
Synthesis, Biological Activity, and Molecular Dynamics Study of Novel Series of a Trimethoprim Analogs as Multi-Targeted Compounds: Dihydrofolate Reductase (DHFR) Inhibitors and DNA-Binding Agents


Abstract
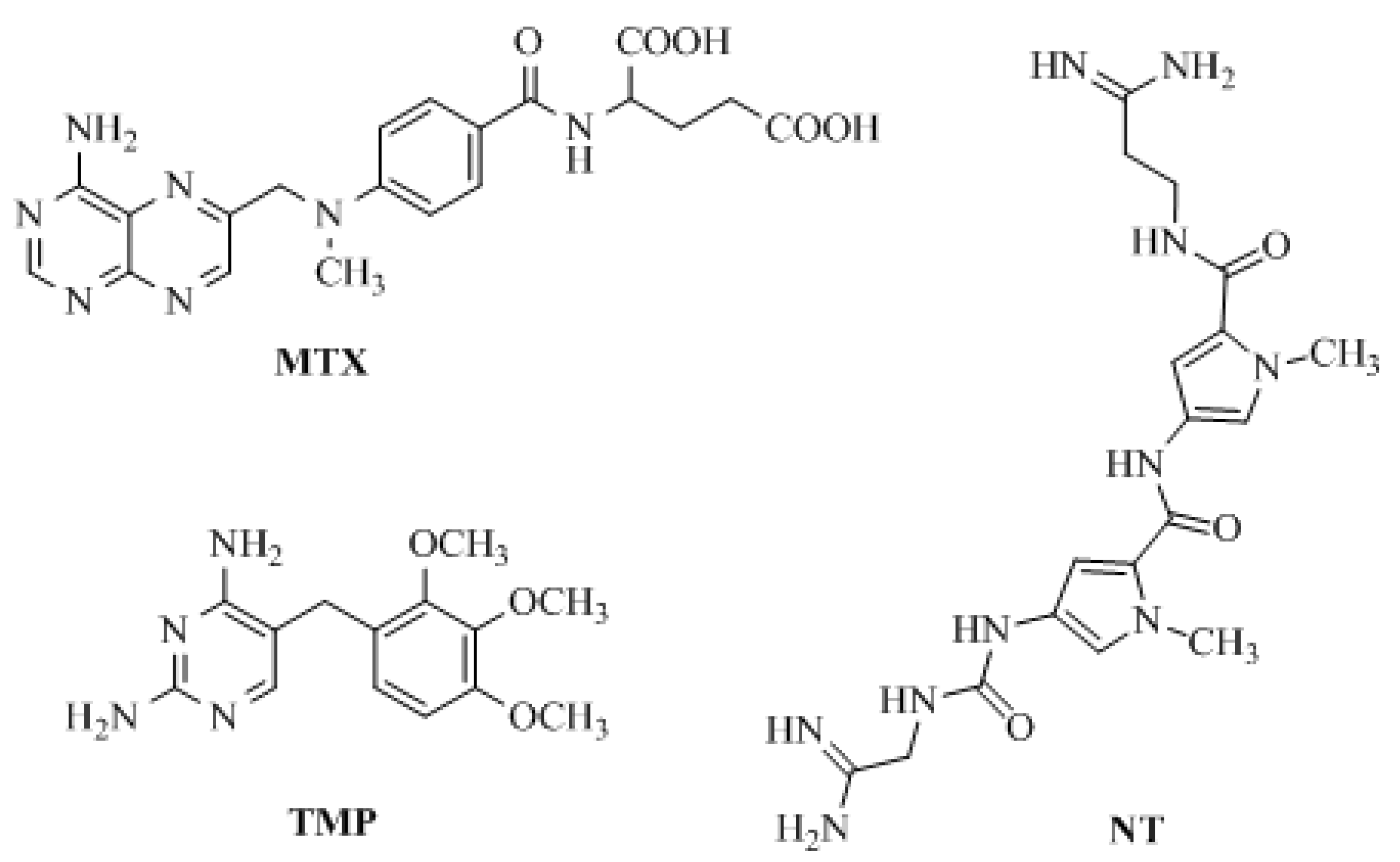
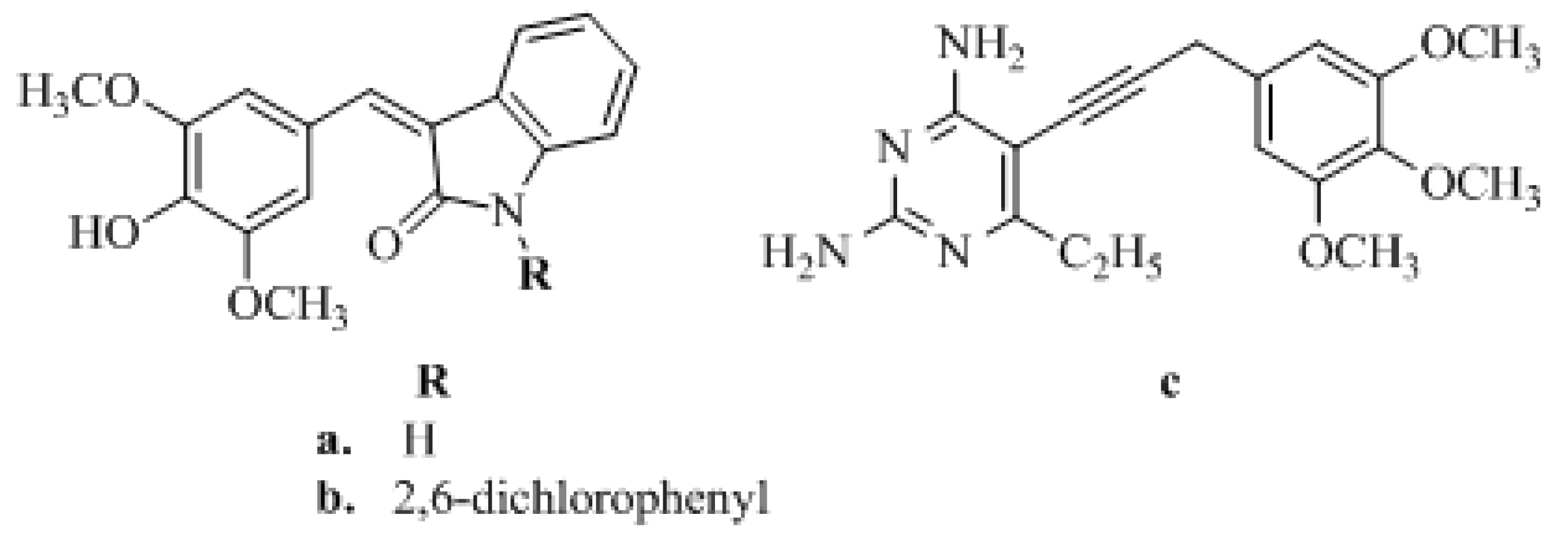
1. Introduction
2. Results and Discussion
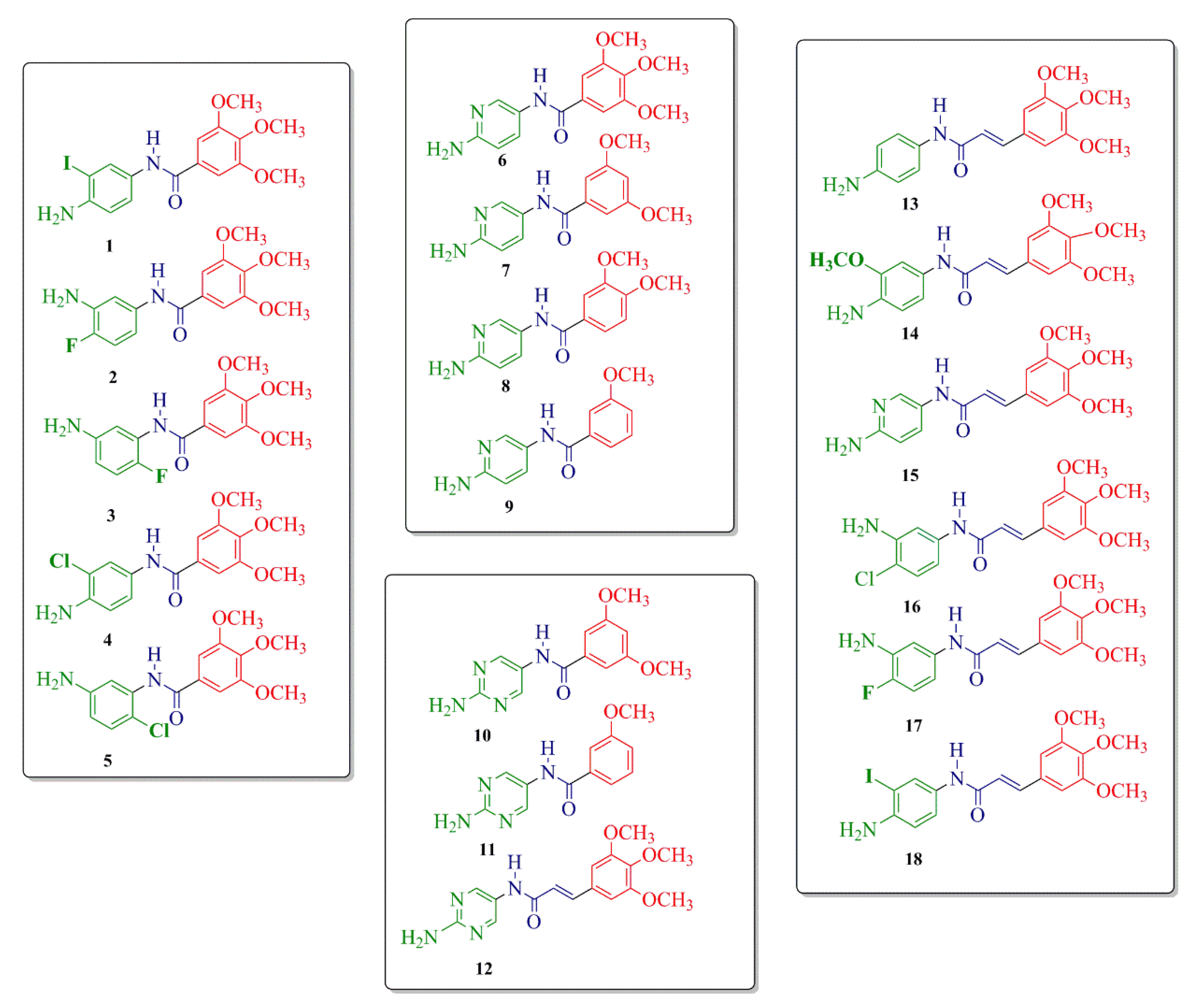
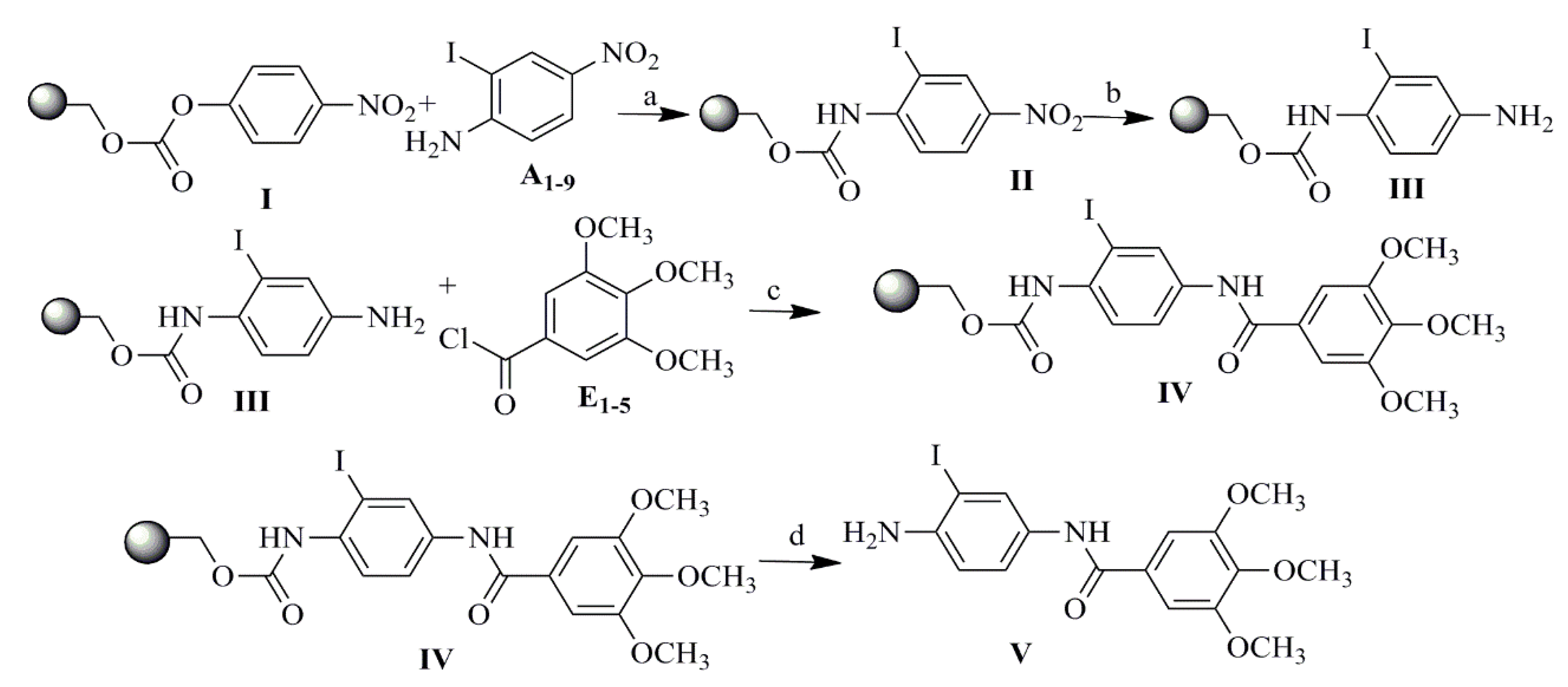
2.1. Compound Design and Synthesis
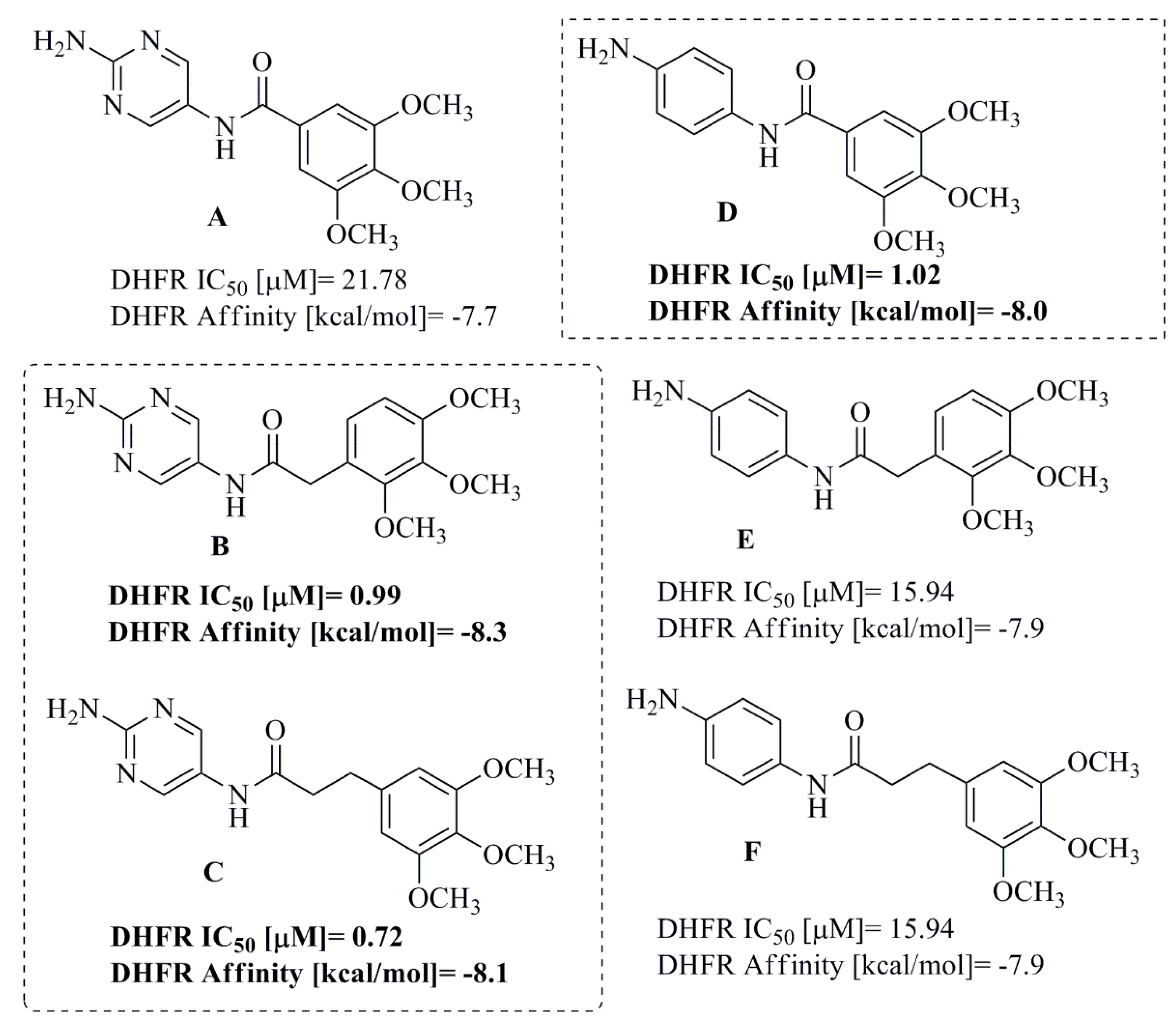
2.2. Biological Activity: DNA-Binding Effects and Dihydrofolate Reductase (DHFR) Inhibition
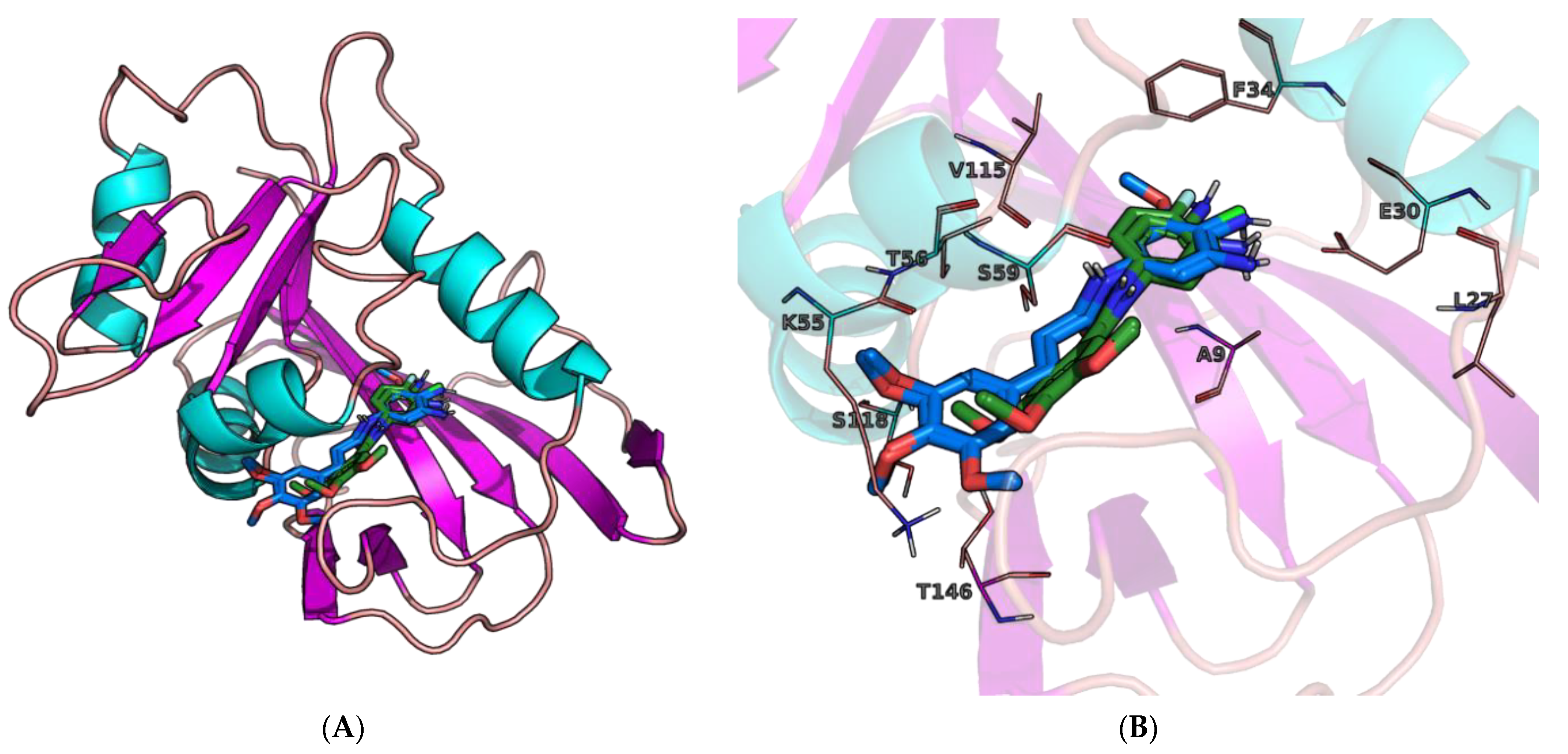
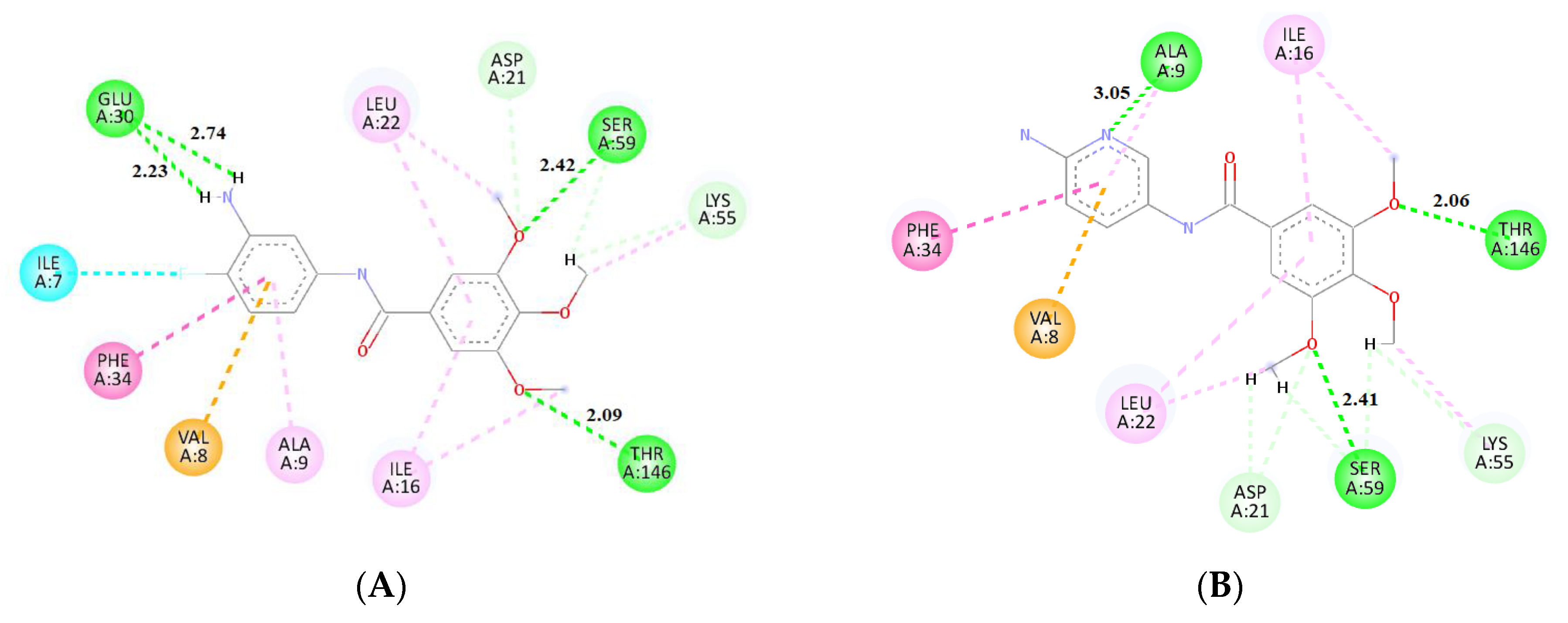
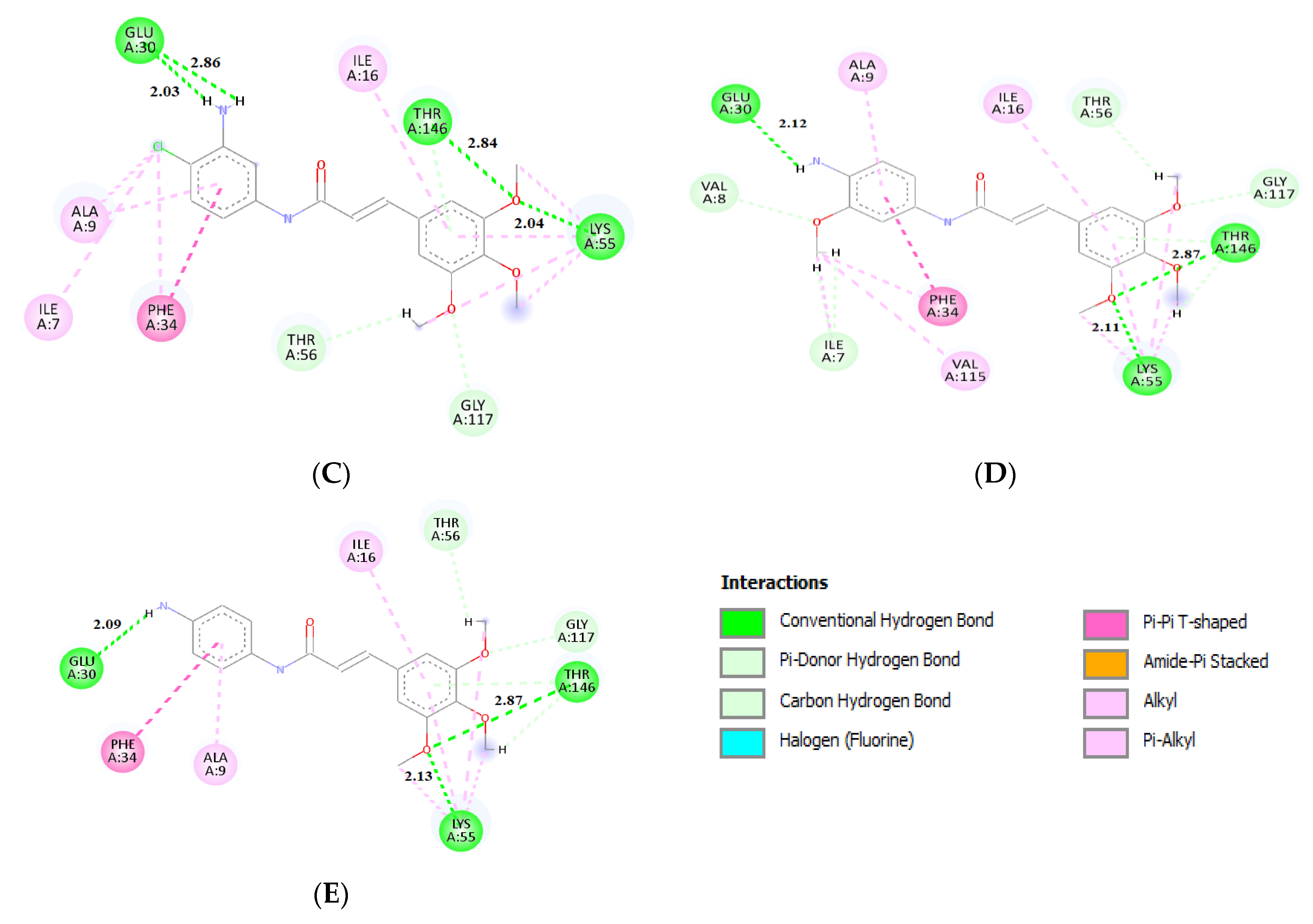
2.3. Molecular Docking
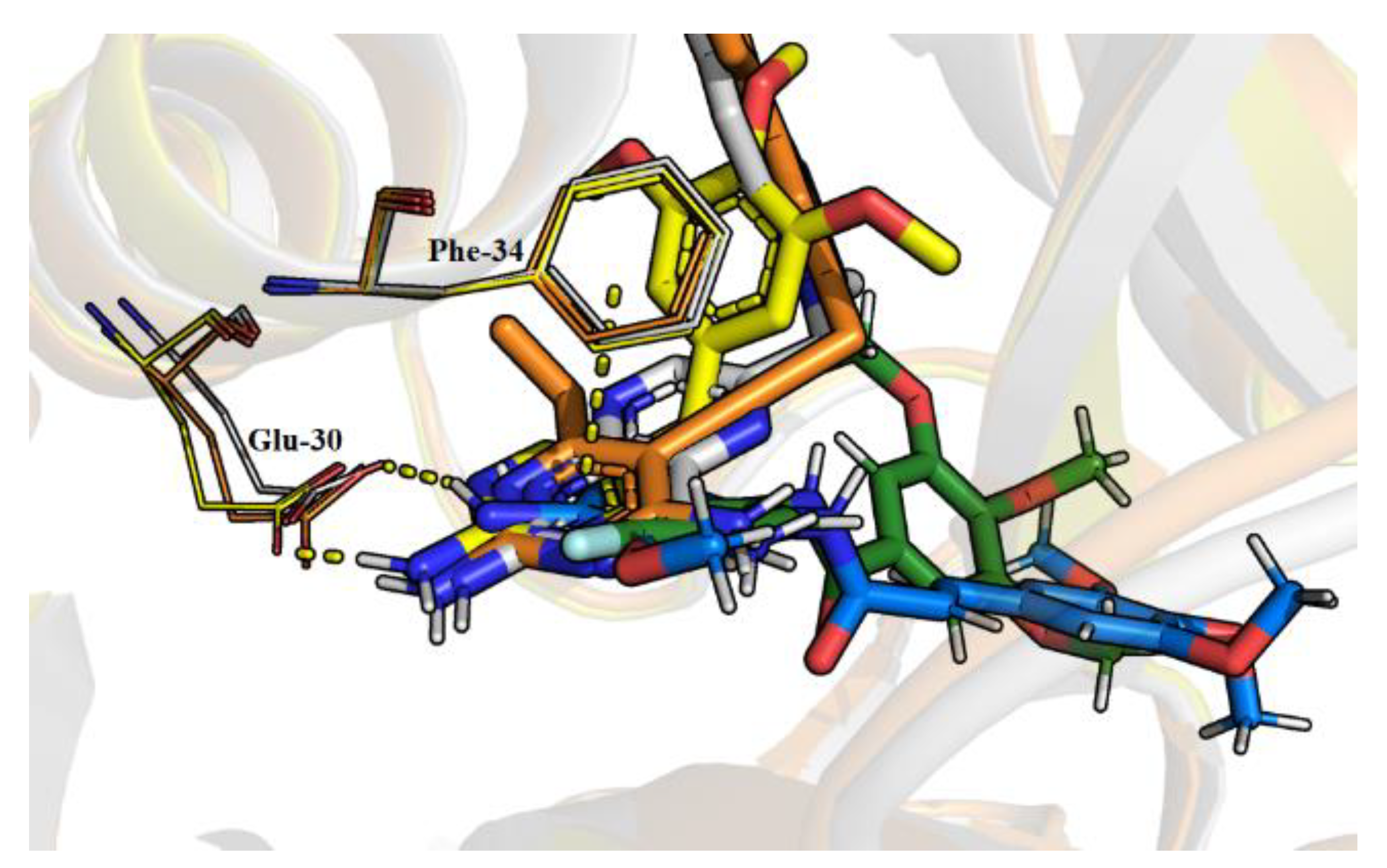
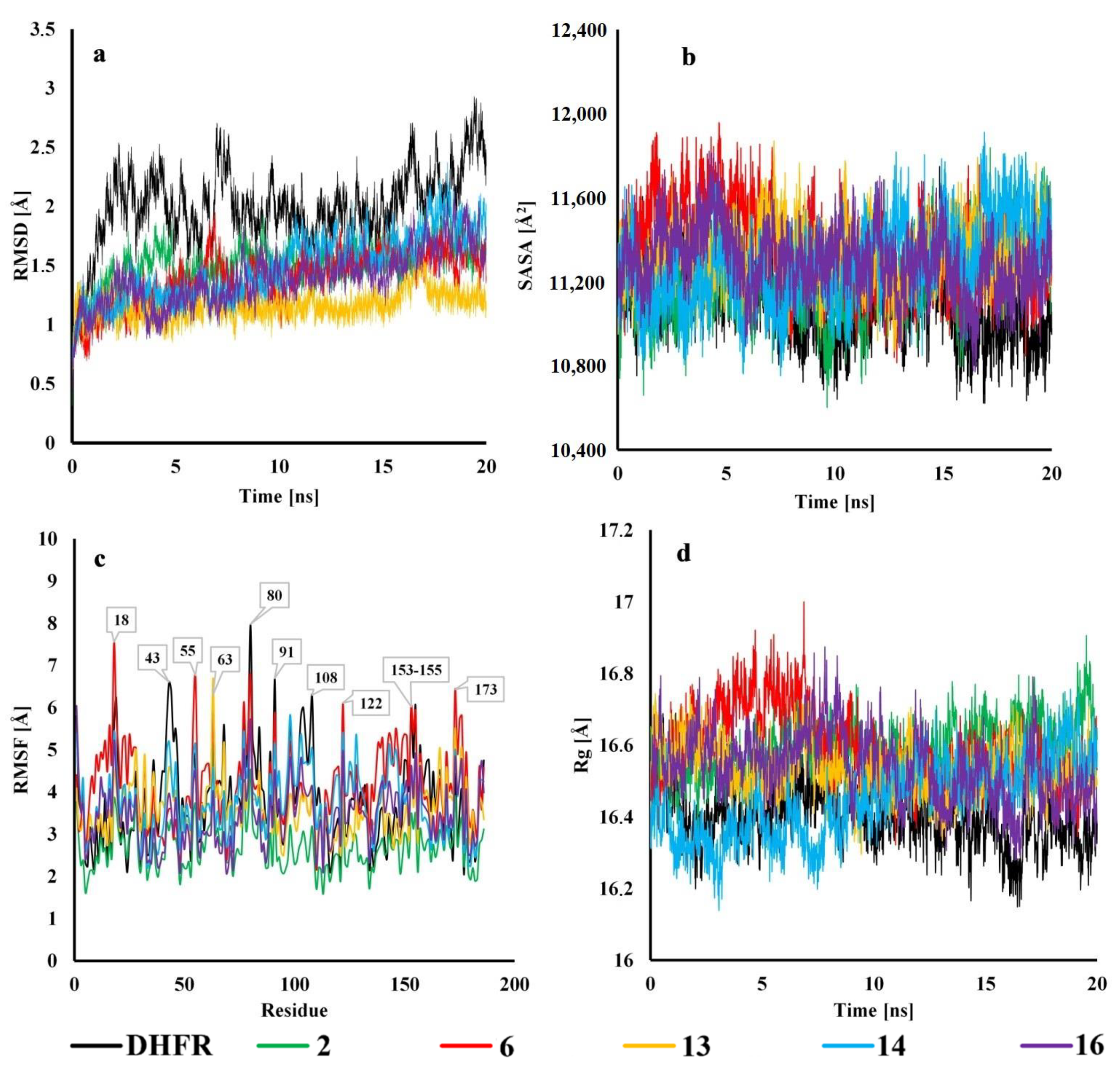
2.4. Molecular Dynamics
3. Materials and Methods
3.1. General Information
3.2. Synthesis
Procedure for the Synthesis of Compounds 1–18
3.3. Ethidium Bromide Assay—DNA-Binding Effects
3.4. Ethidium Displacement Bromide Assay—Determination of DNA-Binding Constants
3.5. Dihydrofolate Reductase (DHFR) Inhibition Assay
3.6. Molecular Docking
3.7. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, D.C.M.; Anderson, A.C. Towards species-specific antifolates. Curr. Med. Chem. 2006, 13, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Champe, P.C.; Harvey, R.A. Lippincott’s Illustrated Reviews, 2nd ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 1994. [Google Scholar]
- Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 2006, 71, 941–948. [Google Scholar] [CrossRef]
- Hussein, E.M.; Al-Rooqi, M.M.; El-Galil, S.M.A.; Ahmed, S.A. Design, synthesis, and biological evaluation of novel N4-substituted sulfonamides: Acetamides derivatives as dihydrofolate reductase (DHFR) inhibitors. BMC Chem. 2019, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Blakley, R.L. Eukaryotic dihydrofolate reductase. Adv. Enzymol. Relat. Areas Mol. Biol. 1995, 70, 23–102. [Google Scholar] [PubMed]
- Heaslet, H.; Harris, M.; Fahnoe, K.; Sarver, R.; Putz, H.; Chang, J.; Subramanyam, C.; Barreiro, G.; Miller, J.R. Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins 2009, 76, 706–717. [Google Scholar] [CrossRef]
- Hong, W.; Wang, Y.; Chang, Z.; Yang, Y.; Pu, J.; Sun, T.; Kaur, S.; Sacchettini, J.C.; Jung, H.; Wong, W.L.; et al. The identification of novel Mycobacterium tuberculosis DHFR inhibitors and the investigation of their binding preferences by using molecular modelling. Sci. Rep. 2015, 5, 15328. [Google Scholar] [CrossRef]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR inhibitors: Reading the past for discovering novel anticancer agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef]
- Reddish, M.J.; Vaughn, M.B.; Fu, R.; Dyer, R.B. Ligand-dependent conformational dynamics of dihydrofolate reductase. Biochemistry 2016, 55, 1485–1493. [Google Scholar] [CrossRef]
- Jackson, R.C.; Niethammer, D. Acquired methotrexate resistance in lymphoblasts resulting from altered kinetic properties of dihydrofolate reductase. Eur. J. Cancer 2017, 13, 567–575. [Google Scholar] [CrossRef]
- Singh, A.; Deshpande, N.; Pramanik, N.; Jhunjhunwala, S.; Rangarajan, A.; Atreya, H.S. Optimized peptide based inhibitors targeting the dihydrofolate reductase pathway in cancer. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Bavetsias, V.; Marriott, J.H.; Melin, C.; Kimbell, R.; Matusiak, Z.S.; Boyle, F.T.; Jackman, A.L. Design and synthesis of cyclopenta[g]quinazoline-based antifolates as inhibitors of thymidylate synthase and potential antitumor agents. J. Med. Chem. 2000, 43, 1910–1926. [Google Scholar] [CrossRef]
- Werbel, L.M.; Degnan, M.J. Antimalarial drugs. 63. Synthesis and antimalarial and antitumor effects of 2-amino-4-(hydrazino and hydroxyamino)-6-[(aryl)thio]quinazolines. J. Med. Chem. 1987, 30, 2151–2154. [Google Scholar] [CrossRef]
- Cao, S.L.; Feng, Y.P.; Jiang, Y.Y.; Liu, S.Y.; Ding, G.Y.; Li, R.Y. Synthesis and in vitro antitumor activity of 4(3H)-quinazolione derivatives with dithiocarbamate side chains. Bioorg. Med. Chem. Lett. 2005, 15, 1915–1917. [Google Scholar] [CrossRef]
- Wyss, P.C.; Gerber, P.; Hartman, P.G.; Hubschwerlen, C.; Locher, H.; Marty, H.-P.; Stahl, M. Novel dihydrofolate reductase inhibitors. Structure-based versus diversity-based library design and high-throughput synthesis and screening. J. Med. Chem. 2003, 46, 2304–2312. [Google Scholar] [CrossRef]
- Berman, E.M.; Werbel, L.M. The renewed potential for folate antagonists in contemporary cancer chemotherapy. J. Med. Chem. 1991, 34, 479–485. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Aune, T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Grivsky, E.M.; Lee, S.; Sigel, C.W.; Duch, D.S.; Nichol, C.A. Synthesis and antitumor activity of 2,4-diamino-6-(2,5-dimethoxybenzyl)-5-methylpyrido 2,3-d]pyrimidine. J. Med. Chem. 1980, 23, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Ackland, S.P.; Beale, P.; Peters, G.J. Thymidylate synthase inhibitors. Cancer Chemother. Biol. Response Modif. 2003, 21, 1–28. [Google Scholar]
- Assaraf, Y.G.; Leamon, C.P.; Reddy, J.A. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist. Updat. 2014, 17, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, A.; Drozdowska, D. Recent design and structure-activity relationship studies on modifications of DHFR inhibitors as anticancer agents. Curr. Med. Chem. 2019, 26, 1. [Google Scholar] [CrossRef]
- Drugs.com Trimethoprim. Available online: https://www.drugs.com/pro/trimethoprim.html (accessed on 20 March 2020).
- Brogden, R.N.; Carmine, A.A.; Heel, R.C.; Speight, T.M.; Avery, G.S. Trimethoprim: A Review of its antibacterial activity, pharmacokinetics and therapeutic use in urinary tract infections. Drugs 1982, 23, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Villa, D.; Aguilar, M.R.; Rojo, L.; Villa, F. Rojo Folic Acid Antagonists: Antimicrobial and Immunomodulating Mechanisms and Applications. Int. J. Mol. Sci. 2019, 20, 4996. [Google Scholar] [CrossRef] [PubMed]
- Gleckman, R.; Blagg, N.; Joubert, D.W. Trimethoprim: Mechanisms of action, antimicrobial activity, bacterial resistance, pharmacokinetics, adverse reactions, and therapeutic indications. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1981, 1, 14–19. [Google Scholar] [CrossRef]
- Wróbel, A.; Arciszewska, K.; Maliszewski, D.; Drozdowska, D. Trimethoprim and other non-classical antifolates an excellent template for searching modifications of dihydrofolate reductase enzyme inhibitors. J. Antibiot. 2019, 73, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kaur, M.; Sachdeva, S. Mechanism Inspired Development of Rationally Designed Dihydrofolate Reductase Inhibitors as Anticancer Agents. J. Med. Chem. 2012, 55, 6381–6390. [Google Scholar] [CrossRef]
- Algul, O.; Paulsen, J.L.; Anderson, A.C. 2,4-Diamino-5-(2′-aryl-propargyl) pyrimidine derivatives as new nonclassical antifolates for human dihydrofolate reductase inhibition. J. Mol. Graph. Model 2011, 9, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M. The benefits of the multi-target approach in drug design and discovery. Bioorg. Med. Chem. 2006, 14, 896–897. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Meegan, M.J. Designed multiple ligands for cancer therapy. Curr. Med. Chem. 2011, 18, 4722–4737. [Google Scholar]
- Arciszewska, K.; Pućkowska, A.; Wróbel, A.; Drozdowska, D. Carbocyclic analogues of distamycin and netropsin. Mini-Reviews Med. Chem. 2018, 19, 98–113. [Google Scholar] [CrossRef]
- Jeon, H.; Nam, H.; Lee, J.B. Sustained release of minor-groove-binding antibiotic netropsin from calcium-coated groove-rich DNA particles. Pharmaceutics 2019, 11, 387. [Google Scholar] [CrossRef]
- Appleman, J.R.; Howell, E.E.; Kraut, J.; Blakely, R.L. Kinetics of the formation and isomerization of methotrexate complexes of recombinant human dihydrofolate reductase. J. Biol. Chem. 1990, 265, 5579–5584. [Google Scholar] [CrossRef]
- Smith, S.L.; Patrick, P.; Stone, D.; Phillips, A.W.; Burchall, J.J. Porcine liver dihydrofolate reductase. Purification, properties, and amino acid sequence. J. Biol. Chem. 1979, 254, 11475–11484. [Google Scholar] [CrossRef]
- Oefner, C.; D’Arcy, A.; Winkler, F.K. Crystal structure of human dihydrofolate reductase complexed with folate. JBIC J. Biol. Inorg. Chem. 1988, 174, 377–385. [Google Scholar] [CrossRef]
- Gready, J.E. Theoretical studies on the activation of the pterin cofactor in the catalytic mechanism of dihydrofolate reductase. Biochemistry 1985, 24, 4761–4766. [Google Scholar] [CrossRef]
- Wan, Q.; Bennett, B.C.; Wilson, M.A.; Kovalevsky, A.; Langan, P.; Howell, E.E.; Dealwis, C. Toward resolving the catalytic mechanism of dihydrofolate reductase using neutron and ultrahigh-resolution X-ray crystallography. Proc. Natl. Acad. Sci. USA 2014, 111, 18225–18230. [Google Scholar] [CrossRef]
- Tosso, R.D.; Andujar, S.A.; Gutierrez, L.; Angelina, E.; Rodríguez, R.; Nogueras, M.; Baldoni, H.A.; Suvire, F.D.; Cobo, J.; Enriz, R.D. Molecular modeling study of dihydrofolate reductase inhibitors. Molecular dynamics simulations, quantum mechanical calculations, and experimental corroboration. J. Chem. Inf. Model. 2013, 53, 2018–2032. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Skacel, N.; Menon, L.G.; Mishra, P.J.; Peters, R.; Banerjee, D.; Bertino, J.R.; Abali, E.E. Identification of amino acids required for the functional up-regulation of human dihydrofolate reductase protein in response to antifolate treatment. J. Biol. Chem. 2005, 280, 22721–22731. [Google Scholar] [CrossRef] [PubMed]
- Francesconi, V.; Giovannini, L.; Santucci, M.; Cichero, E.; Costi, M.P.; Naesens, L.; Giordanetto, F.; Tonelli, M. Synthesis, biological evaluation and molecular modeling of novel azaspiro dihydrotriazines as influenza virus inhibitors targeting the host factor dihydrofolate reductase (DHFR). Eur. J. Med. Chem. 2018, 155, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Rana, R.M.; Rampogu, S.; Zeb, A.; Son, M.; Park, C.; Lee, G.; Yoon, S.; Baek, A.; Parameswaran, S.; Park, S.J.; et al. In silico study probes potential inhibitors of human dihydrofolate reductase for cancer therapeutics. J. Clin. Med. 2019, 8, 233. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, A.; Maliszewski, D.; Baradyn, M.; Drozdowska, D. Trimethoprim: An old antibacterial drug as a template to search for new targets. Synthesis, biological activity and molecular modeling study of novel trimethoprim analogs. Molecules 2019, 25, 116. [Google Scholar] [CrossRef]
- Kerrigan, J.E.; Abali, E.E.; Bertino, J.R. Recent progress in molecular dynamics simulations of dihydrofolate reductase. Curr. Enzym. Inhib. 2012, 8, 140–149. [Google Scholar] [CrossRef]
- Drozdowska, D.; Drozdowska, D. New solid phase synthesis of distamycin analogues. Molecules 2011, 16, 5753. [Google Scholar] [CrossRef]
- Szerszenowicz, J.; Drozdowska, D. Semi-automatic synthesis, antiproliferative activity and dna-binding properties of new netropsin and bis-netropsin analogues. Molecules 2014, 19, 11300–11315. [Google Scholar] [CrossRef]
- Drozdowska, D.; Rusak, M.; Miltyk, W.; Markowska, A.; Samczuki, P. Antiproliferative effects on breast cancer cells and some interactions of new distamycin analogues with dna, endonucleases and dna topoisomerases. Acta Pol. Pharm. Drug Res. 2016, 73, 47–53. [Google Scholar]
- Debart, F.; Periguad, C.; Gosselin, D.; Mrani, D.; Rayner, B.; Ber, P.L.; Auclair, C.; Balzarini, J.; Clercq, E.D.; Paoletti, C. Synthesis, DNA binding and biological evaluation of synthetic precursors and novel analogs of netropsin. J. Med. Chem. 1989, 32, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological functions and molecular mechanisms of antibiotic tigecycline in the treatment of cancers. Int. J. Mol. Sci. 2019, 20, 3577. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical scoring functions for structure-based virtual screening: Applications, critical aspects, and challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systèmes. In Discovery Studio Visualizer. v21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Cody, V.; Luft, J.R.; Pangborn, W. Understanding the role of Leu22 variants in methotrexate resistance: Comparison of wild-type and Leu22Arg variant mouse and human dihydrofolate reductase ternary crystal complexes with methotrexate and NADPH. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 147–155. [Google Scholar] [CrossRef]
- Leung, A.; Reynolds, R.; Borhani, D. Human dihydrofolate reductase complexed with nadph and trimethoprim. Available online: https://www.rcsb.org/structure/2W3A (accessed on 1 March 2021).
- Lamb, K.M.; G-Dayanandan, N.; Wright, D.L.; Anderson, A.C. Elucidating features that drive the design of selective antifolates using crystal structures of human dihydrofolate reductase. Biochemistry 2013, 52, 7318–7326. [Google Scholar] [CrossRef]
- Pedrola, M.; Jorba, M.; Jardas, E.; Jardi, F.; Ghashghaei, O.; Viñas, M.; Lavilla, R. Multicomponent reactions upon the known drug trimethoprim as a source of novel antimicrobial agents. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Pućkowska, A.; Drozdowska, D.; Midura-Nowaczek, K. Carbocyclic analogues of lexitropsin--DNA affinity and endonuclease inhibition. Acta Pol. Pharm. Drug Res. 2007, 64, 115. [Google Scholar]
- Sigma Aldrich. Technical Bulletin. Available online: https://www.sigmaaldrich.com/content/dam/sigmaaldrich/docs/Sigma/Bulletin/cs0340bul.pdf. (accessed on 1 March 2021).
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive charmm all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef]
- The CHARMM General Force Field (CGenFF) Program. Available online: https://cgenff.umaryland.edu (accessed on 1 March 2021).
- Mayne, C.G.; Saam, J.; Schulten, K.; Tajkhorshid, E.; Gumbart, J.C. Rapid parameterization of small molecules using the force field toolkit. J. Comput. Chem. 2013, 34, 2757–2770. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rogošić, M.; Stepto, R. Definition of terms relating to individual macromolecules, macromolecular assemblies, polymer solutions, and amorphous bulk poliymers (IUPAC Recommendations 2014). Pure Appl. Chem. 2015, 87, 71–120. [Google Scholar]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Ali, S.A.; Hassan, I.; Islam, A.; Ahmad, F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Decrease of Fluorescence [%] | Kapp × 105 M−1 | DHFR Affinity (kcal/mol) | DHFR IC50 (µM) | |||
|---|---|---|---|---|---|---|---|
| Calf Thymus DNA | T4 DNA | Poly (dA-dT)2 | poly (dG-dC)2 | ||||
| EtBr | 100 | 100 | 100 | 95b* | 99 | n.d * | n.d |
| 1 | 74.54 | 3.0 | 1.6 | 3.1 | 3.6 | −7.5 | 21.89 ± 0.03 |
| 2 | 52.89 | 2.6 | 2.6 | 1.9 | 3.0 | −8.0 | 1.11 ± 0.02 |
| 3 | 68.85 | 1.7 | 4.1 | 4.3 | 5.3 | −7.9 | 10.13 ± 0.09 |
| 4 | 87.99 | 2.3 | 2.3 | 2.1 | 1.3 | −7.5 | 5.02 ± 0.03 |
| 5 | 72.56 | 2.7 | 1.0 | 1.3 | 2.3 | −7.9 | 30.02 ± 0.02 |
| 6 | 50.44 | 4.0 | 1.3 | 2.5 | 4.8 | −8.0 | 0.99 ± 0.01 |
| 7 | 66.89 | 4.1 | 2.1 | 3.4 | 2.3 | −7.8 | 3.02 ± 0.09 |
| 8 | 56.58 | 2.5 | 2.4 | 3.1 | 3.5 | −7.7 | 10.59 ± 0.06 |
| 9 | 64.89 | 3.9 | 1.2 | 3.9 | 1.5 | −7.6 | 56.05 ± 0.08 |
| 10 | 70.11 | 2.6 | 0.7 | 1.1 | 4.7 | −7.4 | 19.20 ± 0.01 |
| 11 | 54.54 | 1.2 | 3.1 | 2.6 | 2.5 | −7.6 | 55.32 ± 0.02 |
| 12 | 60.11 | 2.1 | 2.8 | 1.5 | 2.1 | −7.5 | 26.01 ± 0.02 |
| 13 | 49.89 | 1.9 | 2.9 | 4.0 | 3.4 | −8.0 | 0.89 ± 0.07 |
| 14 | 43.85 | 4.9 | 2.9 | 8.9 | 4.2 | −8.2 | 0.97 ± 0.02 |
| 15 | 68.82 | 1.9 | 2.3 | 3.1 | 2.4 | −7.7 | 12.01 ± 0.01 |
| 16 | 57.77 | 1.3 | 3.1 | 2.7 | 3.8 | −8.2 | 0.88 ± 0.02 |
| 17 | 41.68 | 1.8 | 2.9 | 0.5 | 2.5 | −8.1 | 1.22 ± 0.05 |
| 18 | 42.99 | 5.8 | 6.4 | 4.5 | 4.6 | −8.0 | 2.09 ± 0.04 |
| TMP | 100 | n.d | n.d | n.d | n.d | −7.5 | 55.26 ± 0.01 |
| NT [45] | 74 | 8.7 | 8.3 | 875 | 2.5 | −9.6 | 20.56 ± 0.01 |
| MTX | 100 | n.d | n.d | n.d | n.d | −9.5 | 0.08 ± 0.003 |
| Molecule | Residue | Type | Occupancy |
|---|---|---|---|
| 2 | Glu-30-S * | D | 71.83% |
| Glu-30-M * | D | 49.66% | |
| Thr-56 | A | 47.15% | |
| Ala-9 | A | 18.35% | |
| Thr-136 | A | 5.52% | |
| Ser-59 | A | 1.62% | |
| 6 | Ala-9 | A | 64.67% |
| Glu-30 | D | 64.10% | |
| Ser-118 | A | 29.82% | |
| Phe-34 | D | 6.53% | |
| Thr-56 | A | 2.33% | |
| Val-115 | D | 1.10% | |
| 13 | Glu-30 | D | 58.02% |
| Val-115 | D | 17.23% | |
| Ser-118 | A | 11.47% | |
| Tyr-121 | D | 1.85% | |
| Ala-9 | A | 1.23% | |
| 14 | Glu-30 | D | 50.48% |
| Ala-9 | A | 28.90% | |
| Ser-118 | A | 9.35% | |
| Tyr-121 | D | 1.48% | |
| 16 | Glu-30 | D | 33.67% |
| Leu-27 | D/A | 19.65% | |
| Ser-59 | D | 15.23% | |
| Thr-56 | A | 7.55% | |
| Ser-118 | A | 5.60% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wróbel, A.; Baradyn, M.; Ratkiewicz, A.; Drozdowska, D. Synthesis, Biological Activity, and Molecular Dynamics Study of Novel Series of a Trimethoprim Analogs as Multi-Targeted Compounds: Dihydrofolate Reductase (DHFR) Inhibitors and DNA-Binding Agents. Int. J. Mol. Sci. 2021, 22, 3685. https://doi.org/10.3390/ijms22073685
Wróbel A, Baradyn M, Ratkiewicz A, Drozdowska D. Synthesis, Biological Activity, and Molecular Dynamics Study of Novel Series of a Trimethoprim Analogs as Multi-Targeted Compounds: Dihydrofolate Reductase (DHFR) Inhibitors and DNA-Binding Agents. International Journal of Molecular Sciences. 2021; 22(7):3685. https://doi.org/10.3390/ijms22073685
Chicago/Turabian StyleWróbel, Agnieszka, Maciej Baradyn, Artur Ratkiewicz, and Danuta Drozdowska. 2021. "Synthesis, Biological Activity, and Molecular Dynamics Study of Novel Series of a Trimethoprim Analogs as Multi-Targeted Compounds: Dihydrofolate Reductase (DHFR) Inhibitors and DNA-Binding Agents" International Journal of Molecular Sciences 22, no. 7: 3685. https://doi.org/10.3390/ijms22073685
APA StyleWróbel, A., Baradyn, M., Ratkiewicz, A., & Drozdowska, D. (2021). Synthesis, Biological Activity, and Molecular Dynamics Study of Novel Series of a Trimethoprim Analogs as Multi-Targeted Compounds: Dihydrofolate Reductase (DHFR) Inhibitors and DNA-Binding Agents. International Journal of Molecular Sciences, 22(7), 3685. https://doi.org/10.3390/ijms22073685