
Changes in Bacterial Endophyte Community Following Aspergillus flavus Infection in Resistant and Susceptible Maize Kernels



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
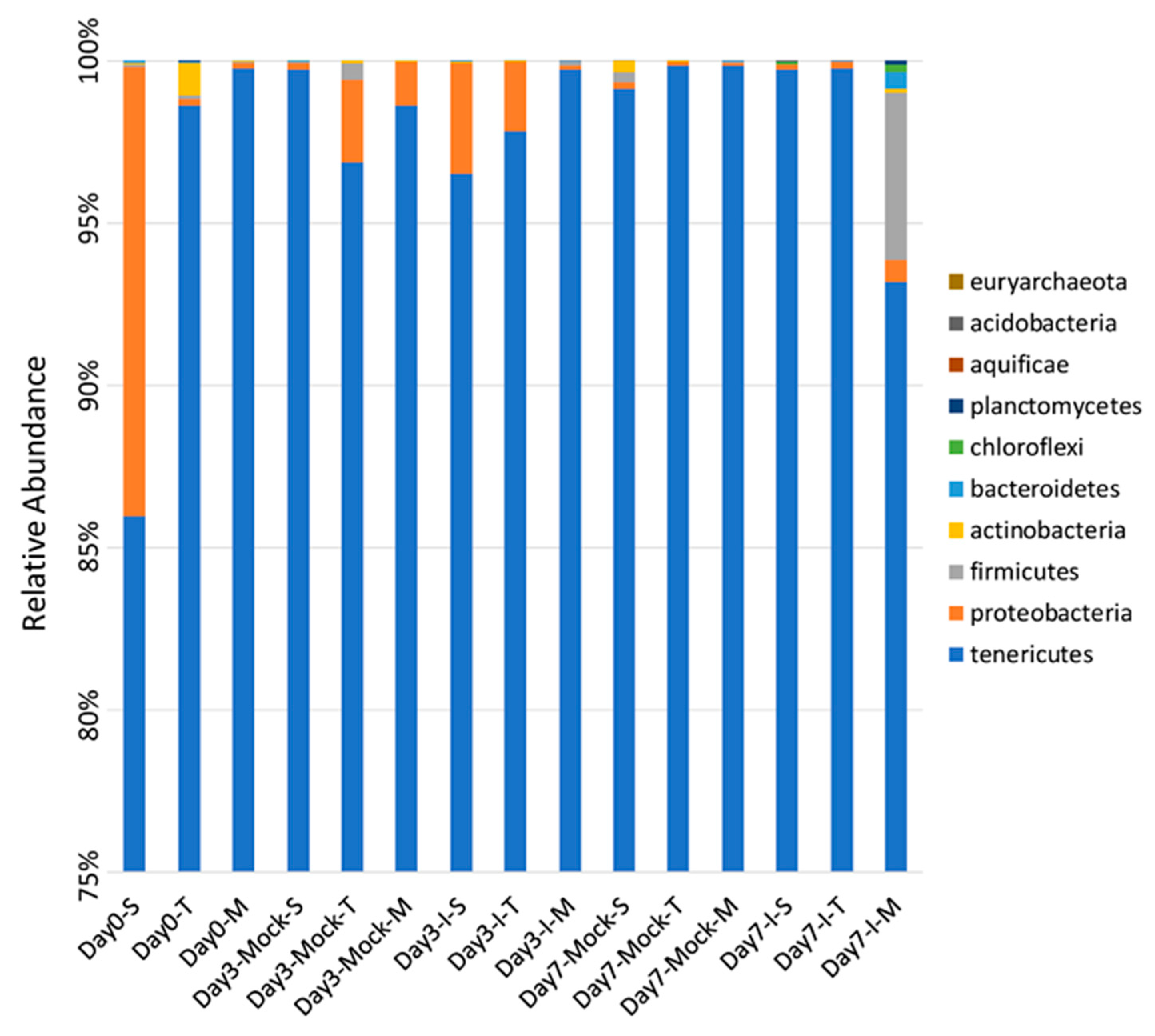
2.1. Significant Differences Observed in Bacterial Phyla between Resistant and Susceptible Lines
2.2. Relatively Fewer Bacterial Genera Varied between Treatment Groups
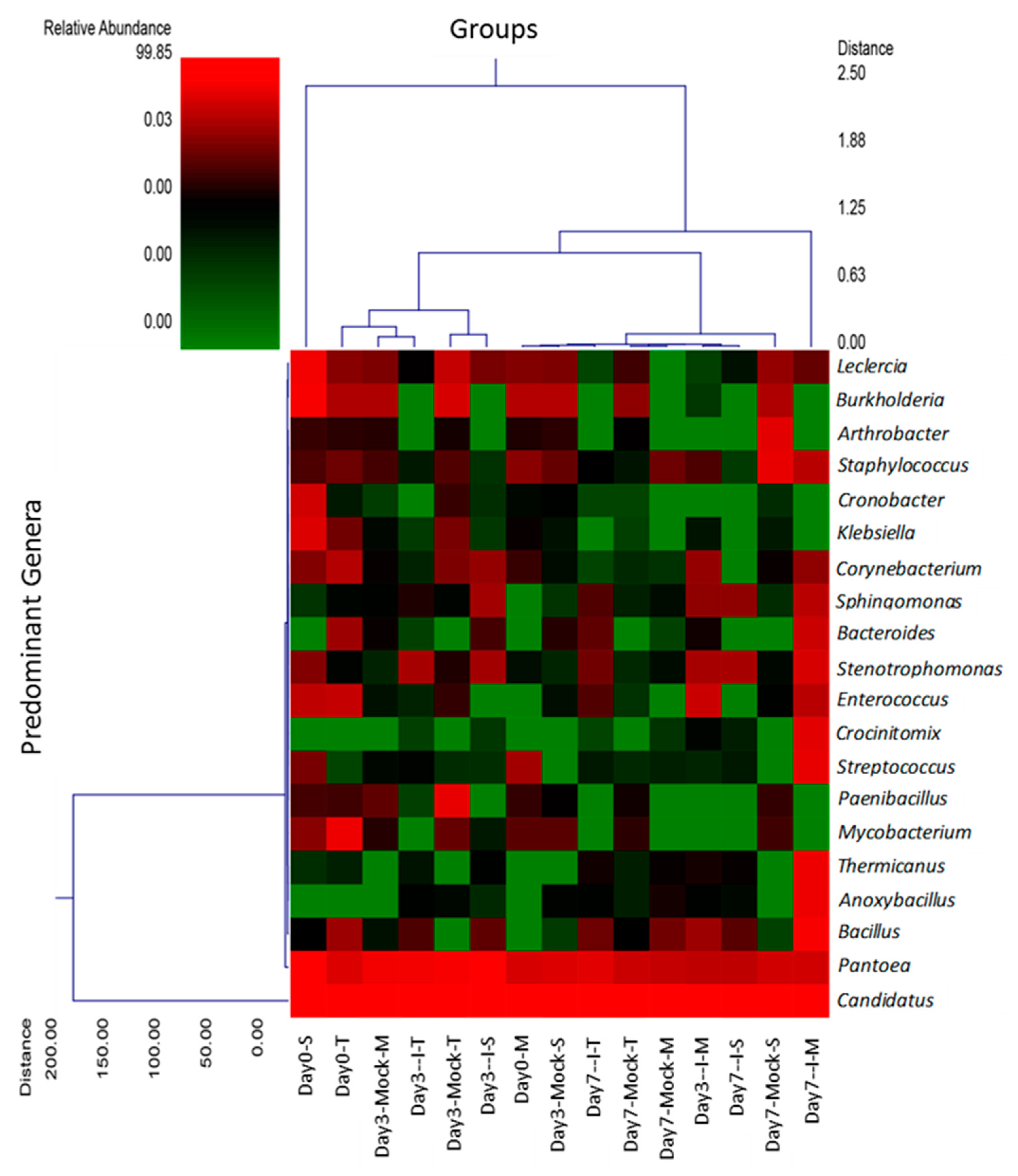
2.3. Hierarchal Clustering Shows Predominant Genera among Different Treatment Groups
2.4. Bacterial Diversity Varied with Fungal Infection in the Resistant and Susceptible Lines
2.4.1. Alpha Diversity of Samples
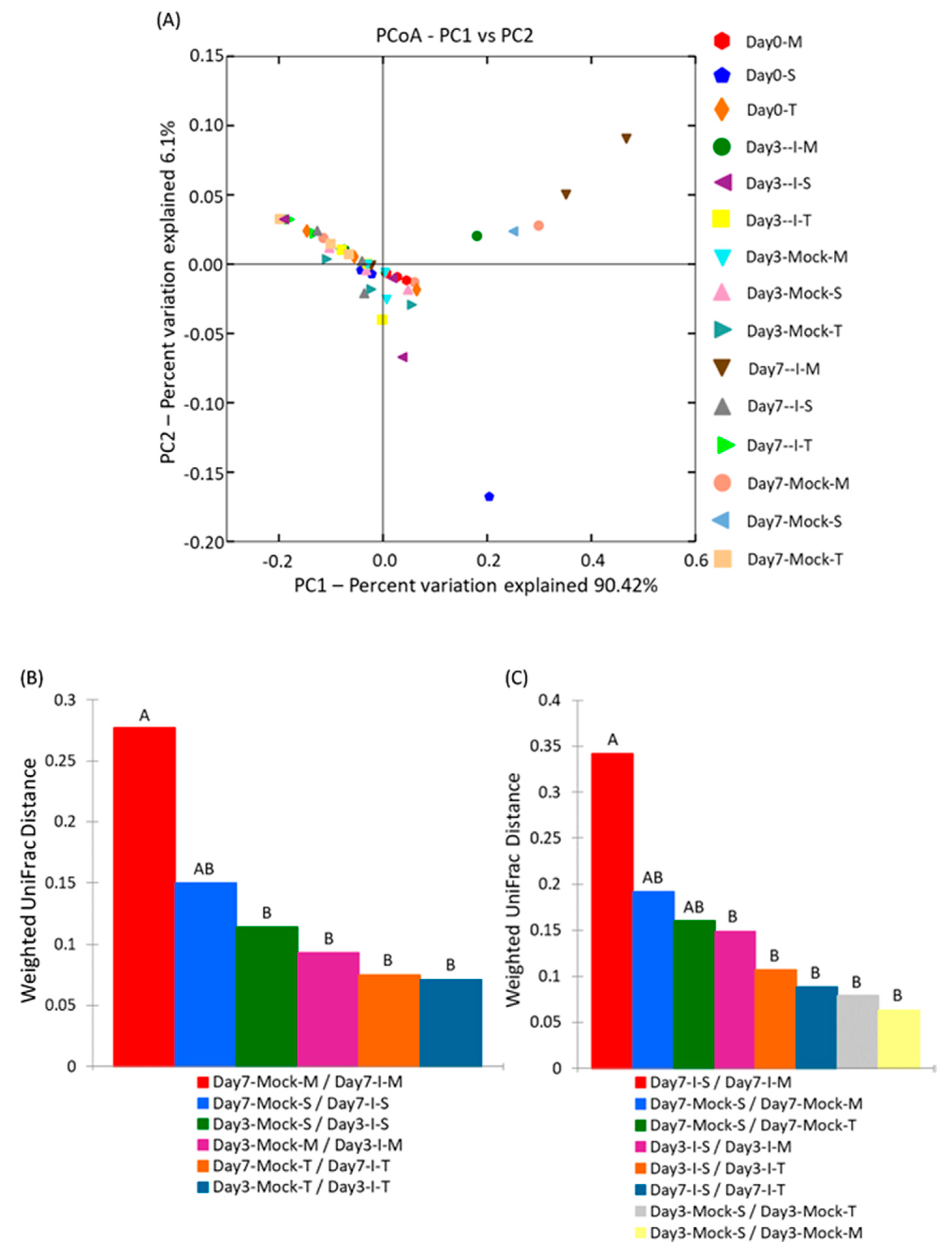
2.4.2. Beta Diversity of Samples
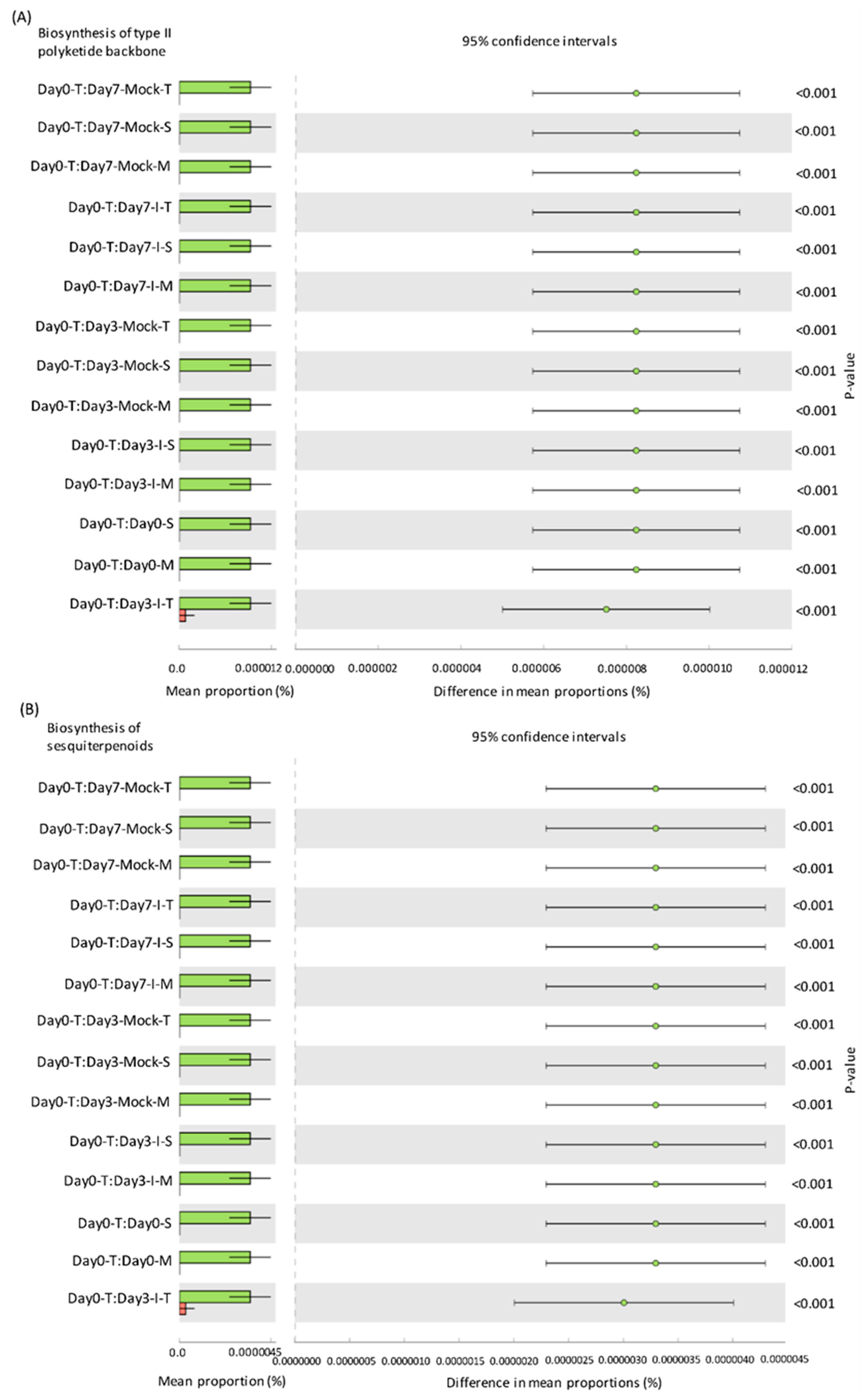
2.5. Predicted KEGG Pathways Were Highly Abundant in the Resistant Lines
3. Discussion
4. Materials and Methods
4.1. Maize Kernel Inoculation and Incubation
4.2. Genomic DNA Extraction, PCR Amplification, and 16S rRNA Sequencing
4.3. Data Processing
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ismaiel, A.; Papenbrock, J. Mycotoxins: Producing fungi and mechanisms of phytotoxicity. Agriculture 2015, 5, 492–537. [Google Scholar] [CrossRef]
- Mitchell, N.; Bowers, E.; Hurburgh, C.; Wu, F. Potential economic losses to the USA corn industry from aflatoxin contamination. Food Addit. Contam. 2016, 33, 540–550. [Google Scholar] [CrossRef]
- Umesha, S.; Manukumar, H.M.G.; Chandrasekhar, B.; Shivakumara, P.; Shiva Kumar, J.; Raghava, S.; Avinash, P.; Shirin, M.; Bharathi, T.R.; Rajini, S.B.; et al. Aflatoxins and food pathogens: Impact of biologically active aflatoxins and their control strategies. J. Sci. Food Agric. 2017, 97, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Mahato, D.K.; Lee, K.E.; Kamle, M.; Devi, S.; Dewangan, K.N.; Kumar, P.; Kang, S.G. Aflatoxins in food and feed: An overview on prevalence, detection and control strategies. Front. Microbiol. 2019, 10, 2266. [Google Scholar] [CrossRef]
- Martinez-Miranda, M.M.; Rosero-Moreano, M.; Taborda-Ocampo, G. Occurrence, dietary exposure and risk assessment of aflatoxins in arepa, bread and rice. Food Control 2019, 98, 359–366. [Google Scholar] [CrossRef]
- Moretti, A.; Pascale, M.; Logrieco, A.F. Mycotoxin risks under a climate change scenario in Europe. Trends Food Sci. Technol. 2019, 84, 38–40. [Google Scholar] [CrossRef]
- Ojiambo, P.S.; Battilani, P.; Cary, J.W.; Blum, B.H.; Carbone, I. Cultural and genetic approaches to manage aflatoxin contamination: Recent insights provide opportunities for improved control. Phytopathology 2018, 108, 1024–1037. [Google Scholar] [CrossRef]
- Lagogianni, C.S.; Tsitsigiannis, D.I. Effective biopesticides and biostimulants to reduce aflatoxins in maize fields. Front. Microbiol. 2019, 10, 2645. [Google Scholar] [CrossRef] [PubMed]
- Accinelli, C.; Abbas, H.K.; Little, N.S.; Kotowicz, J.K.; Mencarelli, M.; Shier, W.T. A liquid bioplastic formulation for film coating of agronomic seeds. Crop Protect. 2016, 89, 123–128. [Google Scholar] [CrossRef]
- Soni, P.; Gangurde, S.S.; Ortega-Beltran, A.; Kumar, R.; Parmar, S.; Sudini, H.K.; Lei, Y.; Ni, X.; Huai, D.; Fountain, J.C.; et al. Functional biology and molecular mechanisms of host-pathogen interactions for aflatoxin contamination in groundnut (Arachis hypogaea L.) and maize (Zea mays L.). Front. Microbiol. 2020, 11, 227. [Google Scholar]
- Drott, M.T.; Debenport, T.; Higgins, S.A.; Buckley, D.H.; Milgroom, M.G. Fitness cost of aflatoxin production in Aspergillus flavus when competing with soil microbes could maintain balancing selection. mBio 2019, 10, e02782-18. [Google Scholar] [CrossRef]
- Liu, H.; Brettell, L.E.; Qiu, Z.; Singh, B.K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef]
- Morelli, M.; Bahar, O.; Papadopoulou, K.K.; Hopkins, D.L.; Obradovic, A. Role of endophytes in plant health and defense against pathogens. Front. Plant Sci. 2020, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant-microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.B. The seed microbiome: Origins, interactions, and impacts. Plant Soil 2018, 422, 7–34. [Google Scholar] [CrossRef]
- Mousa, W.K.; Shearer, C.R.; Limay-Rios, V.; Zhou, T.; Raizada, M.N. Bacterial endophytes from wild maize suppress Fusarium graminearum in modern maize and inhibit mycotoxin accumulation. Front. Plant Sci. 2015, 6, 805. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, R.; Wu, X.; Xu, T.; Ahmad, S.; Zhang, X.; Zhao, J.; Liu, Y. An endophytic strain of the genus Bacillus isolated from the seeds of maize (Zea mays L.) has antagonistic activity against maize pathogenic strains. Microb. Pathog. 2020, 142, 104074. [Google Scholar] [CrossRef]
- Menkir, A.; Brown, R.L.; Bandyopadhyay, R.; Cleveland, T.E. Registration of six tropical maize germplasm lines with resistance to aflatoxin contamination. J. Plant Regist. 2008, 2, 246–250. [Google Scholar] [CrossRef]
- Maupin, L.M.; Clements, M.J.; White, D.G. Evaluation of the MI82 corn line as a source of resistance to aflatoxin in grain and use of BGYF as a selection tool. Plant Dis. 2003, 87, 1059–1066. [Google Scholar] [CrossRef]
- Fadiji, A.E.; Babalola, O.O. Elucidating mechanisms of endophytes used in plant protection and other bioactivities with multifunctional prospects. Front. Bioeng. Biotechnol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Bodhankar, S.; Grover, M.; Hemanth, S.; Reddy, G.; Rasul, S.; Yadav, S.K.; Desai, S.; Mallappa, M.; Mandapaka, M.; Srinivasarao, C. Maize seed endophytic bacteria: Dominance of antagonistic, lytic enzyme-producing Bacillus spp. 3 Biotech 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, R.; Minocha, R.; Lebar, M.D.; Rajasekaran, K.; Long, S.; Carter-Wientjes, C.; Minocha, S.; Cary, J.W. Contribution of maize polyamine and amino acid metabolism toward resistance against Aspergillus flavus infection and aflatoxin production. Front. Plant Sci. 2019, 10, 692. [Google Scholar] [CrossRef] [PubMed]
- Risdian, C.; Mozef, T.; Wink, J. Biosynthesis of polyketides in Streptomyces. Microorganisms 2019, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Zhang, M.Y.; Hou, S.Y.; Chen, J.; Wu, Y.Y.; Zhang, Y.X. Insights into Streptomyces spp. isolated from the rhizospheric soil of Panax notoginseng: Isolation, antimicrobial activity and biosynthetic potential for polyketides and non-ribosomal peptides. BMC Microbiol. 2020, 20, 1–16. [Google Scholar] [CrossRef]
- Mohamad, O.A.; Li, L.; Ma, J.B.; Hatab, S.; Xu, L.; Guo, J.W.; Rasulov, B.A.; Liu, Y.H.; Hedlund, B.P.; Li, W.J. Evaluation of the antimicrobial activity of endophytic bacterial populations from Chinese traditional medicinal plant licorice and characterization of the bioactive secondary metabolites produced by Bacillus atrophaeus against Verticillium dahliae. Front. Microbiol. 2018, 9, 924. [Google Scholar] [CrossRef] [PubMed]
- Ek-Ramos, M.J.; Gomez-Flores, R.; Orozco-Flores, A.A.; Rodriguez-Padilla, C.; Gonzalez-Ochoa, G.; Tamez-Guerra, P. Bioactive products from plant-endophytic Gram-positive bacteria. Front. Microbiol. 2019, 10, 463. [Google Scholar] [CrossRef]
- Etminani, F.; Harighi, B. Isolation and identification of endophytic bacteria with plant growth promoting activity and biocontrol potential from wild pistachio trees. Plant Pathol. J. 2018, 34, 208. [Google Scholar] [CrossRef]
- Pacifico, D.; Squartini, A.; Crucitti, D.; Barizza, E.; Lo Schiavo, F.; Muresu, R.; Carimi, F.; Zottini, M. The role of the endophytic microbiome in the grapevine response to environmental triggers. Front. Plant Sci. 2019, 10, 1256. [Google Scholar] [CrossRef]
- Rahman, L.; Shinwari, Z.K.; Iqrar, I.; Rahman, L.; Tanveer, F. An assessment on the role of endophytic microbes in the therapeutic potential of Fagonia indica. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 1–12. [Google Scholar] [CrossRef]
- Matsumoto, H.; Fan, X.; Wang, Y.; Kusstatscher, P.; Duan, J.; Wu, S.; Chen, S.; Qiao, K.; Wang, Y.; Ma, B.; et al. Bacterial seed endophyte shapes disease resistance in rice. Nat. Plants 2021, 7, 1–13. [Google Scholar] [CrossRef]
- Tiwari, P.; Bae, H. Horizontal gene transfer and endophytes: An implication for the acquisition of novel traits. Plants 2020, 9, 305. [Google Scholar] [CrossRef]
- Mousa, W.K.; Raizada, M.N. The diversity of anti-microbial secondary metabolites produced by fungal endophytes: An interdisciplinary perspective. Front. Microbiol. 2013, 4, 65. [Google Scholar] [CrossRef]
- Lugtenberg, B.J.; Caradus, J.R.; Johnson, L.J. Fungal endophytes for sustainable crop production. FEMS Microbiol. Ecol. 2016, 92, fiw194. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, A.; Singh, R.; Pandey, K.D. Endophytic bacteria: A new source of bioactive compounds. 3 Biotech 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Da Silva, P.H.; Souza, M.P.D.; Bianco, E.A.; da Silva, S.R.; Soares, L.N.; Costa, E.V.; Silva, F.; Barison, A.; Forim, M.R.; Cass, Q.B.; et al. Antifungal polyketides and other compounds from Amazonian endophytic Talaromyces fungi. J. Braz. Chem. Soc. 2018, 29, 622–630. [Google Scholar] [CrossRef]
- Lee, J.; Jo, D.G.; Park, D.; Chung, H.Y.; Mattson, M.P. Adaptive cellular stress pathways as therapeutic targets of dietary phytochemicals: Focus on the nervous system. Pharmacol. Rev. 2014, 66, 815–868. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Williams, W.; Windham, G.; Menkir, A.; Chen, Z.-Y. Evaluation of African-bred maize germplasm lines for resistance to aflatoxin accumulation. Agronomy 2016, 6, 24. [Google Scholar] [CrossRef]
- Rajasekaran, K.; Sickler, C.M.; Brown, R.L.; Cary, J.W.; Bhatnagar, D. Evaluation of resistance to aflatoxin contamination in kernels of maize genotypes using a GFP-expressing Aspergillus flavus strain. World Mycotoxin J. 2013, 6, 151–158. [Google Scholar] [CrossRef]
- Cotty, P.J. Virulence and cultural characteristics of two Aspergillus flavus strains pathogenic on cotton. Phytopathology 1989, 79, 808–814. [Google Scholar] [CrossRef]
- Thijs, S.; Op De Beeck, M.; Beckers, B.; Truyens, S.; Stevens, V.; Van Hamme, J.D.; Weyens, N.; Vangronsveld, J. Comparative evaluation of four bacteria-specific primer pairs for 16S rRNA gene surveys. Front. Microbiol. 2017, 8, 494. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Lu-Irving, P.; Copeland, J.; Guttman, D.S.; Wang, P.W.; Baltrus, D.A.; Dlugosch, K.M.; Johnson, M.T. Chloroplast sequence variation and the efficacy of peptide nucleic acids for blocking host amplification in plant microbiome studies. Microbiome 2018, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.D.; Sun, Y.; McKeehan, T.; Hagevoort, R.G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dowd, S.E.; Sun, Y.; Wolcott, R.D.; Domingo, A.; Carroll, J.A. Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: Bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodborne Pathog. Dis. 2008, 5, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Callaway, T.R.; Dowd, S.E.; Edrington, T.S.; Anderson, R.C.; Krueger, N.; Bauer, N.; Kononoff, P.J.; Nisbet, D.J. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J. Anim. Sci. 2010, 88, 3977–3983. [Google Scholar] [CrossRef]
- Almonacid, D.E.; Kraal, L.; Ossandon, F.J.; Budovskaya, Y.V.; Cardenas, J.P.; Bik, E.M.; Goddard, A.D.; Richman, J.; Apte, Z.S. 16S rRNA gene sequencing and healthy reference ranges for 28 clinically relevant microbial taxa from the human gut microbiome. PLoS ONE 2017, 12, e0176555. [Google Scholar] [CrossRef]
- Jacob, J.H.; Hussein, E.I.; Shakhatreh, M.A.K.; Cornelison, C.T. Microbial community analysis of the hypersaline water of the Dead Sea using high-throughput amplicon sequencing. MicrobiologyOpen 2017, 6, e00500. [Google Scholar] [CrossRef]
- Hussien, E.; Juhmani, A.S.; AlMasri, R.; Al-Horani, F.; Al-Saghir, M. Metagenomic analysis of microbial community associated with coral mucus from the Gulf of Aqaba. Helyon 2019, 5, e02876. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Lin, S.W.; Freedman, N.D.; Shi, J.; Gail, M.H.; Vogtmann, E.; Yu, G.; Klepac-Ceraj, V.; Paster, B.J.; Dye, B.A.; Wang, G.Q.; et al. Beta-diversity metrics of the upper digestive tract microbiome are associated with body mass index. Obesity 2015, 23, 862–869. [Google Scholar] [CrossRef]
- Schlatter, D.C.; Paul, N.C.; Shah, D.H.; Schillinger, W.F.; Bary, A.I.; Sharratt, B.; Paulitz, T.C. Biosolids and tillage practices influence soil bacterial communities in dryland wheat. Microb. Ecol. 2019, 78, 737–752. [Google Scholar] [CrossRef]
- Cephas, K.D.; Kim, J.; Mathai, R.A.; Barry, K.A.; Dowd, S.E.; Meline, B.S.; Swanson, K.S. Comparative analysis of salivary bacterial microbiome diversity in edentulous infants and their mothers or primary care givers using pyrosequencing. PLoS ONE 2011, 6, e23503. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Eren, A.M.; Zozaya, M.; Taylor, C.M.; Dowd, S.E.; Martin, D.H.; Ferris, M.J. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PLoS ONE 2011, 6, e26732. [Google Scholar] [CrossRef]
- Swanson, K.S.; Dowd, S.E.; Suchodolski, J.S.; Middelbos, I.S.; Vester, B.M.; Barry, K.A.; Nelson, K.E.; Torralba, M.; Henrissat, B.; Coutinho, P.M.; et al. Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. ISME J. 2011, 5, 639–649. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majumdar, R.; Kandel, S.L.; Cary, J.W.; Rajasekaran, K. Changes in Bacterial Endophyte Community Following Aspergillus flavus Infection in Resistant and Susceptible Maize Kernels. Int. J. Mol. Sci. 2021, 22, 3747. https://doi.org/10.3390/ijms22073747
Majumdar R, Kandel SL, Cary JW, Rajasekaran K. Changes in Bacterial Endophyte Community Following Aspergillus flavus Infection in Resistant and Susceptible Maize Kernels. International Journal of Molecular Sciences. 2021; 22(7):3747. https://doi.org/10.3390/ijms22073747
Chicago/Turabian StyleMajumdar, Rajtilak, Shyam L. Kandel, Jeffrey W. Cary, and Kanniah Rajasekaran. 2021. "Changes in Bacterial Endophyte Community Following Aspergillus flavus Infection in Resistant and Susceptible Maize Kernels" International Journal of Molecular Sciences 22, no. 7: 3747. https://doi.org/10.3390/ijms22073747
APA StyleMajumdar, R., Kandel, S. L., Cary, J. W., & Rajasekaran, K. (2021). Changes in Bacterial Endophyte Community Following Aspergillus flavus Infection in Resistant and Susceptible Maize Kernels. International Journal of Molecular Sciences, 22(7), 3747. https://doi.org/10.3390/ijms22073747