
Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Results
2.1. Patients Characteristics
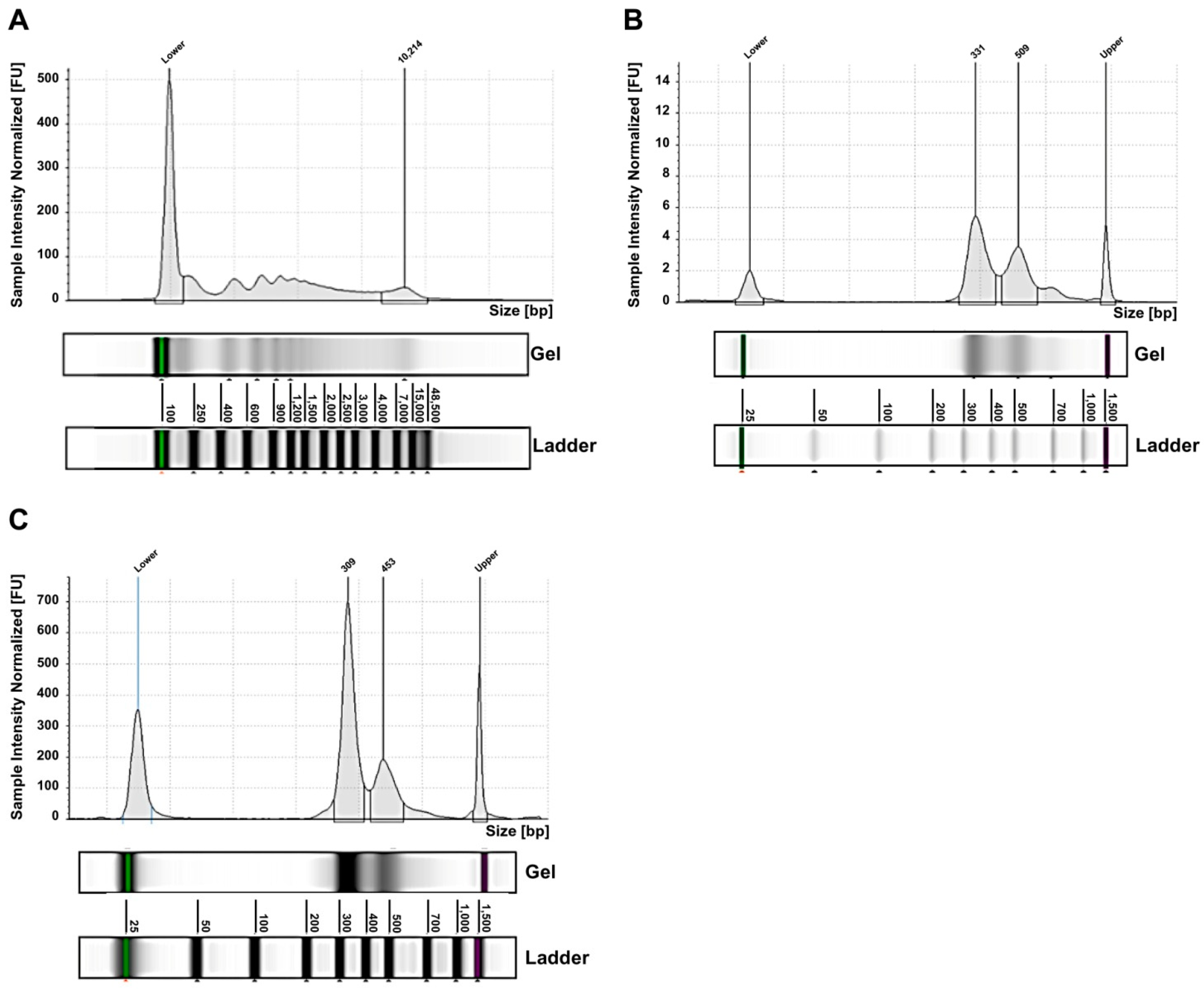
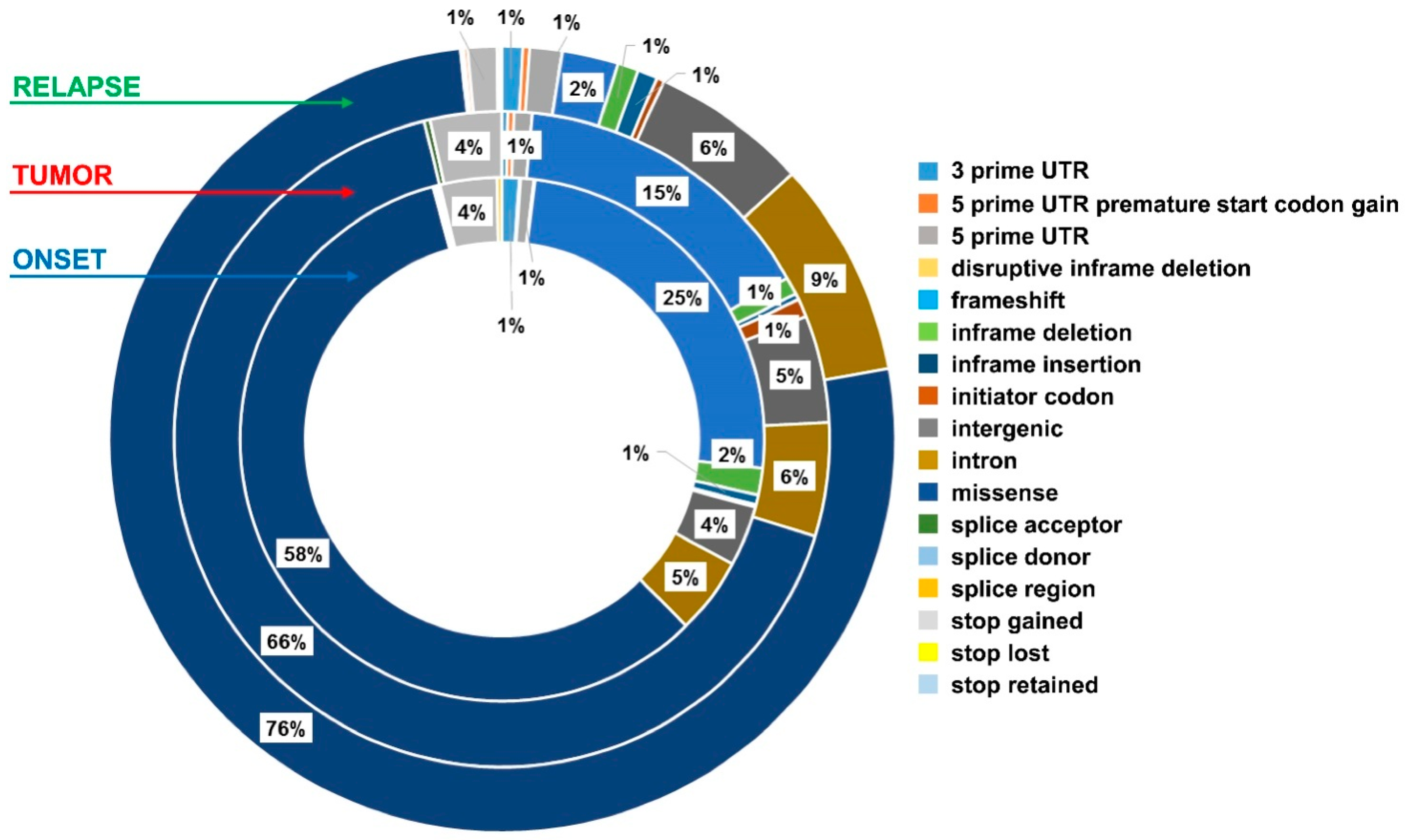
2.2. Exosomes from NB Patients Contain Genomic DsDNA Covering All Chromosomes
2.3. Tumor DNA Content in Exo-DNA Samples
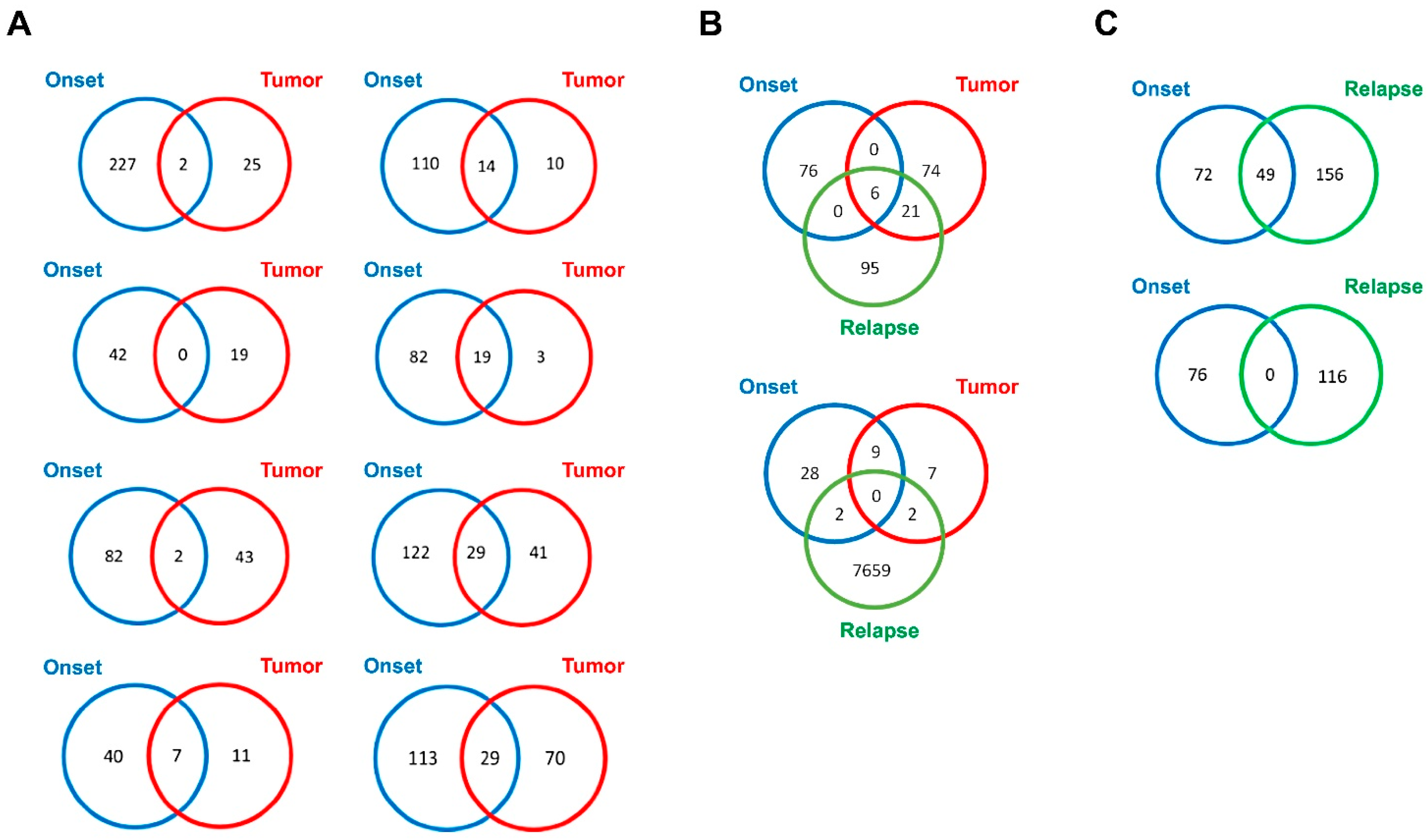
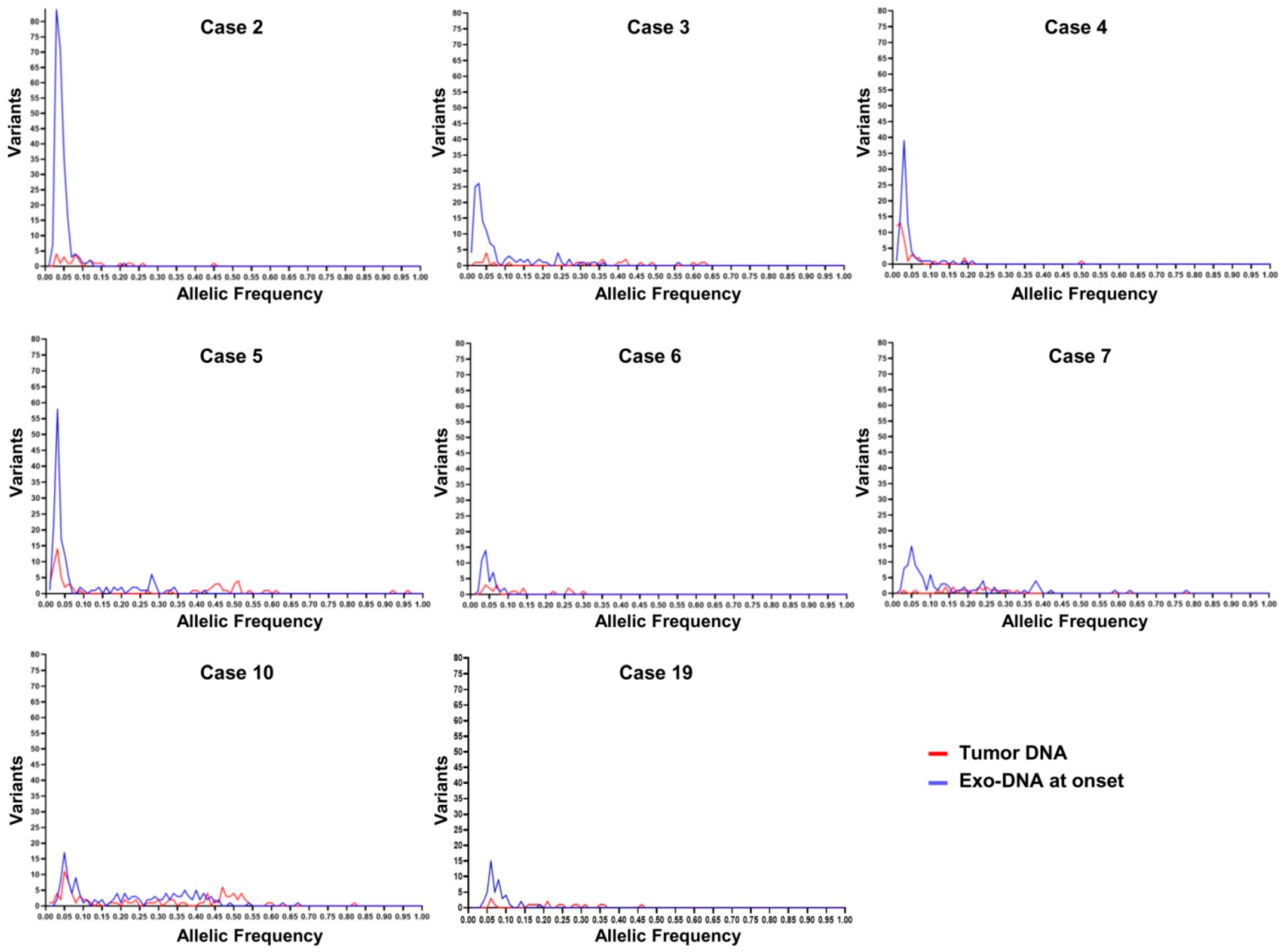
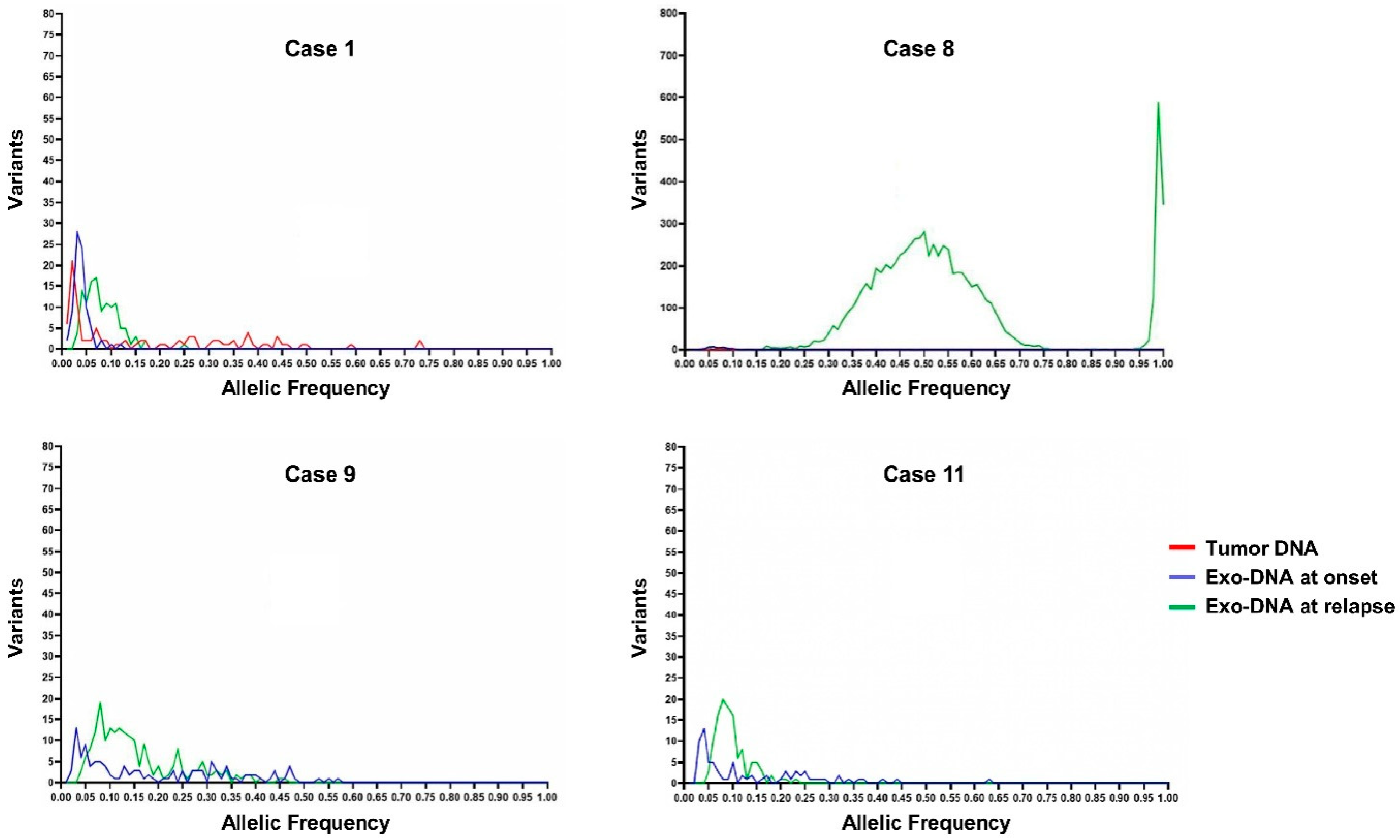
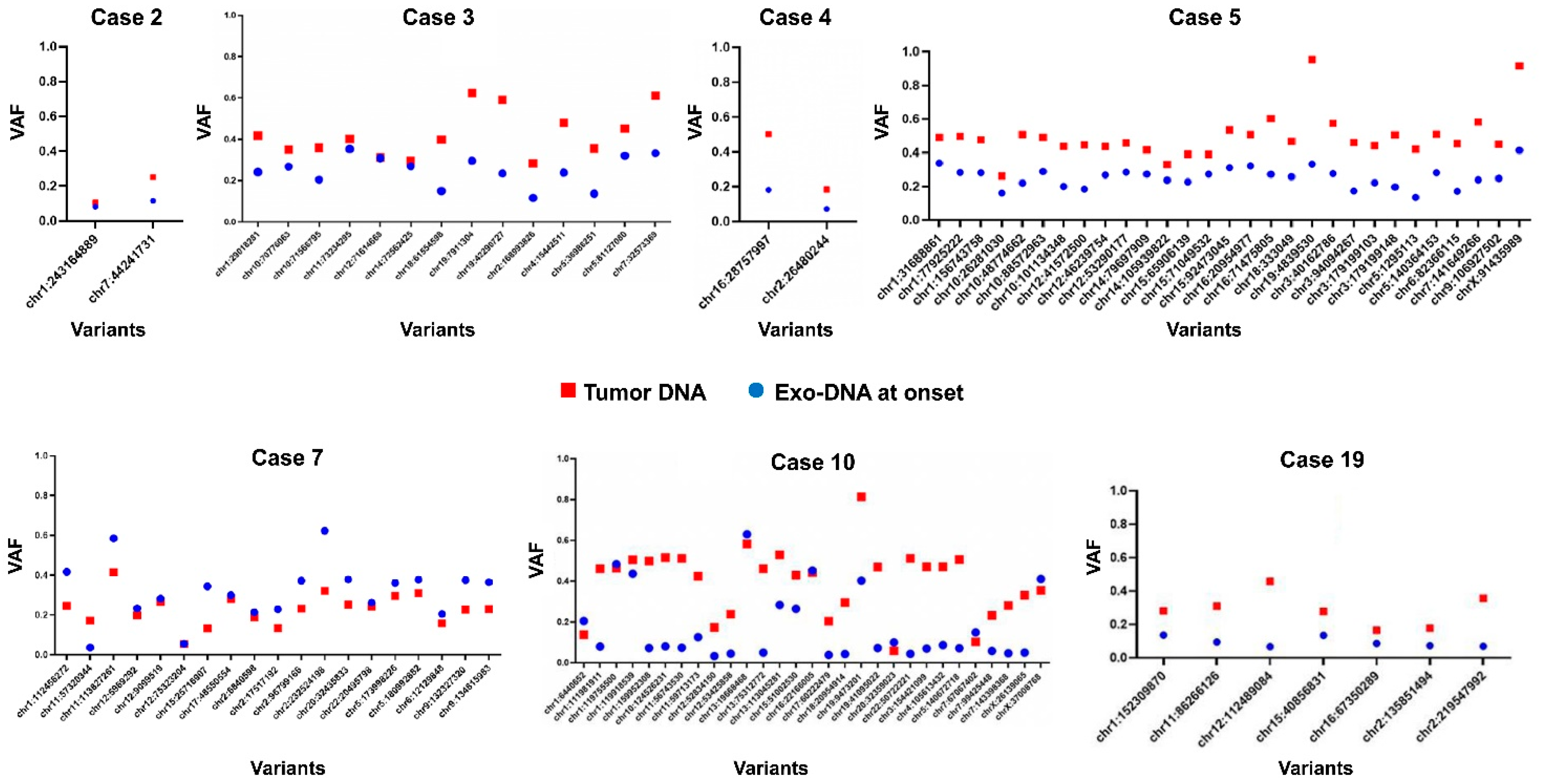
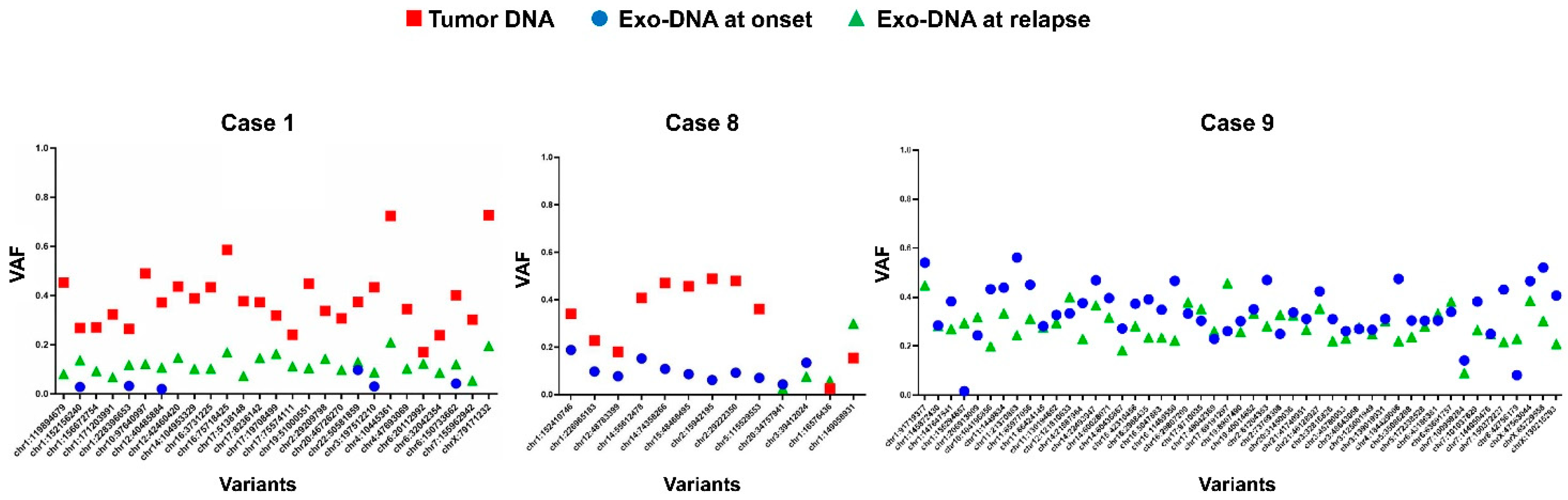
2.4. Exo-DNA-Derived Tumor Mutation Load
2.5. Exo-DNA Contains SNVs of Known Neuroblastoma Driver Genes
2.6. Most Frequently Mutated Genes in Exo-DNA and in Tumor DNA
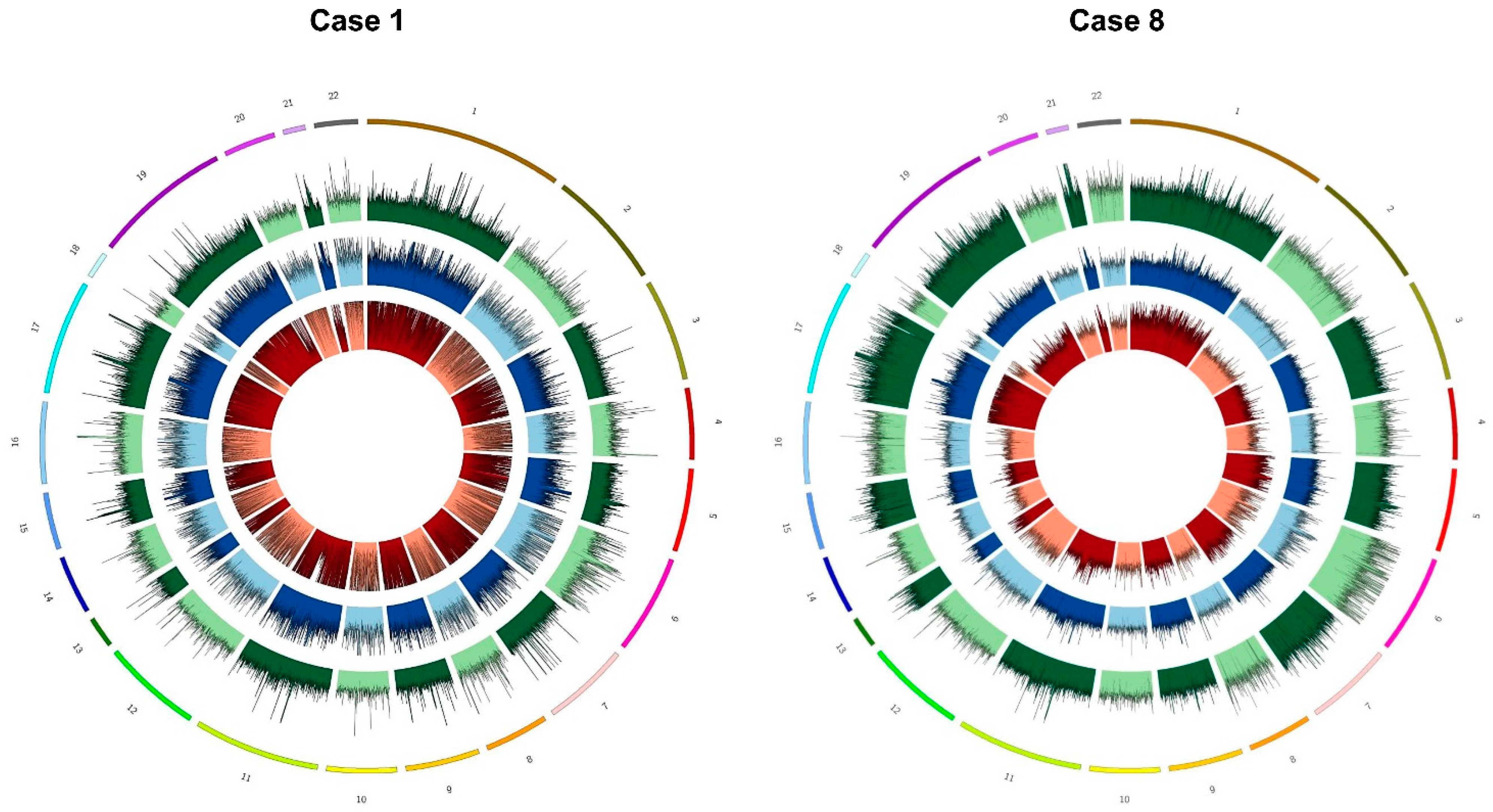
2.7. Identified Copy Number Variations Involved in NB Oncogenesis
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Sample Collection and Processing
4.3. Exo-DNA Purification and Quantification
4.4. Genomic Profiling of NB Primary Tumors
4.5. Library Construction and Exome Capture of Exo-DNA
4.6. Bioinformatics Pipeline
4.6.1. Copy Number Variations (CNVs)
4.6.2. Tumor Mutation Load (TML)
4.6.3. Enrichment Analysis
4.6.4. Concordance Rate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NB | Neuroblastoma |
| MYCN | v-myc myelocytomatosis viral related oncogene, neuroblastoma derived |
| ALK | Anaplastic Limphoma Kinase |
| RAS | Rat Sarcoma |
| INRG | International Neuroblastoma Risk Group |
| a-CGH | Array-Comparative Genome Hybridization |
| chr | Chromosome |
| mAb | Monoclonal antibody |
| dsDNA | Double stranded DNA |
| gDNA | Genomic DNA |
| cfDNA | Cell-free DNA |
| ctDNA | Circulating tumor DNA |
| exo-DNA | Exosomal DNA |
| WES | Whole Exome Sequencing |
| UMI | Unique Molecular Identifier |
| SNV | Single Nucleotide Variant |
| CNV | Copy Number Variation |
| VAF | Variant Allele Frequency |
| TML | Tumor Mutation Load |
| VCF | Variant Call Format |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
References
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Speleman, F.; Park, J.R.; Henderson, T.O. Neuroblastoma: A Tough Nut to Crack. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e548–e557. [Google Scholar] [CrossRef]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. INRG Task Force. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Chicard, M.; Colmet-Daage, L.; Clement, N.; Danzon, A.; Bohec, M.; Bernard, V.; Sylvain Baulande, S.; Bellini, A.; Deveau, P.; Gaëlle Pierron, G.; et al. Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin. Cancer Res. 2018, 24, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C. Liquid Biopsy: Is There an Advantage to Analyzing Circulating Exosomal DNA Compared to cfDNA or Are They the Same? Cancer Res. 2019, 79, 2462–2465. [Google Scholar] [CrossRef]
- Gangoda, L.; Boukouris, S.; Liem, M.; Kalra, H.; Mathivanan, S. Extracellular vesicles including exosomes are mediators of signal transduction: Are they protective or pathogenic? Proteomics 2015, 15, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Morini, M.; Cangelosi, D.; Segalerba, D.; Marimpietri, D.; Raggi, F.; Castellano, A.; Fruci, D.; de Mora, J.F.; Cañete, A.; Yáñez, Y.; et al. Exosomal microRNAs from Longitudinal Liquid Biopsies for the Prediction of Response to Induction Chemotherapy in High-Risk Neuroblastoma Patients: A Proof-of-Concept SIOPEN Study. Cancers 2019, 11, 1476. [Google Scholar] [CrossRef]
- Fonseka, P.; Liem, M.; Ozcitti, C.; Adda, C.G.; Ang, C.S.; Mathivanan, S. Exosomes from N-Myc amplified neuroblastoma cells induce migration and confer chemoresistance to non-N-Myc amplified cells: Implications of intra-tumour heterogeneity. J. Extracell. Vesicles 2019, 8, 1597614. [Google Scholar] [CrossRef]
- Thakur, B.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, O.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double stranded genomic DNA spanning all chromosomes with mutated KRAS and P53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.J.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10, 12564. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2018, 9, 4109. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biol. Sci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Iadarola, B.; Xumerle, L.; Lavezzari, D.; Paterno, M.; Marcolungo, L.; Beltrami, C.; Fortunati, E.; Mei, D.; Vetro, A.; Guerrini, R.; et al. Shedding light on dark genes: Enhanced targeted resequencing by optimizing the combination of enrichment technology and DNA fragment length. Sci. Rep. 2020, 10, 9424. [Google Scholar] [CrossRef]
- Koeppel, F.; Blanchard, S.; Jovelet, C.; Genin, B.; Marcaillou, C.; Martin, E.; Rouleau, E.; Solary, E.; Soria, J.C.; André, F.; et al. Whole exome sequencing for determination of tumor mutation load in liquid biopsy from advanced cancer patients. PLoS ONE 2017, 12, e0188174. [Google Scholar] [CrossRef]
- Möhrmann, L.; Huang, H.J.; Hong, D.S.; Tsimberidou, A.M.; Fu, S.; Piha-Paul, S.A.; Subbiah, V.; Karp, D.D.; Naing, A.; Krug, A.; et al. Liquid Biopsies Using Plasma Exosomal Nucleic Acids and Plasma Cell-Free DNA Compared with Clinical Outcomes of Patients with Advanced Cancers. Clin. Cancer Res. 2018, 24, 181–188. [Google Scholar] [CrossRef]
- Brady, S.W.; Liu, Y.; Xiaotu Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 5183. [Google Scholar] [CrossRef]
- Van Roy, N.; Van Der Linden, M.; Menten, B.; Dheedene, A.; Vandeputte, C.; Van Dorpe, J.; Laureys, G.; Renard, M.; Sante, T.; Lammens, T.; et al. Shallow Whole Genome Sequencing on Circulating Cell-Free DNA Allows Reliable Noninvasive Copy-Number Profiling in Neuroblastoma Patients. Clin. Cancer Res. 2017, 23, 6305–6314. [Google Scholar] [CrossRef]
- Lodrini, M.; Sprüssel, A.; Astrahantseff, K.; Tiburtius, D.; Konschak, R.; Lode, H.N.; Fischer, M.; Keilholz, U.; Eggert, A.; Deubzer, H.E. Using droplet digital PCR to analyze MYCN and ALK copy number in plasma from patients with neuroblastoma. Oncotarget 2017, 8, 85234–85251. [Google Scholar] [CrossRef] [PubMed]
- Colletti, M.; Petretto, A.; Galardi, A.; Di Paolo, V.; Tomao, L.; Lavarello, C.; Inglese, E.; Bruschi, M.; Lopez, A.A.; Pascucci, L.; et al. Proteomic Analysis of Neuroblastoma-Derived Exosomes: New Insights into a Metastatic Signature. Proteomics 2017, 17, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Noma, A.; Suzuki, T. Ribonucleome analysis identified enzyme genes responsible for wybutosine synthesis. Nucleic Acids Symp. Ser. 2006, 50, 65–66. [Google Scholar] [CrossRef]
- Salvador, G.A.; Oteiza, P.I. Iron overload triggers redox-sensitive signals in human IMR-32 neuroblastoma cells. Neurotoxicology 2011, 32, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Maffie, J.K.; Lin, L.; Petralia, R.S.; Rudy, B.; Hoffman, D.A. DPP6 establishes the A-type K (+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 2011, 71, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.R.; Hoxha, E.; Wiech, T.; Schröder, C.; Simon, R.; Stahl, R.A. THSD7A expression in human cancer. Genes Chromosomes Cancer 2017, 56, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Flegel, C.; Manteniotis, S.; Osthold, S.; Hatt, H.; Gisselmann, G. Expression profile of ectopic olfactory receptors determined by deep sequencing. PLoS ONE 2013, 8, e55368. [Google Scholar] [CrossRef]
- Sanz, G.; Leray, I.; Dewaele, A.; Sobilo, J.; Lerondel, S.; Bouet, S.; Grébert, D.; Monnerie, R.; Pajot-Augy, E.; Mir, L.M. Promotion of cancer cell invasiveness and metastasis emergence caused by olfactory receptor stimulation. PLoS ONE 2014, 9, e85110. [Google Scholar] [CrossRef] [PubMed]
- Gerber, T.; Taschner-Mandl, S.; Saloberger-Sindhöringer, L.; Popitsch, N.; Heitzer, E.; Witt, V.; Geyeregger, R.; Hutter, C.; Schwentner, R.; Ambros, I.M.; et al. Assessment of Pre-Analytical Sample Handling Conditions for Comprehensive Liquid Biopsy Analysis. J. Mol. Diagn. 2020, 22, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef] [PubMed]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell Vesicles. 2018, 7, 1505403. [Google Scholar] [CrossRef] [PubMed]
- Ognibene, M.; Morini, M.; Garaventa, A.; Podestà, M.; Pezzolo, A. Identification of a minimal region of loss on chromosome 6q27 associated with poor survival of high-risk neuroblastoma patients. Cancer Biol. Ther. 2020, 21, 391–399. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Risk Group | Age at Diagnosis (months) | Relapse | MYCN Status | Genomic Profile by a-CGH | Chromosome Alterations | Last Follow-Up |
|---|---|---|---|---|---|---|---|
| 1 | M | 61 | Yes | Gain | Segmental | 2p+, +4, +6, +7, +8, +13, +18, 17q+, 11q−, 1q+, 11q+, 12q+, 14q+, +20, 21q−, Xp+, −Y | Alive with evidence of disease |
| 2 | L2 | 34 | No | Single copy | Numerical | −3, +6, −9, −11, +13, −14, −16, +17, +18, −19, −22, −X | Alive with evidence of disease |
| 3 | L2 | 165 | No | Single copy | Segmental | 7p−, 19p−, 19q−, 21q− | Alive |
| 4 | L2 | 1 | No | Single copy | Numerical | −3, −4, +7, −9, −10, −11, +14, −16, +17, +18, −X, −Y | Alive |
| 5 | M | 118 | No | Single copy | Segmental | 4q+, 10p+, 17p−, 17q+, 19p+, 22q− | Alive with evidence of disease |
| 6 | L2 | 28 | No | Single copy | Numerical | +6, + 7, +13, +15, +17 | Alive |
| 7 | M | 45 | No | Amplified | Segmental | 1p−, 2p+, 4p−, 12q+, 17q+, 19p− | Dead |
| 8 | M | 17 | Yes | Single copy | Segmental | 4p−, +6, +7, +13, −9, −10, −11, −12, −14, +15, 16p−, 16q−, +17, −X, +Y | Dead |
| 9 | M | 88 | Yes | Amplified | Segmental | 2q−, 7p+, 7q+, 11q−, 11q+, 12q+, 17q+, 19q−, Xp+ | Dead |
| 10 | M | 64 | No | Amplified | Segmental | +1, 2p+, +3, +7, −5, −8, 17q+, 11q− | Dead |
| 11 | M | 45 | Yes | Single copy | Not performed | - | Dead |
| 12 | M | 70 | No | Amplified | Not performed | - | Dead |
| 13 | M | 58 | No | Gain | Segmental | 1p+, 2p+, 3p−, 5q+, 7q+, 13q+,14q−, 17q+, 11q−, 18p−, 19q−, −Y | Alive with evidence of disease |
| 14 | M | 134 | No | Single copy | Segmental | 1q+, +5, +7, +12, +13, +17, 17q+, +18 | Alive with evidence of disease |
| 15 | M | 24 | No | Amplified | Not performed | - | Dead |
| 16 | M | 212 | No | Single copy | Segmental | −1, 2p+, −3, 9p−, +10, +11, +12, 14q− | Alive with evidence of disease |
| 17 | M | 82 | No | Single copy | Not performed | - | Alive |
| 18 | M | 46 | No | Gain | Segmental | 2p+, 2q+, 3p−, 4p−, +7, 7q+, −8, 8p−, 9p+, +12, 17q+, 11q−, +18, 19p−, 22q+, −X, Xq− | Alive with evidence of disease |
| 19 | Ms | 10 | Yes | Single copy | Segmental | 1p−, −14, 18p−, 19q−, −Y | Alive with evidence of disease (progression and relapse) |
| Chromosome | Genomic DNA | Tumor DNA | Exo-DNA at Onset | Exo-DNAat Relapse |
|---|---|---|---|---|
| %PASS | %PASS | %PASS | %PASS | |
| Chr1 | 83.30 | 87.42 | 89.21 | 82.26 |
| Chr2 | 84.21 | 88.53 | 90.26 | 82.35 |
| Chr3 | 89.54 | 90.65 | 90.51 | 87.66 |
| Chr4 | 91.15 | 92.44 | 93.57 | 90.54 |
| Chr5 | 79.09 | 81.07 | 80.79 | 78.28 |
| Chr6 | 93.58 | 94.86 | 95.61 | 93.53 |
| Chr7 | 85.87 | 88.75 | 89.79 | 84.72 |
| Chr8 | 78.87 | 79.86 | 81.1 | 77.98 |
| Chr9 | 83.75 | 85.78 | 86.69 | 82.88 |
| Chr10 | 89.93 | 93.23 | 95.11 | 89.18 |
| Chr11 | 91.73 | 93.98 | 96.04 | 91.00 |
| Chr12 | 92.21 | 94.27 | 96.4 | 91.40 |
| Chr13 | 90.75 | 92.76 | 97.75 | 90.81 |
| Chr14 | 94.87 | 97.14 | 97.61 | 93.77 |
| Chr15 | 78.69 | 82.83 | 83.75 | 77.87 |
| Chr16 | 77.94 | 81.04 | 82.63 | 76.47 |
| Chr17 | 91.57 | 93.52 | 94.11 | 90.38 |
| Chr18 | 88.03 | 91.49 | 96.52 | 88.92 |
| Chr19 | 97.02 | 97.21 | 97.2 | 96.36 |
| Chr20 | 98.48 | 98.42 | 98.26 | 97.91 |
| Chr21 | 71.48 | 72.96 | 73.41 | 70.49 |
| Chr22 | 87.22 | 89.93 | 92.67 | 87.08 |
| ChrX | 83.30 | 88.83 | 89.27 | 88.11 |
| N° Cases | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genes of interest | |||||||||||||||||||
| ALK c.3824 G > A | X | X | X | ||||||||||||||||
| ALK c.3522 C > A | X | X | X | ||||||||||||||||
| ALK c.4587 C > G | |||||||||||||||||||
| ALK c.4338 G > T | |||||||||||||||||||
| ALK c.3509 T > A | X | ||||||||||||||||||
| ALK c.1260 T > C | |||||||||||||||||||
| ALK c.1853 G > A | |||||||||||||||||||
| ALK c.1827 G > A | |||||||||||||||||||
| CHD5 c.4789 G > A | |||||||||||||||||||
| CHD5 c.1857 C > T | |||||||||||||||||||
| CHD5 c.635 C > T | |||||||||||||||||||
| CDKN1B c.326 T > G | |||||||||||||||||||
| PHOX2B c.752 C > T | |||||||||||||||||||
| PHOX2B c.722_738 del | |||||||||||||||||||
| DENND3 c.2053 G > T | X | ||||||||||||||||||
| TP53 c.811 G > A | |||||||||||||||||||
| ATRX c.3028 G > T | |||||||||||||||||||
| TERT c.510 G > T | |||||||||||||||||||
| TERT c.2297_2299 del | |||||||||||||||||||
| BRAF c.1803 A > T | |||||||||||||||||||
| BRAF c.1741 A > T | |||||||||||||||||||
| MYCN c.131 C > T | X | ||||||||||||||||||
| SHANK2 c.2900 A > G | |||||||||||||||||||
| SHANK2 c.1175-5810 | |||||||||||||||||||
| SHANK2 c.5404 G > A | |||||||||||||||||||
| SHANK2 c.122 C > A | |||||||||||||||||||
| PTPRD c.1335 T > C | |||||||||||||||||||
| FGFR1 c.1625 del A | |||||||||||||||||||
| FGFR1 c.1638 C > G | X | ||||||||||||||||||
| PTPN11 c.1508G > T | |||||||||||||||||||
| KIT c.2394 C > T | X | ||||||||||||||||||
| PLAG1 c.1372 C > A | X | ||||||||||||||||||
| KCNJ12 c.167 A > C | X | ||||||||||||||||||
 Mutation detected only in tumor DNA;
Mutation detected only in tumor DNA;  Mutation detected only in exo-DNA at onset;
Mutation detected only in exo-DNA at onset;  Mutation detected only in exo-DNA at relapse;
Mutation detected only in exo-DNA at relapse;  Concordant mutation in tumor DNA and in exo-DNA at onset;
Concordant mutation in tumor DNA and in exo-DNA at onset;  Concordant mutation in tumor DNA and in exo-DNA at relapse; X: SNVs frequently associated with NB.
Concordant mutation in tumor DNA and in exo-DNA at relapse; X: SNVs frequently associated with NB.| Exo-DNA at Onset | Exo-DNA at Relapse | Tumor DNA | |||
|---|---|---|---|---|---|
| Gene | FDR_BH p-Value | Gene | FDR_BH p-Value | Gene | FDR_BH p-Value |
| TYW1 | 9.79 × 10−29 | AHNAK2 | 0.00075 | ALK | 4.18 × 10−8 |
| ALK | 3.02 × 10−9 | OR52R1 | 0.00075 | PRDM2 | 4.7 × 10−5 |
| MUC16 | 7.44 × 10−9 | OR10H3 | 0.00075 | NRXN3 | 4.7 × 10−5 |
| DPP6 | 9.93 × 10−9 | C17orf78 | 0.00075 | SI | 4.7 × 10−5 |
| OR8G5 | 3.26 × 10−8 | DEGS1 | 0.00075 | OTOF | 0.000119 |
| ALPK1 | 5.92 × 10−6 | C5orf38 | 0.00075 | MXI1 | 0.00134 |
| MYH4 | 6.75 × 10−6 | THEGL | 0.00114 | C5orf47 | 0.00134 |
| LOC105370980 | 7.87 × 10−6 | CPO | 0.00114 | TMEM198 | 0.00134 |
| NCKAP5L | 9.28 × 10−6 | CYP3A43 | 0.00114 | ASCL2 | 0.00134 |
| FLG | 1.99 × 10−5 | RETSAT | 0.00114 | C1QTNF7 | 0.00134 |
| TRIOBP | 1.99 × 10−5 | C16orf89 | 0.00114 | AP1M1 | 0.00134 |
| NPIPB2 | 1.99 × 10−5 | GTPBP4 | 0.00114 | ENO3 | 0.00134 |
| DTD2 | 1.99 × 10−5 | TYR | 0.00114 | CLEC9A | 0.00134 |
| PSG9 | 2.35 × 10−5 | SLC2A10 | 0.00114 | EED | 0.00134 |
| FRAS1 | 3.18 × 10−5 | C14orf39 | 0.00165 | SRL | 0.00134 |
| HYDIN | 3.55 × 10−5 | TAP1 | 0.00185 | NPTXR | 0.00134 |
| MUC4 | 3.55 × 10−5 | NUAK1 | 0.00185 | P2RY2 | 0.00134 |
| TMEM14B | 4.46 × 10−5 | DNAI1 | 0.00197 | LCE3D | 0.00134 |
| ASB11 | 4.55 × 10−5 | DTNA | 0.00205 | LCE2D | 0.00134 |
| TAF11 | 4.55 × 10−5 | NPAS4 | 0.00222 | MYCN | 0.00134 |
| Exo-DNA at Onset | FDR_BH p-Value |
|---|---|
| chr6:41198279-41198279 (TREML2 gene) | 3.35 × 10−17 |
| chr7:154053011-154053011 (DPP6 gene) | 3.33 × 10−18 |
| chr7:154052929-154052929 (DPP6 gene) | 3.87 × 10−18 |
| chr7:11831836-11831843 (THSD7A gene) | 1.94 × 10−10 |
| chr7:67067402-67067402 (TYW1 gene) | 9.61 × 10−59 |
| chr11:124265668-124265676 (OR8G5 gene) | 2.85 × 10−16 |
| chr12:123937252-123937252 (CCDC92 gene) | 1.2 × 10−17 |
| chr14:31457378-31457378 (DTD2 gene) | 1.2 × 10−17 |
| chr19:42740350-42740352 (PSG3 gene) | 1.14 × 10−10 |
| Gene. | Exo-DNA at Onset | Exo-DNA at Relapse | Tumor DNA |
|---|---|---|---|
| NF1 | 7 | 3 | 2 |
| BRAF | 5 | 1 | 3 |
| MYCN | 6 | 1 | 2 |
| ALK | 5 | 1 | 2 |
| SHANK2 | 6 | 0 | 2 |
| PTPN11 | 4 | 0 | 3 |
| TP53 | 3 | 0 | 4 |
| ATRX | 1 | 1 | 4 |
| CDKN1B | 4 | 0 | 2 |
| KRAS | 3 | 0 | 2 |
| TERT | 3 | 1 | 1 |
| CHD5 | 2 | 1 | 0 |
| PHOX2B | 1 | 0 | 2 |
| PTPRD | 1 | 0 | 1 |
| SMARCA4 | 1 | 0 | 1 |
| FGFR1 | 1 | 0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degli Esposti, C.; Iadarola, B.; Maestri, S.; Beltrami, C.; Lavezzari, D.; Morini, M.; De Marco, P.; Erminio, G.; Garaventa, A.; Zara, F.; et al. Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells. Int. J. Mol. Sci. 2021, 22, 3667. https://doi.org/10.3390/ijms22073667
Degli Esposti C, Iadarola B, Maestri S, Beltrami C, Lavezzari D, Morini M, De Marco P, Erminio G, Garaventa A, Zara F, et al. Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells. International Journal of Molecular Sciences. 2021; 22(7):3667. https://doi.org/10.3390/ijms22073667
Chicago/Turabian StyleDegli Esposti, Chiara, Barbara Iadarola, Simone Maestri, Cristina Beltrami, Denise Lavezzari, Martina Morini, Patrizia De Marco, Giovanni Erminio, Alberto Garaventa, Federico Zara, and et al. 2021. "Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells" International Journal of Molecular Sciences 22, no. 7: 3667. https://doi.org/10.3390/ijms22073667
APA StyleDegli Esposti, C., Iadarola, B., Maestri, S., Beltrami, C., Lavezzari, D., Morini, M., De Marco, P., Erminio, G., Garaventa, A., Zara, F., Delledonne, M., Ognibene, M., & Pezzolo, A. (2021). Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells. International Journal of Molecular Sciences, 22(7), 3667. https://doi.org/10.3390/ijms22073667