
Interplay between Histone and DNA Methylation Seen through Comparative Methylomes in Rare Mendelian Disorders


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
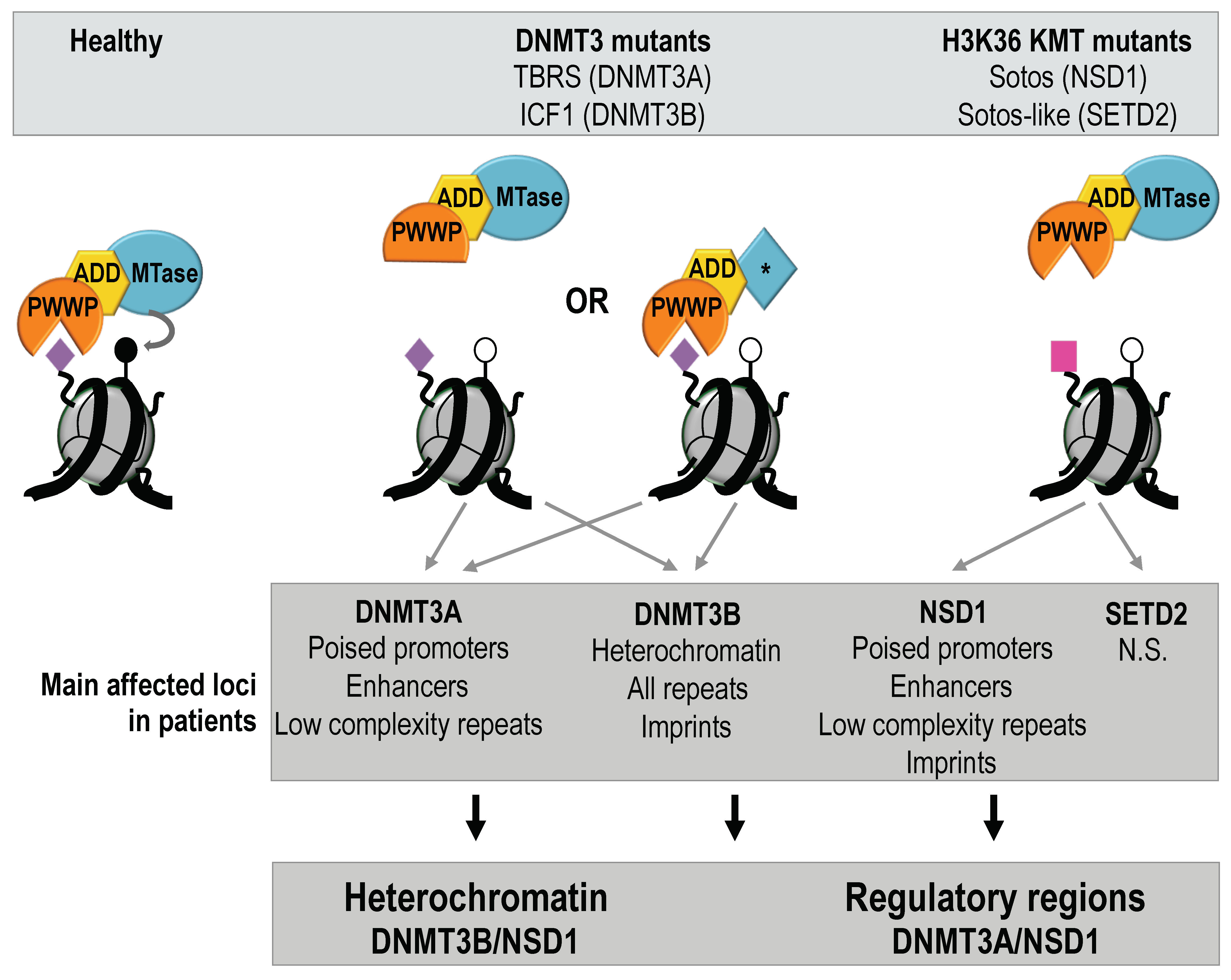
Abstract
1. Introduction
2. Results
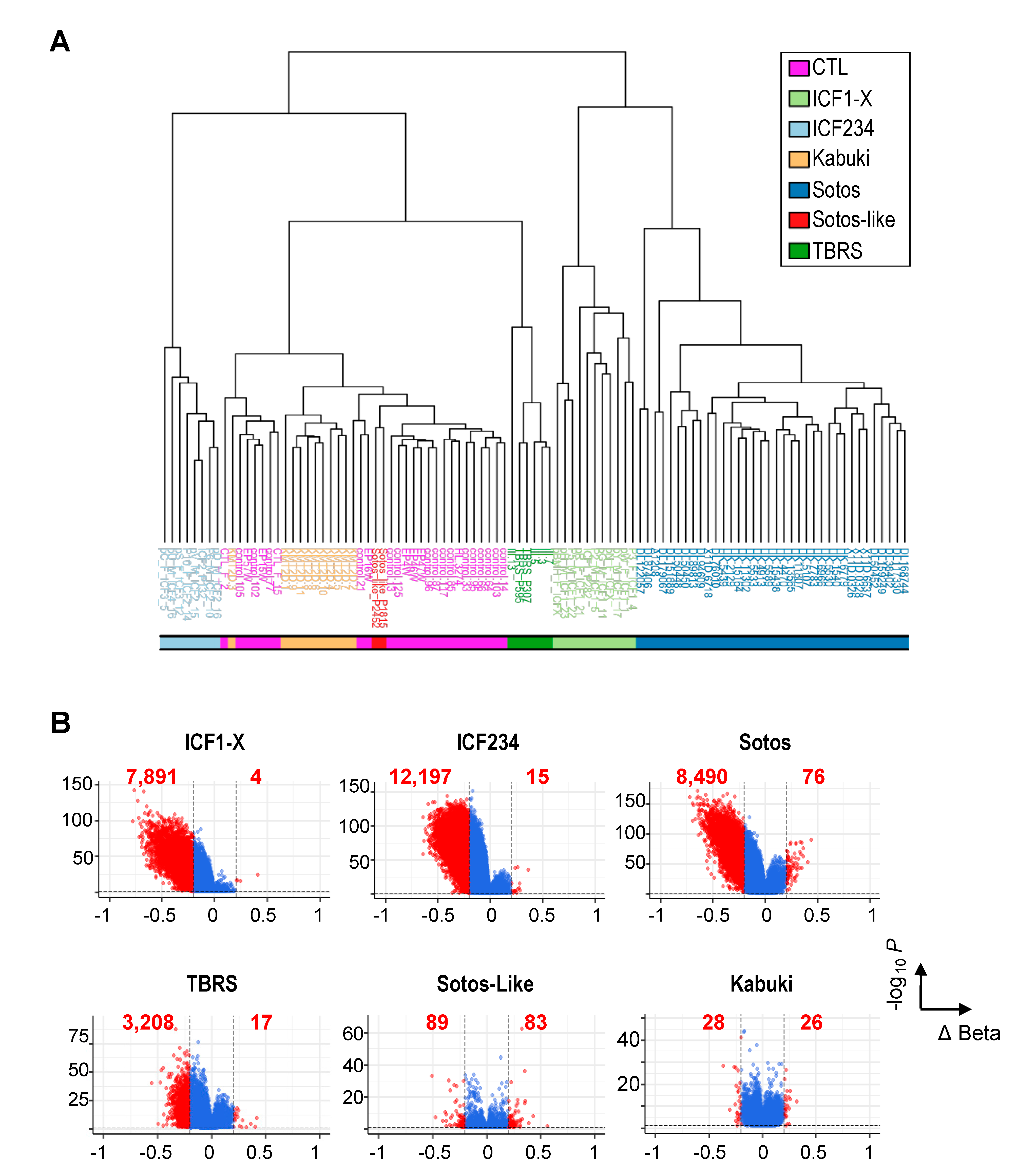
2.1. Non-Redundant Functions of De Novo DNMTs or H3K36 KMTs Are Reflected in Distinct DNAme Alterations Downstream of Their Mutations
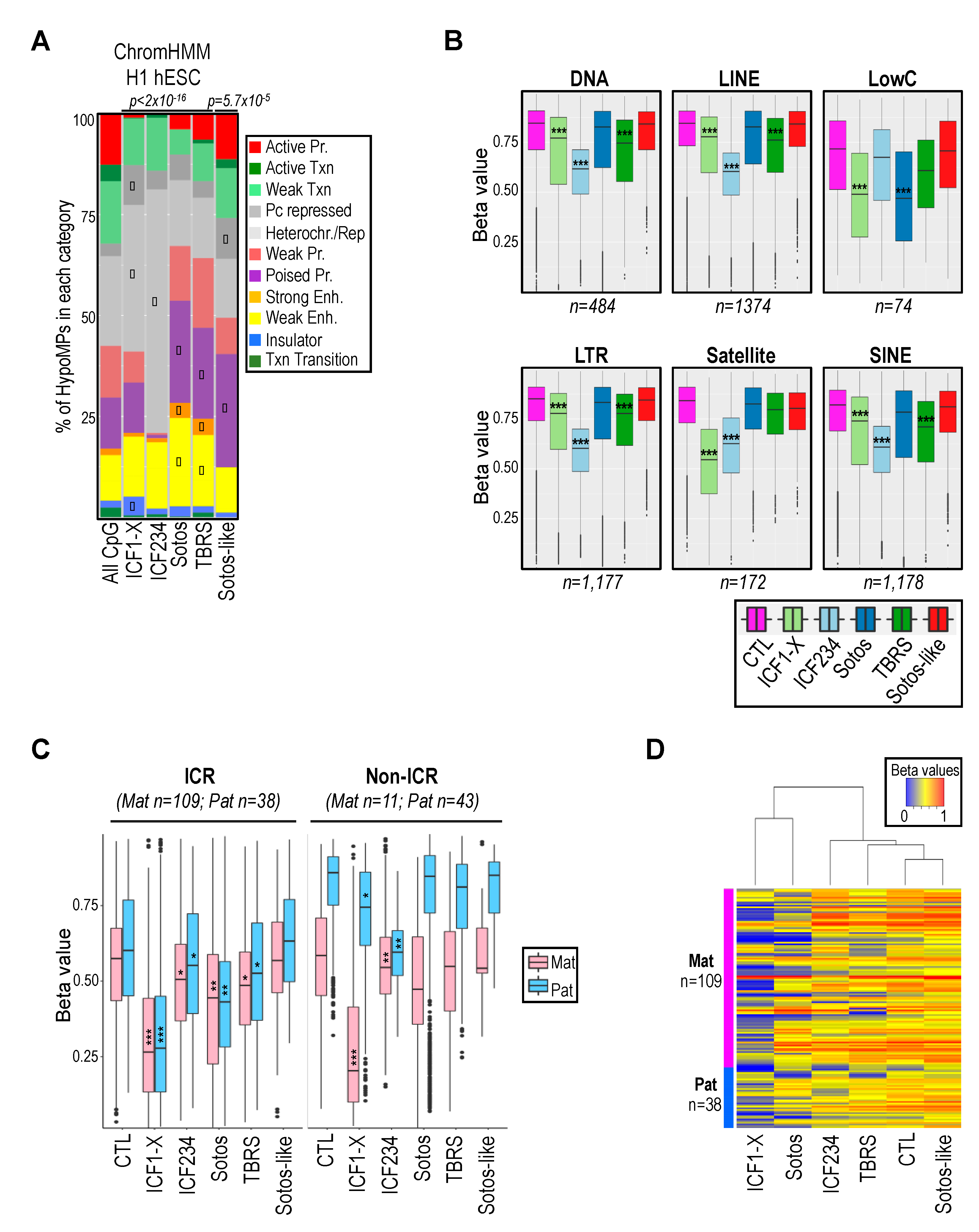
2.2. Hypomethylation of Satellite Repeats and Parental Imprints Are Hallmarks of ICF Patients but Not Patients with Overgrowth Syndromes
2.3. Hypomethylation of Developmentally Regulated Genes Is Shared between ICF, TBRS and Sotos Patients
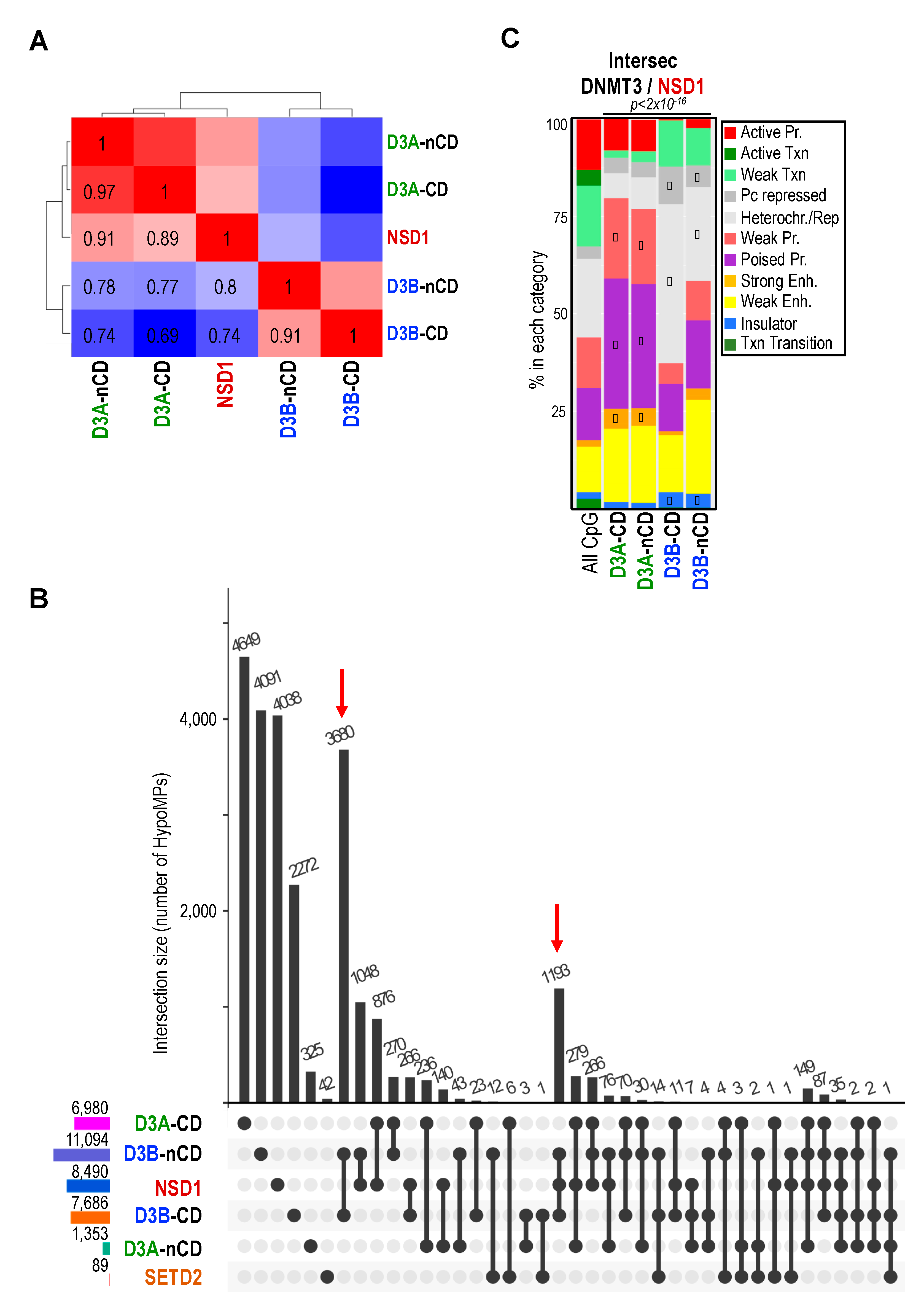
2.4. DNMT3 and NSD1 Mutations Have Overlapping Consequences for DNAme Alterations in Patients
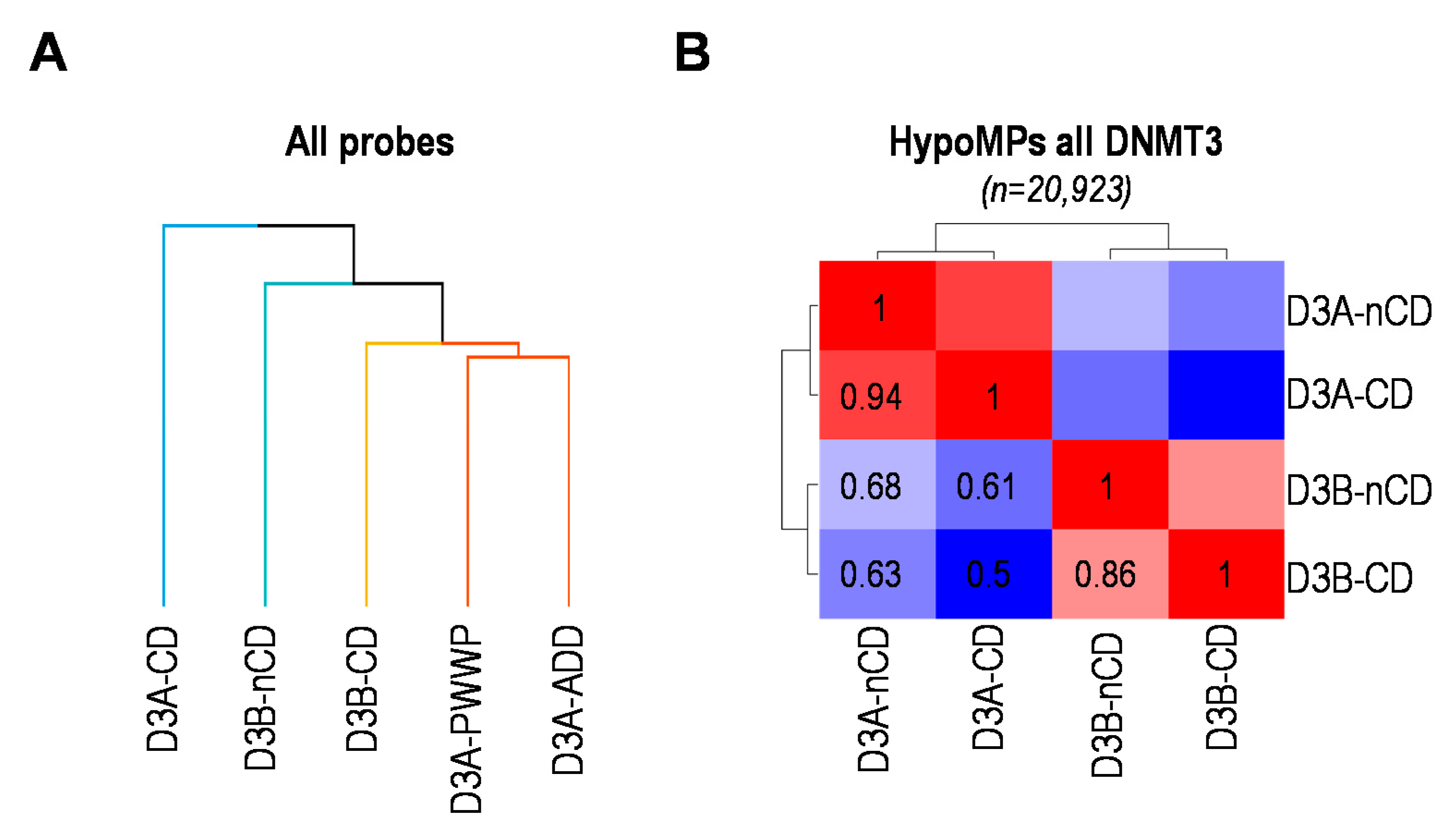
2.5. Catalytic or Non-Catalytic Mutations of DNMT3 Enzymes Lead to Overlapping DNAme Alterations
2.6. NSD1 and DNMT3B Mutations Lead to Overlapping DNAme Alterations at Heterochromatin Domains
3. Discussion
3.1. DNA Methylation Profiling for Diagnosis and Understanding of Clinical Manifestations
3.2. DNA Methylation Profiling in Patients to Predict Determinants of DNAme in Humans
3.3. Conclusions
4. Materials and Methods
4.1. Sample Preparation for Genome-Wide DNA Methylation Analysis and Quality Controls
4.2. Datasets of DNA Methylation Arrays Used
4.3. Processing of DNA Methylation Array Data
4.4. Visualization of DNA Methylation Data
4.5. Statistical Analysis
4.6. Annotation of DMPs and Databases Used
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Dahlet, T.; Argueso Lleida, A.; Al Adhami, H.; Dumas, M.; Bender, A.; Ngondo, R.P.; Tanguy, M.; Vallet, J.; Auclair, G.; Bardet, A.F.; et al. Genome-wide analysis in the mouse embryo reveals the importance of DNA methylation for transcription integrity. Nat. Commun. 2020, 11, 3153. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Defossez, P.A. Reading DNA Modifications. J. Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Marchal, C.; Miotto, B. Emerging concept in DNA methylation: Role of transcription factors in shaping DNA methylation patterns. J. Cell Physiol. 2015, 230, 743–751. [Google Scholar] [CrossRef]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schubeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Muller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef]
- Jeltsch, A.; Jurkowska, R.Z. Allosteric control of mammalian DNA methyltransferases—A new regulatory paradigm. Nucleic Acids Res. 2016, 44, 8556–8575. [Google Scholar] [CrossRef]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Parry, A.; Rulands, S.; Reik, W. Active turnover of DNA methylation during cell fate decisions. Nat. Rev. Genet. 2020. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef] [PubMed]
- Suelves, M.; Carrio, E.; Nunez-Alvarez, Y.; Peinado, M.A. DNA methylation dynamics in cellular commitment and differentiation. Brief. Funct. Genom. 2016, 15, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Cariaga-Martinez, A.E.; Gutierrez, K.J.; Alelu-Paz, R. The Vast Complexity of the Epigenetic Landscape during Neurodevelopment: An Open Frame to Understanding Brain Function. Int. J. Mol. Sci. 2018, 19, 1333. [Google Scholar] [CrossRef]
- Celik, H.; Kramer, A.; Challen, G.A. DNA methylation in normal and malignant hematopoiesis. Int. J. Hematol. 2016, 103, 617–626. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Genetic syndromes caused by mutations in epigenetic genes. Hum. Genet. 2013, 132, 359–383. [Google Scholar] [CrossRef]
- Velasco, G.; Francastel, C. Genetics meets DNA methylation in rare diseases. Clin. Genet. 2019, 95, 210–220. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian disorders of the epigenetic machinery: Tipping the balance of chromatin states. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Groupe, D.I.F.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Douglas, J.; Hanks, S.; Temple, I.K.; Davies, S.; Murray, A.; Upadhyaya, M.; Tomkins, S.; Hughes, H.E.; Cole, T.R.; Rahman, N. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am. J. Hum. Genet. 2003, 72, 132–143. [Google Scholar] [CrossRef]
- Tlemsani, C.; Luscan, A.; Leulliot, N.; Bieth, E.; Afenjar, A.; Baujat, G.; Doco-Fenzy, M.; Goldenberg, A.; Lacombe, D.; Lambert, L.; et al. SETD2 and DNMT3A screen in the Sotos-like syndrome French cohort. J. Med. Genet. 2016, 53, 743–751. [Google Scholar] [CrossRef]
- Luscan, A.; Laurendeau, I.; Malan, V.; Francannet, C.; Odent, S.; Giuliano, F.; Lacombe, D.; Touraine, R.; Vidaud, M.; Pasmant, E.; et al. Mutations in SETD2 cause a novel overgrowth condition. J. Med. Genet. 2014, 51, 512–517. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef]
- Velasco, G.; Grillo, G.; Touleimat, N.; Ferry, L.; Ivkovic, I.; Ribierre, F.; Deleuze, J.F.; Chantalat, S.; Picard, C.; Francastel, C. Comparative methylome analysis of ICF patients identifies heterochromatin loci that require ZBTB24, CDCA7 and HELLS for their methylated state. Hum. Mol. Genet. 2018, 27, 2409–2424. [Google Scholar] [CrossRef]
- Jeffries, A.R.; Maroofian, R.; Salter, C.G.; Chioza, B.A.; Cross, H.E.; Patton, M.A.; Dempster, E.; Temple, I.K.; Mackay, D.J.G.; Rezwan, F.I.; et al. Growth disrupting mutations in epigenetic regulatory molecules are associated with abnormalities of epigenetic aging. Genome Res. 2019, 29, 1057–1066. [Google Scholar] [CrossRef]
- Unoki, M.; Funabiki, H.; Velasco, G.; Francastel, C.; Sasaki, H. CDCA7 and HELLS mutations undermine nonhomologous end joining in centromeric instability syndrome. J. Clin. Investig. 2019, 129, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Rigolet, M.; Bourc’his, D.; Nigon, F.; Bokesoy, I.; Fryns, J.P.; Hulten, M.; Jonveaux, P.; Maraschio, P.; Megarbane, A.; et al. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum. Mutat. 2005, 25, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Walton, E.L.; Sterlin, D.; Hedouin, S.; Nitta, H.; Ito, Y.; Fouyssac, F.; Megarbane, A.; Sasaki, H.; Picard, C.; et al. Germline genes hypomethylation and expression define a molecular signature in peripheral blood of ICF patients: Implications for diagnosis and etiology. Orphanet J. Rare Dis. 2014, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Chan, M.M.; Humm, K.C.; Karnik, R.; Mekhoubad, S.; Regev, A.; Eggan, K.; Meissner, A. DNA methylation dynamics of the human preimplantation embryo. Nature 2014, 511, 611–615. [Google Scholar] [CrossRef]
- Francastel, C.; Magdinier, F. DNA methylation in satellite repeats disorders. Essays Biochem. 2019, 63, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Bobek, M.P.; Kuick, R.; Lamb, B.; Zhu, X.; Narayan, A.; Bourc’his, D.; Viegas-Pequignot, E.; Ehrlich, M.; Hanash, S.M. Whole-genome methylation scan in ICF syndrome: Hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum. Mol. Genet. 2000, 9, 597–604. [Google Scholar] [CrossRef]
- Miniou, P.; Bourc’his, D.; Molina Gomes, D.; Jeanpierre, M.; Viegas-Pequignot, E. Undermethylation of Alu sequences in ICF syndrome: Molecular and in situ analysis. Cytogenet. Cell Genet. 1997, 77, 308–313. [Google Scholar] [CrossRef]
- Bjornsson, H.T. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef]
- Court, F.; Tayama, C.; Romanelli, V.; Martin-Trujillo, A.; Iglesias-Platas, I.; Okamura, K.; Sugahara, N.; Simon, C.; Moore, H.; Harness, J.V.; et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014, 24, 554–569. [Google Scholar] [CrossRef]
- Joshi, R.S.; Garg, P.; Zaitlen, N.; Lappalainen, T.; Watson, C.T.; Azam, N.; Ho, D.; Li, X.; Antonarakis, S.E.; Brunner, H.G.; et al. DNA Methylation Profiling of Uniparental Disomy Subjects Provides a Map of Parental Epigenetic Bias in the Human Genome. Am. J. Hum. Genet. 2016, 99, 555–566. [Google Scholar] [CrossRef]
- Plasschaert, R.N.; Bartolomei, M.S. Genomic imprinting in development, growth, behavior and stem cells. Development 2014, 141, 1805–1813. [Google Scholar] [CrossRef]
- Gao, L.; Emperle, M.; Guo, Y.; Grimm, S.A.; Ren, W.; Adam, S.; Uryu, H.; Zhang, Z.M.; Chen, D.; Yin, J.; et al. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat. Commun. 2020, 11, 3355. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.N.; Papillon-Cavanagh, S.; Chen, H.; Yue, Y.; Chen, X.; Rajagopalan, K.N.; Horth, C.; McGuire, J.T.; Xu, X.; Nikbakht, H.; et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019, 573, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, J.; Rong, B.; Zhao, B.; Wang, M.; Dai, R.; Chen, Q.; Liu, H.; Gu, Z.; Liu, S.; et al. DNMT3A reads and connects histone H3K36me2 to DNA methylation. Protein Cell 2020, 11, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Haghshenas, S.; Bhai, P.; Aref-Eshghi, E.; Sadikovic, B. Diagnostic Utility of Genome-Wide DNA Methylation Analysis in Mendelian Neurodevelopmental Disorders. Int. J. Mol. Sci. 2020, 21, 9303. [Google Scholar] [CrossRef]
- Aygun, D.; Bjornsson, H.T. Clinical epigenetics: A primer for the practitioner. Dev. Med. Child. Neurol. 2020, 62, 192–200. [Google Scholar] [CrossRef]
- Velasco, G.; Hube, F.; Rollin, J.; Neuillet, D.; Philippe, C.; Bouzinba-Segard, H.; Galvani, A.; Viegas-Pequignot, E.; Francastel, C. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc. Natl. Acad. Sci. USA 2010, 107, 9281–9286. [Google Scholar] [CrossRef]
- Ehrlich, M. The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin. Immunol. 2003, 109, 17–28. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Weemaes, C.M.; van Tol, M.J.; Wang, J.; van Ostaijen-ten Dam, M.M.; van Eggermond, M.C.; Thijssen, P.E.; Aytekin, C.; Brunetti-Pierri, N.; van der Burg, M.; Graham Davies, E.; et al. Heterogeneous clinical presentation in ICF syndrome: Correlation with underlying gene defects. Eur. J. Hum. Genet. 2013, 21, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Rajshekar, S.; Yao, J.; Arnold, P.K.; Payne, S.G.; Zhang, Y.; Bowman, T.V.; Schmitz, R.J.; Edwards, J.R.; Goll, M. Pericentromeric hypomethylation elicits an interferon response in an animal model of ICF syndrome. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Sterlin, D.; Velasco, G.; Moshous, D.; Touzot, F.; Mahlaoui, N.; Fischer, A.; Suarez, F.; Francastel, C.; Picard, C. Genetic, Cellular and Clinical Features of ICF Syndrome: A French National Survey. J. Clin. Immunol. 2016, 36, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Helfricht, A.; Thijssen, P.E.; Rother, M.B.; Shah, R.G.; Du, L.; Takada, S.; Rogier, M.; Moritz, J.; IJspeert, H.; Stoepker, C.; et al. Loss of ZBTB24 impairs nonhomologous end-joining and class-switch recombination in patients with ICF syndrome. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Choufani, S.; Cytrynbaum, C.; Chung, B.H.; Turinsky, A.L.; Grafodatskaya, D.; Chen, Y.A.; Cohen, A.S.; Dupuis, L.; Butcher, D.T.; Siu, M.T.; et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 2015, 6, 10207. [Google Scholar] [CrossRef]
- Sendzikaite, G.; Hanna, C.W.; Stewart-Morgan, K.R.; Ivanova, E.; Kelsey, G. A DNMT3A PWWP mutation leads to methylation of bivalent chromatin and growth retardation in mice. Nat. Commun. 2019, 10, 1884. [Google Scholar] [CrossRef]
- Heyn, P.; Logan, C.V.; Fluteau, A.; Challis, R.C.; Auchynnikava, T.; Martin, C.A.; Marsh, J.A.; Taglini, F.; Kilanowski, F.; Parry, D.A.; et al. Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat. Genet. 2019, 51, 96–105. [Google Scholar] [CrossRef]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef]
- Smith, F.M.; Garfield, A.S.; Ward, A. Regulation of growth and metabolism by imprinted genes. Cytogenet. Genome Res. 2006, 113, 279–291. [Google Scholar] [CrossRef]
- Wilkinson, L.S.; Davies, W.; Isles, A.R. Genomic imprinting effects on brain development and function. Nat. Rev. Neurosci. 2007, 8, 832–843. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef]
- Watanabe, H.; Higashimoto, K.; Miyake, N.; Morita, S.; Horii, T.; Kimura, M.; Suzuki, T.; Maeda, T.; Hidaka, H.; Aoki, S.; et al. DNA methylation analysis of multiple imprinted DMRs in Sotos syndrome reveals IGF2-DMR0 as a DNA methylation-dependent, P0 promoter-specific enhancer. FASEB J. 2020, 34, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Shirane, K.; Miura, F.; Ito, T.; Lorincz, M.C. NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat. Genet. 2020, 52, 1088–1098. [Google Scholar] [CrossRef]
- Baujat, G.; Rio, M.; Rossignol, S.; Sanlaville, D.; Lyonnet, S.; Le Merrer, M.; Munnich, A.; Gicquel, C.; Cormier-Daire, V.; Colleaux, L. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am. J. Hum. Genet. 2004, 74, 715–720. [Google Scholar] [CrossRef]
- Hutter, B.; Helms, V.; Paulsen, M. Tandem repeats in the CpG islands of imprinted genes. Genomics 2006, 88, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Chiaradia, I.; Lancaster, M.A. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat. Neurosci. 2020, 23, 1496–1508. [Google Scholar] [CrossRef]
- de Greef, J.C.; Wang, J.; Balog, J.; den Dunnen, J.T.; Frants, R.R.; Straasheijm, K.R.; Aytekin, C.; van der Burg, M.; Duprez, L.; Ferster, A.; et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011, 88, 796–804. [Google Scholar] [CrossRef]
- Thijssen, P.E.; Ito, Y.; Grillo, G.; Wang, J.; Velasco, G.; Nitta, H.; Unoki, M.; Yoshihara, M.; Suyama, M.; Sun, Y.; et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 2015, 6, 7870. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Li, J.; Cao, Y.; Huang, Y.; Li, W.; Zhu, H.; Zhao, Q.; Han, J.J.; Wu, Q.; Li, J.; et al. A role for LSH in facilitating DNA methylation by DNMT1 through enhancing UHRF1 chromatin association. Nucleic Acids Res. 2020, 48, 12116–12134. [Google Scholar] [CrossRef]
- Yu, W.; McIntosh, C.; Lister, R.; Zhu, I.; Han, Y.; Ren, J.; Landsman, D.; Lee, E.; Briones, V.; Terashima, M.; et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014, 24, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Hardikar, S.; Ying, Z.; Zeng, Y.; Zhao, H.; Liu, B.; Veland, N.; McBride, K.; Cheng, X.; Chen, T. The ZBTB24-CDCA7 axis regulates HELLS enrichment at centromeric satellite repeats to facilitate DNA methylation. Protein Cell 2020, 11, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Jenness, C.; Giunta, S.; Muller, M.M.; Kimura, H.; Muir, T.W.; Funabiki, H. HELLS and CDCA7 comprise a bipartite nucleosome remodeling complex defective in ICF syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E876–E885. [Google Scholar] [CrossRef] [PubMed]
- Shirohzu, H.; Kubota, T.; Kumazawa, A.; Sado, T.; Chijiwa, T.; Inagaki, K.; Suetake, I.; Tajima, S.; Wakui, K.; Miki, Y.; et al. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 2002, 112, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef] [PubMed]
- Morselli, M.; Pastor, W.A.; Montanini, B.; Nee, K.; Ferrari, R.; Fu, K.; Bonora, G.; Rubbi, L.; Clark, A.T.; Ottonello, S.; et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. Elife 2015, 4, e06205. [Google Scholar] [CrossRef]
- Rondelet, G.; Dal Maso, T.; Willems, L.; Wouters, J. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J. Struct. Biol. 2016, 194, 357–367. [Google Scholar] [CrossRef]
- Guo, X.; Wang, L.; Li, J.; Ding, Z.; Xiao, J.; Yin, X.; He, S.; Shi, P.; Dong, L.; Li, G.; et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 2015, 517, 640–644. [Google Scholar] [CrossRef]
- Li, B.Z.; Huang, Z.; Cui, Q.Y.; Song, X.H.; Du, L.; Jeltsch, A.; Chen, P.; Li, G.; Li, E.; Xu, G.L. Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res. 2011, 21, 1172–1181. [Google Scholar] [CrossRef]
- Otani, J.; Nankumo, T.; Arita, K.; Inamoto, S.; Ariyoshi, M.; Shirakawa, M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009, 10, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Auclair, G.; Guibert, S.; Bender, A.; Weber, M. Ontogeny of CpG island methylation and specificity of DNMT3 methyltransferases during embryonic development in the mouse. Genome Biol. 2014, 15, 545. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Kabata, M.; Tanaka, A.; Ukai, T.; Ohta, S.; Nakabayashi, K.; Shimizu, M.; Hata, K.; Meissner, A.; Yamamoto, T.; et al. Identification of distinct loci for de novo DNA methylation by DNMT3A and DNMT3B during mammalian development. Nat. Commun. 2020, 11, 3199. [Google Scholar] [CrossRef]
- Lees-Murdock, D.J.; Shovlin, T.C.; Gardiner, T.; De Felici, M.; Walsh, C.P. DNA methyltransferase expression in the mouse germ line during periods of de novo methylation. Dev. Dyn. 2005, 232, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Sessa, A.; Fagnocchi, L.; Mastrototaro, G.; Massimino, L.; Zaghi, M.; Indrigo, M.; Cattaneo, S.; Martini, D.; Gabellini, C.; Pucci, C.; et al. SETD5 Regulates Chromatin Methylation State and Preserves Global Transcriptional Fidelity during Brain Development and Neuronal Wiring. Neuron 2019, 104, 271–289.e213. [Google Scholar] [CrossRef]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Parker, M.J.; Archer, H.; Firth, H.V.; Park, S.M.; Canham, N.; Holder, S.E.; Wilson, M.; et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 2014, 94, 618–624. [Google Scholar] [CrossRef]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef]
- Rajagopalan, K.N.; Chen, X.; Weinberg, D.N.; Chen, H.; Majewski, J.; Allis, C.D.; Lu, C. Depletion of H3K36me2 recapitulates epigenomic and phenotypic changes induced by the H3.3K36M oncohistone mutation. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Ropero, S.; Setien, F.; Fraga, M.F.; Lapunzina, P.; Losson, R.; Alaminos, M.; Cheung, N.K.; Rahman, N.; Esteller, M. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl. Acad. Sci. USA 2009, 106, 21830–21835. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Min, J. Structure and function of the nucleosome-binding PWWP domain. Trends Biochem. Sci. 2014, 39, 536–547. [Google Scholar] [CrossRef]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.; Backlin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.L.; Flaegstad, T.; Forestier, E.; Frost, B.M.; Harila-Saari, A.; et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velasco, G.; Ulveling, D.; Rondeau, S.; Marzin, P.; Unoki, M.; Cormier-Daire, V.; Francastel, C. Interplay between Histone and DNA Methylation Seen through Comparative Methylomes in Rare Mendelian Disorders. Int. J. Mol. Sci. 2021, 22, 3735. https://doi.org/10.3390/ijms22073735
Velasco G, Ulveling D, Rondeau S, Marzin P, Unoki M, Cormier-Daire V, Francastel C. Interplay between Histone and DNA Methylation Seen through Comparative Methylomes in Rare Mendelian Disorders. International Journal of Molecular Sciences. 2021; 22(7):3735. https://doi.org/10.3390/ijms22073735
Chicago/Turabian StyleVelasco, Guillaume, Damien Ulveling, Sophie Rondeau, Pauline Marzin, Motoko Unoki, Valérie Cormier-Daire, and Claire Francastel. 2021. "Interplay between Histone and DNA Methylation Seen through Comparative Methylomes in Rare Mendelian Disorders" International Journal of Molecular Sciences 22, no. 7: 3735. https://doi.org/10.3390/ijms22073735
APA StyleVelasco, G., Ulveling, D., Rondeau, S., Marzin, P., Unoki, M., Cormier-Daire, V., & Francastel, C. (2021). Interplay between Histone and DNA Methylation Seen through Comparative Methylomes in Rare Mendelian Disorders. International Journal of Molecular Sciences, 22(7), 3735. https://doi.org/10.3390/ijms22073735