. Genome-Wide Prediction of Transcription Factor Binding Sites Using an Integrated Model. Genome Biol. 2010, 11, R7. [Google Scholar] [CrossRef]
- Zehnder, T.; Benner, P.; Vingron, M. Predicting Enhancers in Mammalian Genomes Using Supervised Hidden Markov Models. BMC Bioinform. 2019, 20, 157. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating Chromatin-State Discovery and Characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef]
- Zacher, B.; Michel, M.; Schwalb, B.; Cramer, P.; Tresch, A.; Gagneur, J. Accurate Promoter and Enhancer Identification in 127 ENCODE and Roadmap Epigenomics Cell Types and Tissues by GenoSTAN. PLoS ONE 2017, 12, 1–25. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised Pattern Discovery in Human Chromatin Structure through Genomic Segmentation. In BCB’13: Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics; Association for Computing Machinery: New York, NY, USA, 2013; Volume 9, pp. 813–814. [Google Scholar]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative Annotation of Chromatin Elements from ENCODE Data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Firpi, H.A.; Ucar, D.; Tan, K. Discover Regulatory DNA Elements Using Chromatin Signatures and Artificial Neural Network. Bioinformatics 2010, 26, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, N.; Xie, W.; Li, Y.; Wagner, U.; Wang, W.; Stamatoyannopoulos, J.; Ernst, J.; Kellis, M.; Ren, B. RFECS: A Random-Forest Based Algorithm for Enhancer Identification from Chromatin State. PLoS Comput. Biol. 2013, 9, e1002968. [Google Scholar] [CrossRef]
- Fernández, M.; Miranda-Saavedra, D. Genome-Wide Enhancer Prediction from Epigenetic Signatures Using Genetic Algorithm-Optimized Support Vector Machines. Nucleic Acids Res. 2012, 40, e77. [Google Scholar] [CrossRef]
- Erwin, G.D.; Oksenberg, N.; Truty, R.M.; Kostka, D.; Murphy, K.K.; Ahituv, N.; Pollard, K.S.; Capra, J.A. Integrating Diverse Datasets Improves Developmental Enhancer Prediction. PLoS Comput. Biol. 2014, 10, e1003677. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser—A Database of Tissue-Specific Human Enhancers. Nucleic Acids Res. 2007, 35, 88–92. [Google Scholar] [CrossRef]
- Kleftogiannis, D.; Kalnis, P.; Bajic, V.B. DEEP: A General Computational Framework for Predicting Enhancers. Nucleic Acids Res. 2015, 43, e6. [Google Scholar] [CrossRef] [PubMed]
- Bravo González-Blas, C.; Minnoye, L.; Papasokrati, D.; Aibar, S.; Hulselmans, G.; Christiaens, V.; Davie, K.; Wouters, J.; Aerts, S. CisTopic: Cis-Regulatory Topic Modeling on Single-Cell ATAC-Seq Data. Nat. Methods 2019, 16, 397–400. [Google Scholar] [CrossRef]
- Liu, B.; Fang, L.; Long, R.; Lan, X.; Chou, K.C. IEnhancer-2L: A Two-Layer Predictor for Identifying Enhancers and Their Strength by Pseudo k-Tuple Nucleotide Composition. Bioinformatics 2016, 32, 362–369. [Google Scholar] [CrossRef]
- Jia, C.; He, W. EnhancerPred: A Predictor for Discovering Enhancers Based on the Combination and Selection of Multiple Features. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef]
- He, W.; Jia, C. EnhancerPred2.0: Predicting Enhancers and Their Strength Based on Position-Specific Trinucleotide Propensity and Electron-Ion Interaction Potential Feature Selection. Mol. Biosyst. 2017, 13, 767–774. [Google Scholar] [CrossRef]
- Liu, B.; Li, K.; Huang, D.S.; Chou, K.C. IEnhancer-EL: Identifying Enhancers and Their Strength with Ensemble Learning Approach. Bioinformatics 2018, 34, 3835–3842. [Google Scholar] [CrossRef]
- Zhang, T.; Flores, M.; Huang, Y. ES-ARCNN: Predicting Enhancer Strength by Using Data Augmentation and Residual Convolutional Neural Network. Anal. Biochem. 2021, 618, 114120. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Gorkin, D.U.; Baker, M.; Strober, B.J.; Asoni, A.L.; McCallion, A.S.; Beer, M.A. A Method to Predict the Impact of Regulatory Variants from DNA Sequence. Nat. Genet. 2015, 47, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, A.; Uyar, A.; Khetan, S.; Stitzel, M.L.; Ucar, D. A Neural Network Based Model Effectively Predicts Enhancers from Clinical ATAC-Seq Samples. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Smemo, S.; Campos, L.C.; Moskowitz, I.P.; Krieger, J.E.; Pereira, A.C.; Nobrega, M.A. Regulatory Variation in a TBX5 Enhancer Leads to Isolated Congenital Heart Disease. Hum. Mol. Genet. 2012, 21, 3255–3263. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Xing, Q.; Ma, L.; Meng, H.; Liu, Y.; Pang, S.; Yan, B. Genetic Analysis of an Enhancer of the NKX2-5 Gene in Ventricular Septal Defects. Gene 2012, 508, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Meng, H.; Qiao, Y.; Pang, S.; Chen, D.; Yan, B. Two Novel and Functional DNA Sequence Variants within an Upstream Enhancer of the Human NKX2-5 Gene in Ventricular Septal Defects. Gene 2013, 524, 152–155. [Google Scholar] [CrossRef]
- Lettice, L.A.; Heaney, S.J.H.; Purdie, L.A.; Li, L.; de Beer, P.; Oostra, B.A.; Goode, D.; Elgar, G.; Hill, R.E.; de Graaff, E. A Long-Range Shh Enhancer Regulates Expression in the Developing Limb and Fin and Is Associated with Preaxial Polydactyly. Hum. Mol. Genet. 2003, 12, 1725–1735. [Google Scholar] [CrossRef]
- Mongin, E.; Dewar, K.; Blanchette, M. Mapping Association between Long-Range Cis-Regulatory Regions and Their Target Genes Using Synteny. J. Comput. Biol. 2011, 18, 1115–1130. [Google Scholar] [CrossRef]
- Clément, Y.; Torbey, P.; Gilardi-Hebenstreit, P.; Crollius, H.R. Enhancer-Gene Maps in the Human and Zebrafish Genomes Using Evolutionary Linkage Conservation. Nucleic Acids Res. 2020, 48, 2357–2371. [Google Scholar] [CrossRef]
- Bertero, A.; Rosa-Garrido, M. Three-Dimensional Chromatin Organization in Cardiac Development and Disease. J. Mol. Cell. Cardiol. 2021, 151, 89–105. [Google Scholar] [CrossRef]
- Lu, L.; Liu, X.; Huang, W.-K.; Giusti-Rodríguez, P.; Cui, J.; Zhang, S.; Xu, W.; Wen, Z.; Ma, S.; Rosen, J.D.; et al. Robust Hi-C Maps of Enhancer-Promoter Interactions Reveal the Function of Non-Coding Genome in Neural Development and Diseases. Mol. Cell 2020, 79, 521–534.e15. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, G.; Lopes, R.; Ugalde, A.P.; Nevedomskaya, E.; Han, R.; Myacheva, K.; Zwart, W.; Elkon, R.; Agami, R. Functional Genetic Screens for Enhancer Elements in the Human Genome Using CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, Y.; Cao, H.; Zhang, Y.; Gu, Z.; Liu, X.; Yu, A.; Kaphle, P.; Dickerson, K.E.; Ni, M.; et al. Interrogation of Enhancer Function by Enhancer-Targeting CRISPR Epigenetic Editing. Nat. Commun. 2020, 11, 485. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Approaches | Work-Principle | Reference |
|---|---|---|
| Enhancer-Deletion Approach | Deletion of non-coding cis-regulatory DNA elements can severely disrupt the systemic functions. | [83] |
| Enhancer-Trap Assay | Through microinjection of embryos random integration of a vector-construct with minimal promoter and reporter gene, driving expression if enhancer is present. | [84,85,86,87,88] |
| Transient Transfection Assay | Luciferase reporter plasmid constructs containing promoter and 5′-flanking DNA sequence increases the luciferase expression in the presence of enhancer region. | [81] |
| High-Throughput Techniques | ||
| DNase I-seq | DNase I digestion and DHS fragments mostly comprises cis-regulatory regions (e.g., enhancers). | [89] |
| Epigenomic Profiling (ChIP-Seq) | Enrichment of H3K4me1, H3K4ac, and P300 histone modifications determines the active enhancers. | [20,55,56,60,90,91,92,93] |
| CAGE | High resolution map of TSS and bidirectional transcription patterns defines the precise location of enhancers. | [94,95] |
| NET-CAGE | Capturing 5′-ends of nascent transcripts by fusing two technologies helps to identify unstable transcripts (eRNA). | [8] |
| ATAC-Seq | Accessible chromatin regions encompass enhancer elements. | [58,96] |
| Mathematical Model | Algorithm | Reference | Link |
|---|---|---|---|
| Supervised Machine Learning (Probabilistic Graphical Models) | |||
| HMM-SA | An HMM-based classifier obtained for enhancer, promoter, and background. Individual log-odd score measurement for each classifier for a genomic region of interest and score is averaged over three quantified scores. Simulated annealing algorithm implementation to obtain best combination of histone modification marks defining enhancers. | [167] | http://http:/nash.ucsd.edu/chromatin.tar.gz (accessed on 9 April 2021) |
| CHROMatin based Integrated Approach (Chromia) | Parallel HMM model combines histone modifications data and genomic sequence (motif information) to perform predictions. Computation of position specific scoring matrices (PSSM Scores). | [168] | http://wanglab.ucsd.edu/star/ (accessed on 9 April 2021) |
| enhancer-HMM | A probabilistic model based on HMM. Training is performed with histone modification marks data (ChIP-Seq) and chromatin accessibility data (ATAC-Seq). | [169] | https://github.com/tobiaszehnder/ehmm (accessed on 9 April 2021) |
| Unsupervised Machine Learning | |||
| ChromHMM | Application of multivariate HMM to train the classification model. Training is performed on histone modification marks data. | [170] | http://compbio.mit.edu/ChromHMM/ (accessed on 9 April 2021) |
| GenoStan | Genome segmentation-based method with HMM application. Read counts modeled with Poisson lognormal and negative binomial distribution approaches. Model’s parameter training solely relies upon the given raw data without automation on chromatin states (manual parameter). Model training using ChIP-Seq and DNase I-Seq chromatin marks. | [171] | http://bioconductor.org/packages/3.4/bioc/html/STAN.html http://i12g-gagneurweb.in.tum.de/public/paper/GenoSTAN (accessed on 9 April 2021) |
| Segway | An application of unsupervised genome segmentation approach using dynamic Bayesian network algorithm. Integration of ChIP-seq, DNase I-Seq, transcription factor and FAIRE-Seq data. Model training on 1% of human genome with ChIP-Seq, Dnase I-Seq and FAIRE-Seq data from ENCODE pilot project. Viterbi decoding helped to identify genome segments of 2 Mb size. | [172] | https://pmgenomics.ca/hoffmanlab/proj/segway/ (accessed on 9 April 2021) |
| cisTopic | Model training on single-cell ATAC-Seq data using unsupervised Bayesian framework. Probabilistic modeling with latent Dirichlet allocation with a collapsed Gibbs sampler. | [180] | http://github.com/aertslab/cistopic (accessed on 9 April 2021) |
| Artificial Neural Networks (ANN) | |||
| Chromatin Signature Identification by Artificial Neural Network (CSI-ANN) | Time-delay neural network was applied for feature classification task.Mathematical functions: Mean and Energy are utilized to transform genome-wide data. Fisher discriminant analysis is performed to convert the high dimensionality of data to enhance the accuracy of classification model. Model is trained on histone modifications data. | [174] | http://www.medicine.uiowa.edu/Labs/tan/CSIANNsoft.zip (accessed on 9 April 2021) |
| Random Forest based Enhancer identification from Chromatin States (RFECS) | Random forest-based mathematical model is utilized to classify features. Model is trained on ENCODE chromatin modifications data and DNase I-Seq data. | [175] | http://enhancer.ucsd.edu/renlab/RFECS_enhancer_prediction/Training (accessed on 9 April 2021) |
| Support Vector Machine | |||
| ChromaGenSVM | Chromatin state detection using support vector machines in combination with genetic algorithm optimization. Model is trained with ChIP-chip data from ENCODE project and ChIP-Seq data containing DNA-methylation and acetylation marks. | [176] | http://sysimm.ifrec.osaka-u.ac.jp/download/Diego/ (accessed on 9 April 2021) |
| EnhancerFinder (multiple kernel learning) | It works by incorporating multiple datatypes in the prediction process such as chromatin modification marks, sequence-level conservation, and DNA sequence motifs. Model is trained by using developmental enhancers from VISTA enhancer browser. | [177] | Putative enhancer elements are available at UCSC genome browser (accessed on 9 April 2021) |
| DEEP | It comprises three main components: DEEP-ENCODE, DEEP-FANTOM5 and DEEP-VISTA. Application of both SVM and ANN to train the prediction model. | [179] | http://cbrc.kaust.edu.sa/deep/ (accessed on 9 April 2021) |
| TF Footprinting | Nucleosome bound DNA restricts its cleavage, producing low signal. Similarly, open chromatin regions (with high signal) are bound by TFs tend to restrict cleavage, generating weak signal. These regions are referred to as “footprints”, representing the presence of enhancer elements occupied by TFs. | [159,160,161,162,163,164,165,166] | |
| Sequence-based Evolutionary Conservation | Developmental enhancers are known to be conserved among cross-species genomic sequences. | [103,104,105,106,107,108,109] | |
| Enhancers Strength Prediction | |||
| iEnhancer-2L | A SVM-based model trained on histone modification data. Use of pseudo k-tuple nucleotide sequence composition. | [181] | http://bioinformatics.hitsz.edu.cn/iEnhancer-2L/ (accessed on 9 April 2021) |
| EnhancerPred | Model is trained on chromatin states. Implementation of Bi-profile Bayes to obtain nucleotide sequence features. Rank the predictions based on F-score. | [182] | http://server.malab.cn/EnhancerPRED/ (accessed on 9 April 2021) |
| EnhancerPred2.0 | A SVM based classification model trained on chromatin modifications data. Integration of position-specific trinucleotide propensity and electron ion-interaction pseudopotential of DNA sequence. Computation of F-score to rank predictions. | [183] | |
| iEnhancer-EL | A SVM based model. DNA sequence composition and nucleotide frequencies are obtained using Kmer, subsequence and pseudo k-tuple methods. | [184] | http://bioinformatics.hitsz.edu.cn/iEnhancer-EL/ (accessed on 9 April 2021) |
| enhancer sequences by implementing Augmented data and Residual Convolutional Neural Network (ES-ARCNN) | Implementation of residual convolution neural network to train the classification model. Enlarging the input data using reverse complement and shifting method to gain better predictions. | [185] | http://compgenomics.utsa.edu/ES-ARCNN/ (accessed on 9 April 2021) |
| Sequence-level Variation within Enhancers | |||
| DeltaSVM | Prediction of the impact of sequence variation in enhancer activity. Model is trained with DNase I-seq and ChIP-seq data. | [186] | http://www.beerlab.org/deltasvm (accessed on 9 April 2021) |
| Predicting Enhancers from ATAC-Seq data (PEAS) | Implementation of neural networks model. Integration of chromatin accessibility data with nucleotide sequence composition (e.g., GC%). | [187] | https://github.com/UcarLab/PEAS (accessed on 9 April 2021) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parisi, C.; Vashisht, S.; Winata, C.L. Fish-Ing for Enhancers in the Heart. Int. J. Mol. Sci. 2021, 22, 3914. https://doi.org/10.3390/ijms22083914
Parisi C, Vashisht S, Winata CL. Fish-Ing for Enhancers in the Heart. International Journal of Molecular Sciences. 2021; 22(8):3914. https://doi.org/10.3390/ijms22083914
Chicago/Turabian StyleParisi, Costantino, Shikha Vashisht, and Cecilia Lanny Winata. 2021. "Fish-Ing for Enhancers in the Heart" International Journal of Molecular Sciences 22, no. 8: 3914. https://doi.org/10.3390/ijms22083914
APA StyleParisi, C., Vashisht, S., & Winata, C. L. (2021). Fish-Ing for Enhancers in the Heart. International Journal of Molecular Sciences, 22(8), 3914. https://doi.org/10.3390/ijms22083914