
Elucidation of the Clustered Nano-Architecture of Radiation-Induced DNA Damage Sites and Surrounding Chromatin in Cancer Cells: A Single Molecule Localization Microscopy Approach

 ,
,  ,
,  , ,
, ,
Abstract
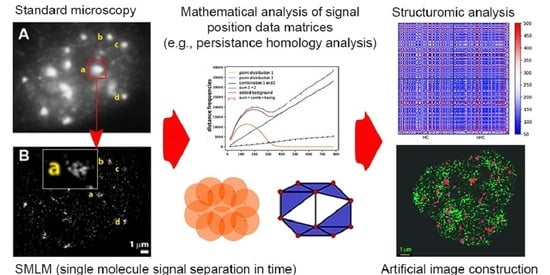
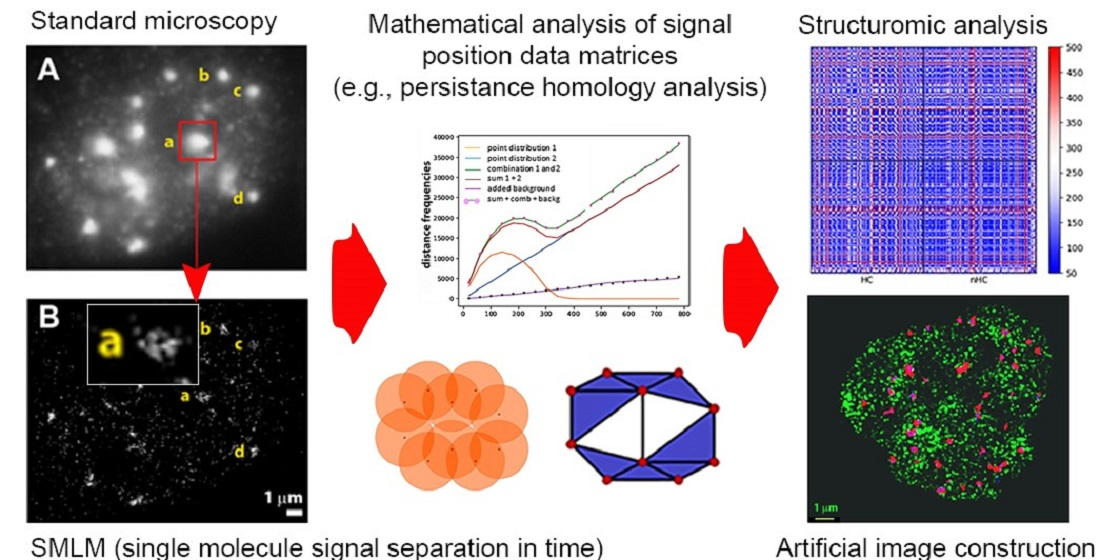{kind=link}
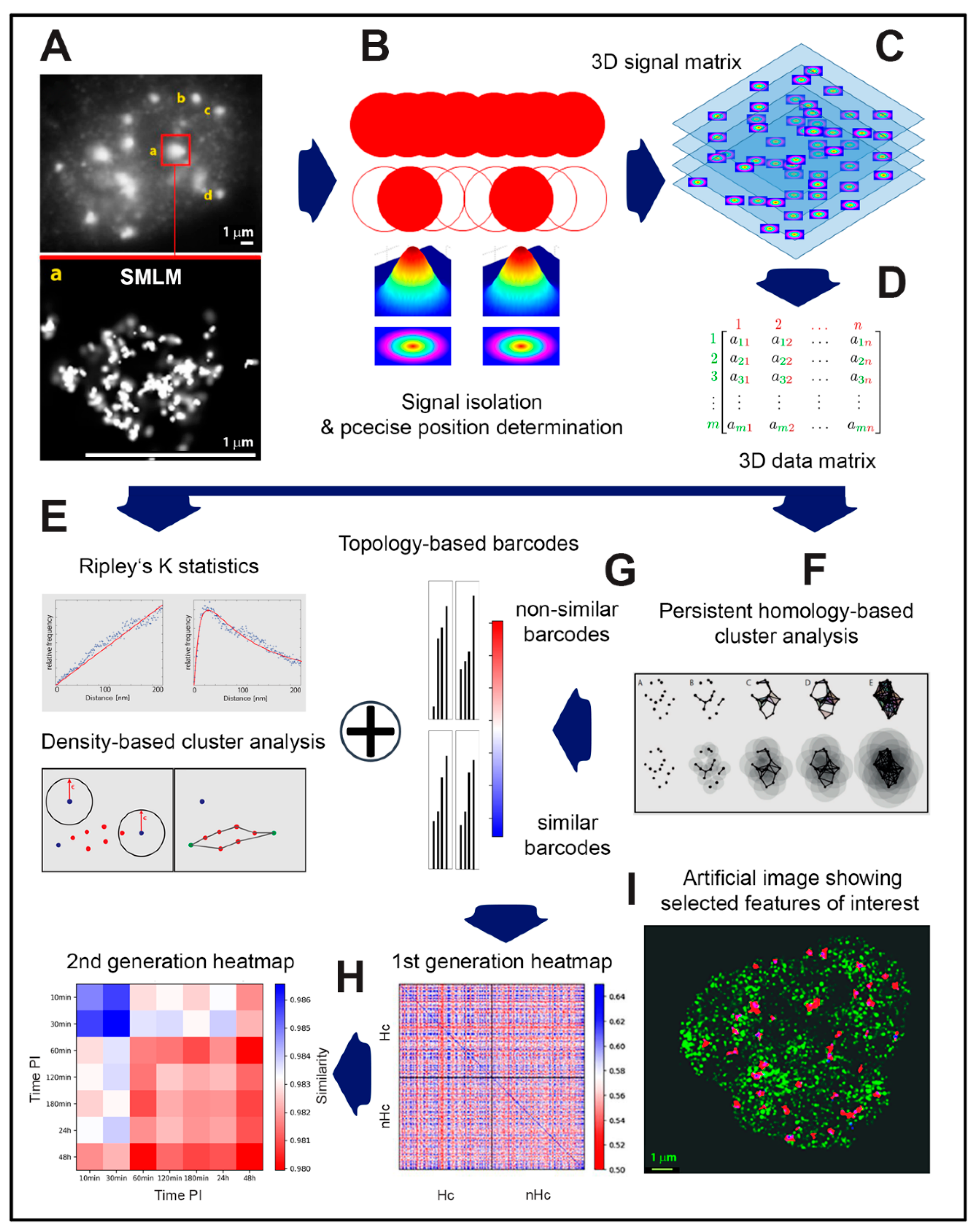{kind=link}
{kind=link}
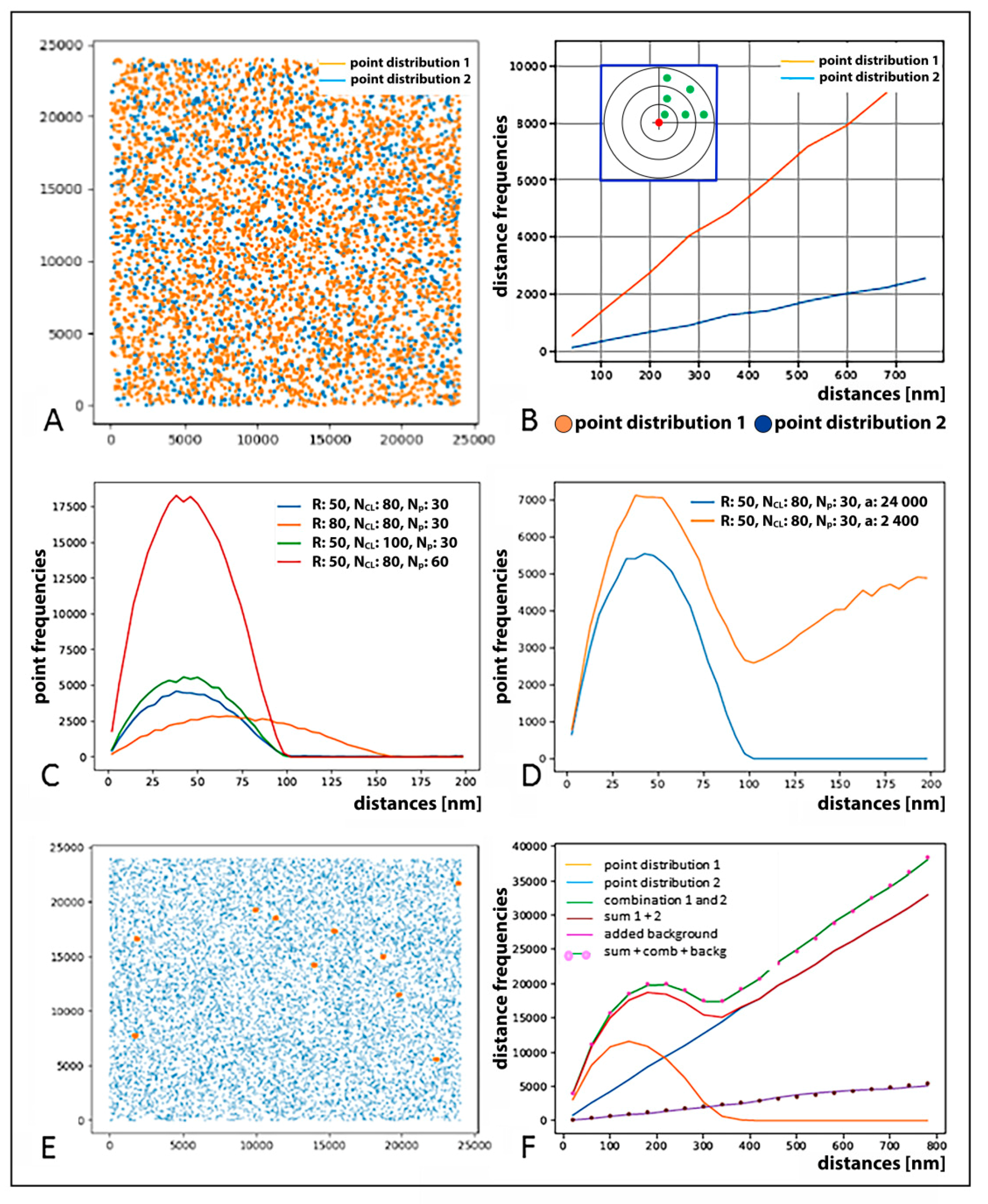{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
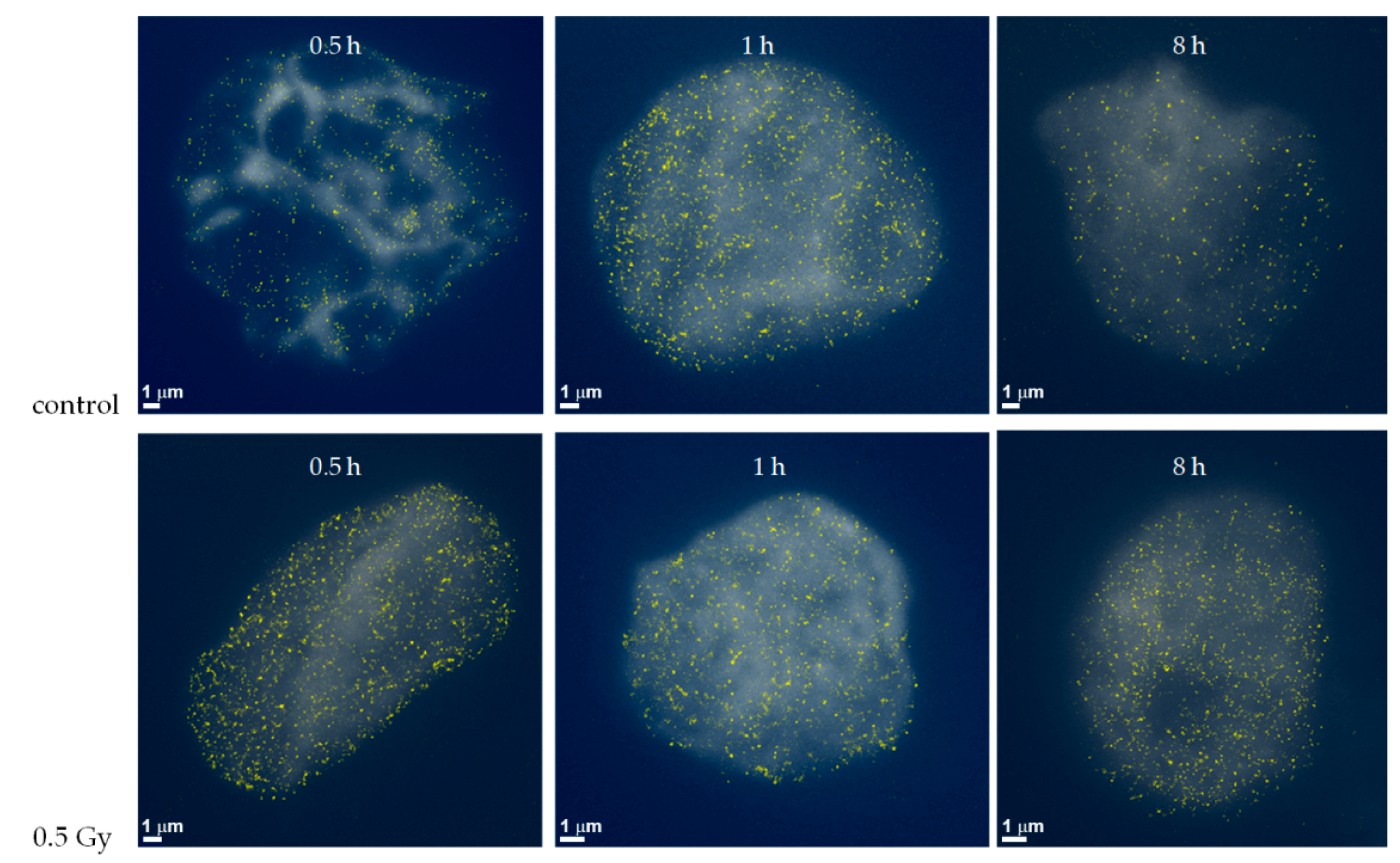
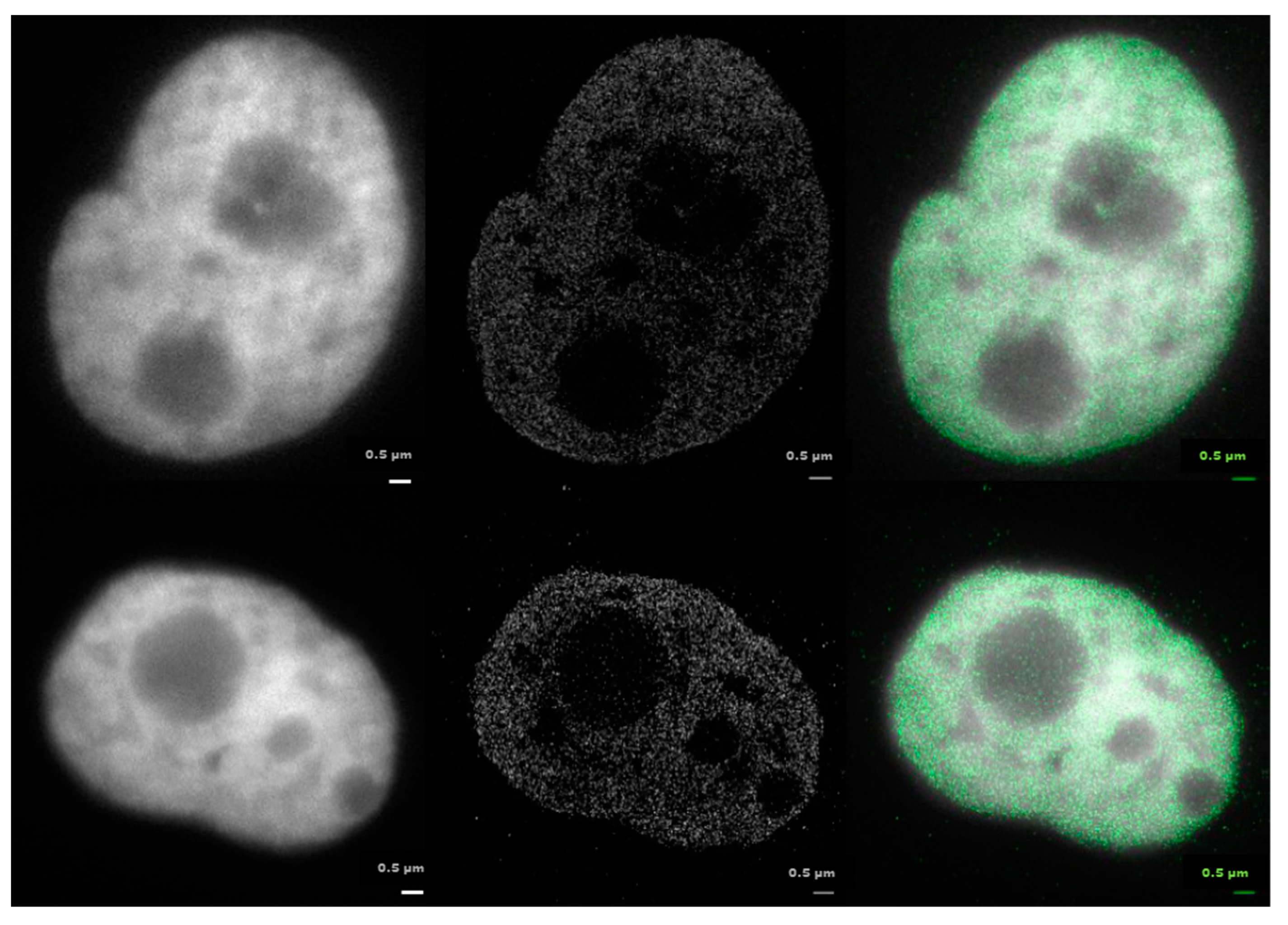
2.1. From Widefield Light Microscopy to SMLM Data Acquisition
2.2. SMLM Data Simulations and Interpretation
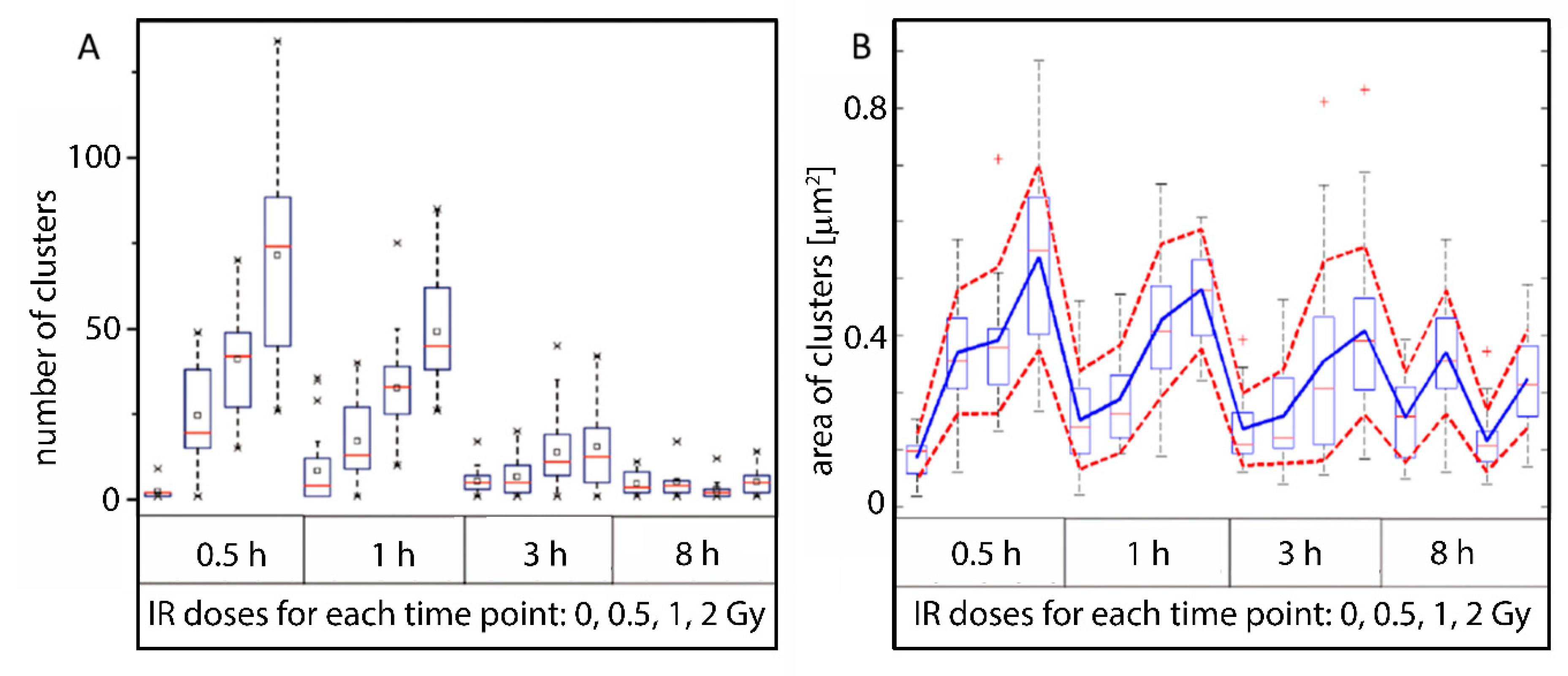
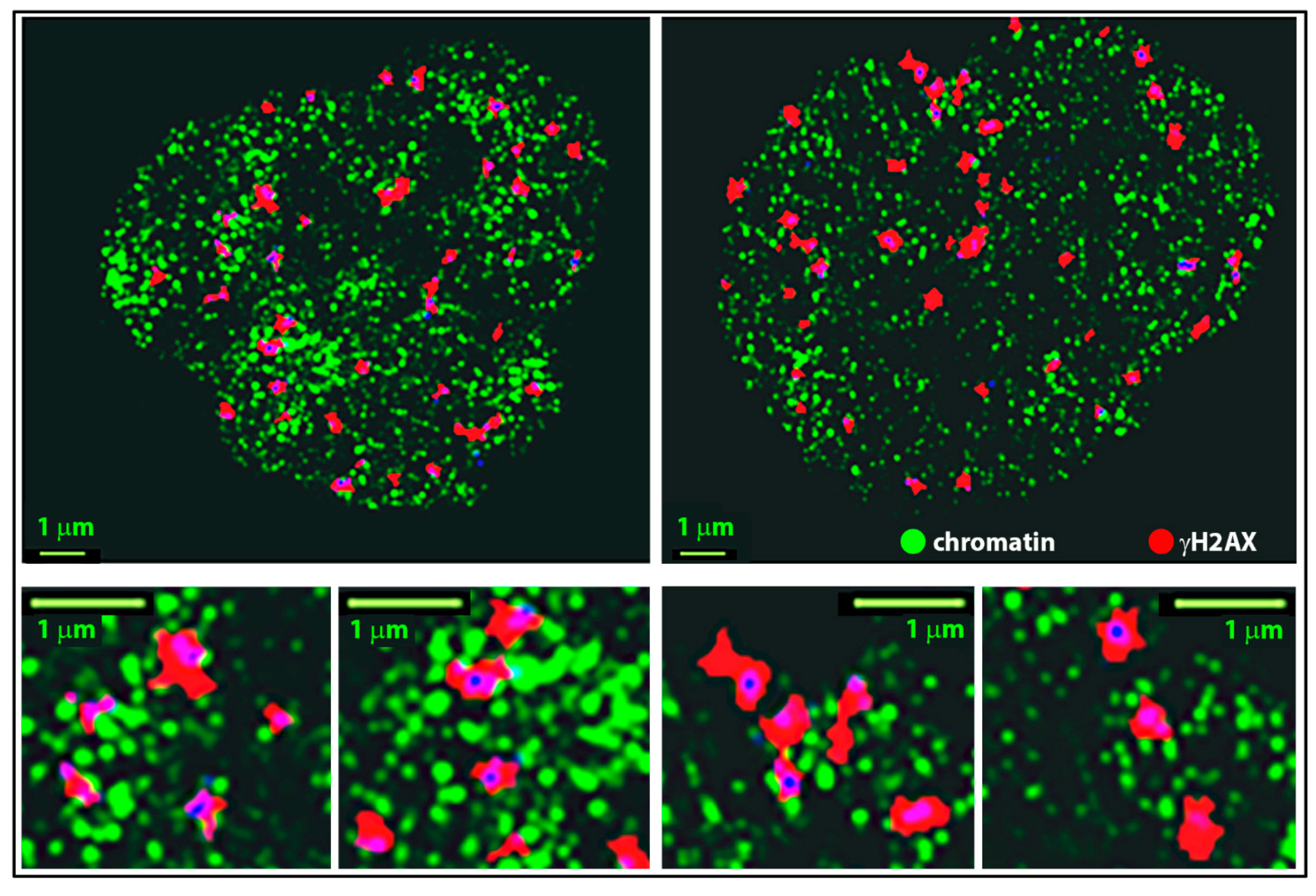
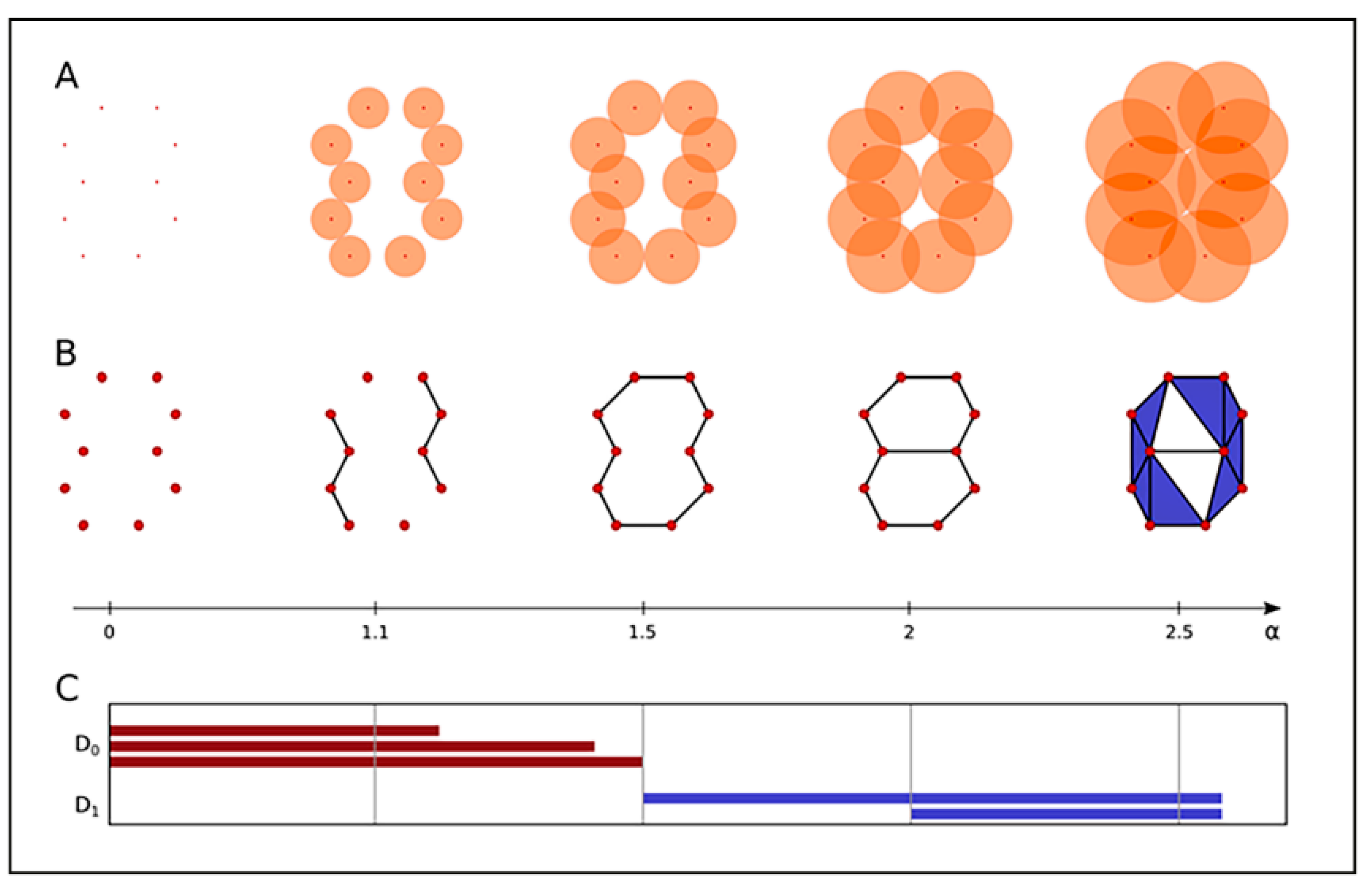
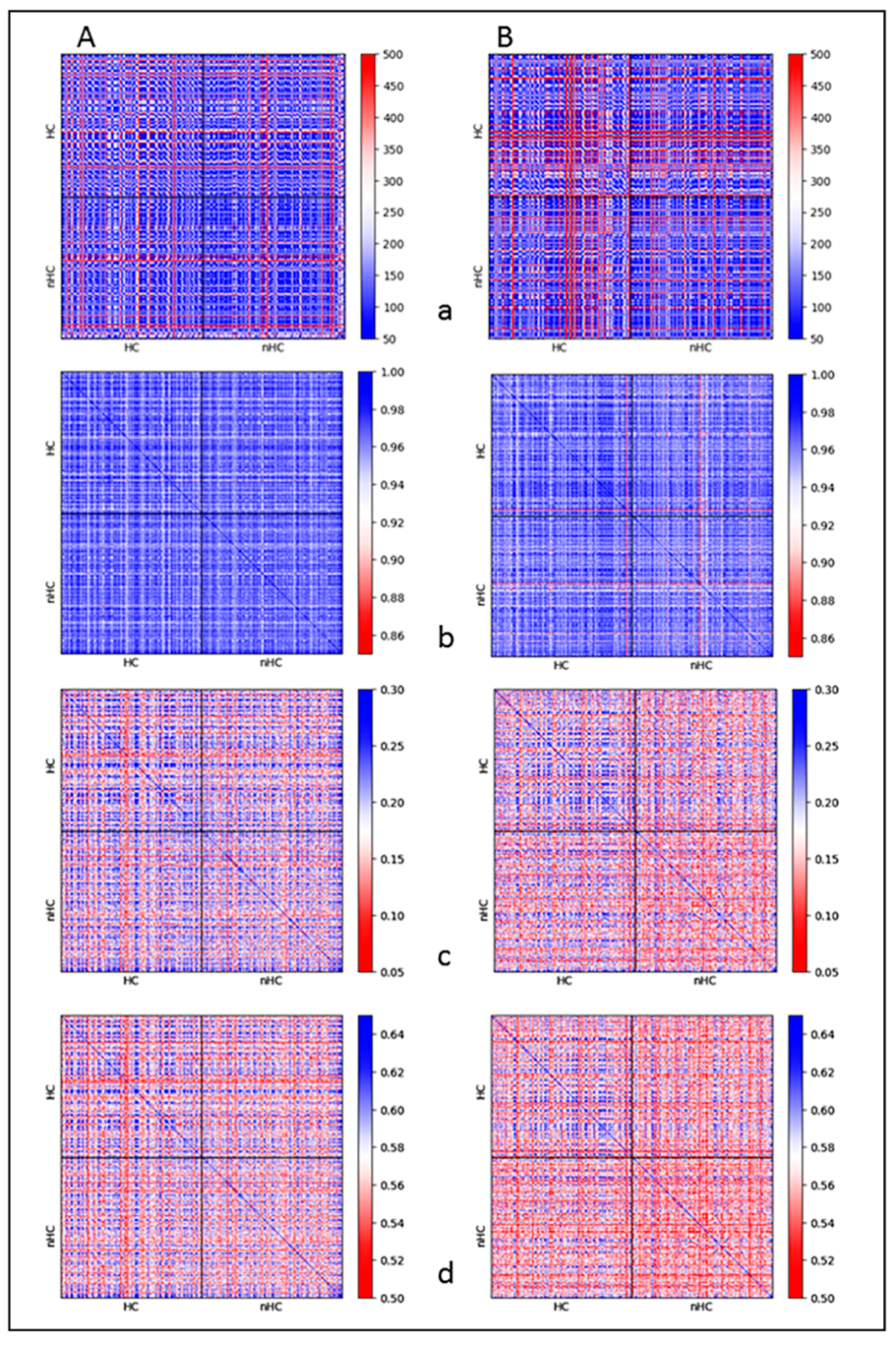
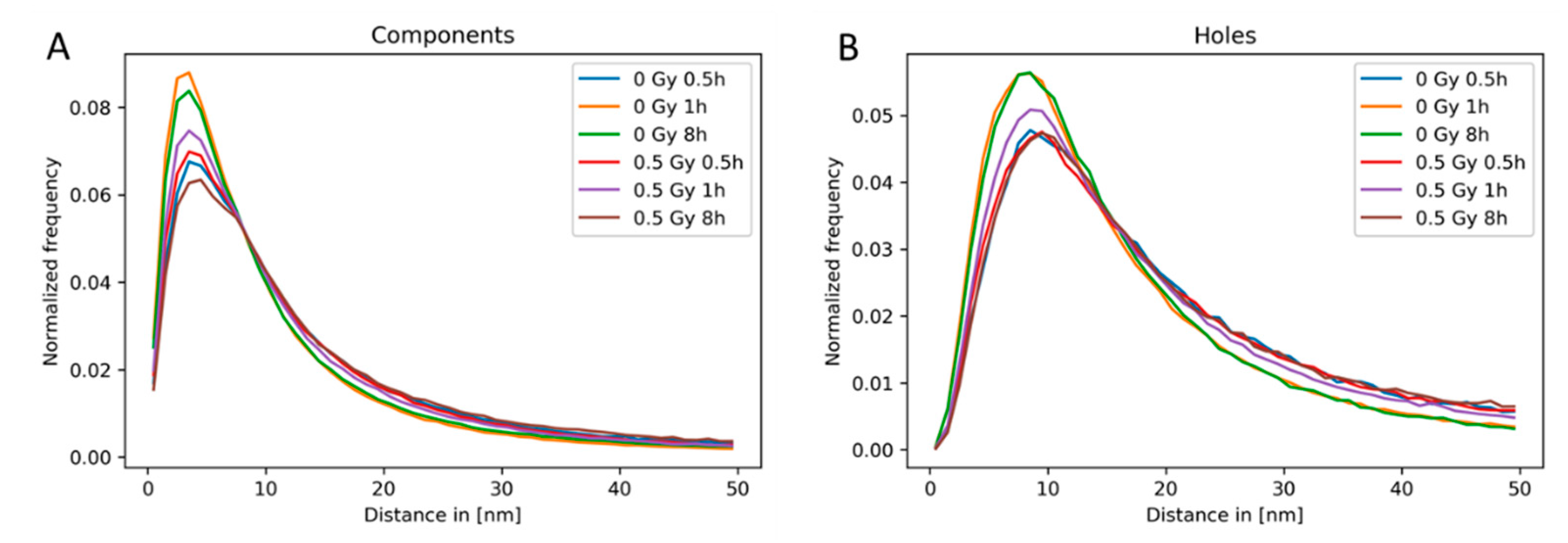
2.3. Clustering and Persistent Homologies of γH2AX Foci and Heterochromatin
2.4. Clustering of Nucleosomes upon Cell Exposure to Different Radiation Quality
3. Discussion
4. Materials and Methods
4.1. Cell Culturing and Preparation for Irradiation
4.2. Irradiation
4.3. Single Molecule Localization Microscopy (SMLM)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woods, D.; Turchi, J.J. Chemotherapy Induced DNA Damage Response: Convergence of Drugs and Pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef]
- Monajembashi, S.; Rapp, A.; Schmitt, E.; Dittmar, H.; Greulich, K.-O.; Hausmann, M. Spatial Association of Homologous Pericentric Regions in Human Lymphocyte Nuclei during Repair. Biophys. J. 2005, 88, 2309–2322. [Google Scholar] [CrossRef]
- Rittich, B.; Spanová, A.; Falk, M.; Benes, M.J.; Hrubý, M. Cleavage of Double Stranded Plasmid DNA by Lanthanide Complexes. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 800, 169–173. [Google Scholar] [CrossRef]
- Falk, M.; Falková, I.; Kopečná, O.; Bačíková, A.; Pagáčová, E.; Šimek, D.; Golan, M.; Kozubek, S.; Pekarová, M.; Follett, S.E.; et al. Chromatin Architecture Changes and DNA Replication Fork Collapse Are Critical Features in Cryopreserved Cells That Are Differentially Controlled by Cryoprotectants. Sci. Rep. 2018, 8, 14694. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Arnaudeau, C.; Lundin, C.; Helleday, T. DNA Double-Strand Breaks Associated with Replication Forks Are Predominantly Repaired by Homologous Recombination Involving an Exchange Mechanism in Mammalian Cells11Edited by J. Karn. J. Mol. Biol. 2001, 307, 1235–1245. [Google Scholar] [CrossRef]
- Lukášová, E.; Kovarˇík, A.; Bacˇíková, A.; Falk, M.; Kozubek, S. Loss of Lamin B Receptor Is Necessary to Induce Cellular Senescence. Biochem. J. 2017, 474, 281–300. [Google Scholar] [CrossRef]
- Pilarczyk, G.; Papenfuß, F.; Bestvater, F.; Hausmann, M. Spatial Arrangements of Connexin43 in Cancer Related Cells and Re-Arrangements under Treatment Conditions: Investigations on the Nano-Scale by Super-Resolution Localization Light Microscopy. Cancers 2019, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Pilarczyk, G.; Nesnidal, I.; Gunkel, M.; Bach, M.; Bestvater, F.; Hausmann, M. Localisation Microscopy of Breast Epithelial ErbB-2 Receptors and Gap Junctions: Trafficking after γ-Irradiation, Neuregulin-1β, and Trastuzumab Application. Int. J. Mol. Sci. 2017, 18, 362. [Google Scholar] [CrossRef]
- Falk, M.; Lukasova, E.; Kozubek, S. Higher-Order Chromatin Structure in DSB Induction, Repair and Misrepair. Mutat. Res. 2010, 704, 88–100. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA Double-Strand Break Repair during the Mammalian Cell Cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB Repair Pathway Choice: An Orchestrated Handover Mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; et al. Factors Determining DNA Double-Strand Break Repair Pathway Choice in G2 Phase: DSB Repair Pathway Choice in G2 Phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA Repair Mechanisms. Maturitas 2001, 38, 17–22; discussion 22–23. [Google Scholar] [CrossRef]
- Brandsma, I.; Gent, D.C. Pathway Choice in DNA Double Strand Break Repair: Observations of a Balancing Act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA Double Strand Break Repair via Non-Homologous End-Joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Falk, M.; Hausmann, M. A Paradigm Revolution or Just Better Resolution–Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation? Cancers 2021, 13, 18. [Google Scholar] [CrossRef]
- Yang, K.; Guo, R.; Xu, D. Non-Homologous End Joining: Advances and Frontiers. Acta Biochim. Biophys. Sin. 2016, 48, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Krasieva, T.B.; Kurumizaka, H.; Chen, D.J.; Taylor, A.M.R.; Yokomori, K. Independent and Sequential Recruitment of NHEJ and HR Factors to DNA Damage Sites in Mammalian Cells. J. Cell Biol. 2005, 170, 341–347. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.A. Canonical DNA Non-Homologous End-Joining; Capacity versus Fidelity. Br. J. Radiol. 2020, 93, 20190966. [Google Scholar] [CrossRef] [PubMed]
- Groth, P.; Orta, M.L.; Elvers, I.; Majumder, M.M.; Lagerqvist, A.; Helleday, T. Homologous Recombination Repairs Secondary Replication Induced DNA Double-Strand Breaks after Ionizing Radiation. Nucleic Acids Res. 2012, 40, 6585–6594. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Cromie, G.A.; Connelly, J.C.; Leach, D.R. Recombination at Double-Strand Breaks and DNA Ends: Conserved Mechanisms from Phage to Humans. Mol. Cell 2001, 8, 1163–1174. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Alternative Pathways of Non-Homologous End Joining (NHEJ) in Genomic Instability and Cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar] [CrossRef]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhász, S.; Künzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Löbrich, M. DNA Double-Strand Break Resection Occurs during Non-Homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef]
- Decottignies, A. Alternative End-Joining Mechanisms: A Historical Perspective. Front. Genet. 2013, 4, 48. [Google Scholar] [CrossRef]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G. Backup Pathways of NHEJ in Cells of Higher Eukaryotes: Cell Cycle Dependence. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2009, 92, 310–315. [Google Scholar] [CrossRef]
- Seol, J.-H.; Shim, E.Y.; Lee, S.E. Microhomology-Mediated End Joining: Good, Bad and Ugly. Mutat. Res. Mol. Mech. Mutagen. 2018, 809, 81–87. [Google Scholar] [CrossRef]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular Basis of Microhomology-Mediated End-Joining by Purified Full-Length Polθ. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Janssen, A.; Colmenares, S.U.; Lee, T.; Karpen, G.H. Timely Double-Strand Break Repair and Pathway Choice in Pericentromeric Heterochromatin Depend on the Histone Demethylase DKDM4A. Genes Dev. 2019, 33, 103–115. [Google Scholar] [CrossRef]
- Clouaire, T.; Legube, G. DNA Double Strand Break Repair Pathway Choice: A Chromatin Based Decision? Nucleus 2015, 6, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Baer, R.; Gautier, J. DNA Double-Strand Break Repair Pathway Choice and Cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef]
- Schwarz, B.; Friedl, A.A.; Girst, S.; Dollinger, G.; Reindl, J. Nanoscopic Analysis of 53BP1, BRCA1 and Rad51 Reveals New Insights in Temporal Progression of DNA-Repair and Pathway Choice. Mutat. Res. 2019, 816–818, 111675. [Google Scholar] [CrossRef]
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef]
- Jachimowicz, R.D.; Goergens, J.; Reinhardt, H.C. DNA Double-Strand Break Repair Pathway Choice-from Basic Biology to Clinical Exploitation. Cell Cycle 2019, 18, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, S. Targeting DNA Double-Strand Break (DSB) Repair to Counteract Tumor Radio-Resistance. Curr. Drug Targets 2019, 20, 891–902. [Google Scholar] [CrossRef]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation Induced Chromatin Conformation Changes Analysed by Fluorescent Localization Microscopy, Statistical Physics, and Graph Theory. PLoS ONE 2015, 10, e0128555. [Google Scholar] [CrossRef]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome Maintenance in Response to DNA Damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef] [PubMed]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Epimutation and Cancer: A New Carcinogenic Mechanism of Lynch Syndrome. Int. J. Oncol. 2012, 41, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Machnik, M.; Oleksiewicz, U. Dynamic Signatures of the Epigenome: Friend or Foe? Cells 2020, 9, 653. [Google Scholar] [CrossRef]
- Ježková, L.; Falk, M.; Falková, I.; Davídková, M.; Bačíková, A.; Štefančíková, L.; Vachelová, J.; Michaelidesová, A.; Lukášová, E.; Boreyko, A.; et al. Function of Chromatin Structure and Dynamics in DNA Damage, Repair and Misrepair: γ-Rays and Protons in Action. Appl. Radiat. Isot. Data Instrum. Methods Use Agric. Ind. Med. 2014, 83 Pt B, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA Damage Concentrated in Particle Trajectories Causes Persistent Large-Scale Rearrangements in Chromatin Architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef]
- Jakob, B.; Dubiak-Szepietowska, M.; Janiel, E.; Schmidt, A.; Durante, M.; Taucher-Scholz, G. Differential Repair Protein Recruitment at Sites of Clustered and Isolated DNA Double-Strand Breaks Produced by High-Energy Heavy Ions. Sci. Rep. 2020, 10, 1443. [Google Scholar] [CrossRef]
- Falk, M.; Hausmann, M.; Lukášová, E.; Biswas, A.; Hildenbrand, G.; Davídková, M.; Krasavin, E.; Kleibl, Z.; Falková, I.; Ježková, L.; et al. Determining Omics Spatiotemporal Dimensions Using Exciting New Nanoscopy Techniques to Assess Complex Cell Responses to DNA Damage: Part A—Radiomics. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Hausmann, M.; Lukášová, E.; Biswas, A.; Hildenbrand, G.; Davídková, M.; Krasavin, E.; Kleibl, Z.; Falková, I.; Ježková, L.; et al. Determining Omics Spatiotemporal Dimensions Using Exciting New Nanoscopy Techniques to Assess Complex Cell Responses to DNA Damage: Part B—Structuromics. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 225–247. [Google Scholar] [CrossRef]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Local Changes of Higher-Order Chromatin Structure during DSB-Repair. J. Phys. Conf. Ser. 2008. [Google Scholar] [CrossRef]
- Depes, D.; Lee, J.-H.; Bobkova, E.; Jezkova, L.; Falkova, I.; Bestvater, F.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; et al. Single-Molecule Localization Microscopy as a Promising Tool for ΓH2AX/53BP1 Foci Exploration. Eur. Phys. J. D 2018, 72, 1–11. [Google Scholar] [CrossRef]
- Bobkova, E.; Depes, D.; Lee, J.-H.; Jezkova, L.; Falkova, I.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; Kulikova, E.; et al. Recruitment of 53BP1 Proteins for DNA Repair and Persistence of Repair Clusters Differ for Cell Types as Detected by Single Molecule Localization Microscopy. Int. J. Mol. Sci. 2018, 19, 3713. [Google Scholar] [CrossRef]
- Fabre, E.; Zimmer, C. From Dynamic Chromatin Architecture to DNA Damage Repair and Back. Nucleus 2018, 9, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Eberle, J.P.; Rapp, A.; Krufczik, M.; Eryilmaz, M.; Gunkel, M.; Erfle, H.; Hausmann, M. Super-Resolution Microscopy Techniques and Their Potential for Applications in Radiation Biophysics; Methods in Molecular Biology; Humana Press Inc. of Springer Science and Business Media: Totowa, NJ, USA, 2017; Volume 1663, ISBN 10643745 (ISSN). [Google Scholar]
- Hausmann, M.; Wagner, E.; Lee, J.-H.; Schrock, G.; Schaufler, W.; Krufczik, M.; Papenfuß, F.; Port, M.; Bestvater, F.; Scherthan, H. Super-Resolution Localization Microscopy of Radiation-Induced Histone H2AX-Phosphorylation in Relation to H3K9-Trimethylation in HeLa Cells. Nanoscale 2018, 10, 4320–4331. [Google Scholar] [CrossRef]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 Recognition by BRCA1–BARD1 Directs Homologous Recombination to Sister Chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Krigerts, J.; Salmina, K.; Freivalds, T.; Zayakin, P.; Rumnieks, F.; Inashkina, I.; Giuliani, A.; Hausmann, M.; Erenpreisa, J. Differentiating breast cancer cells reveal early large-scale genome regulation by pericentric domains. Biophys. J. 2021, 120, 711–724. [Google Scholar] [CrossRef]
- Hausmann, M.; Neitzel, C.; Bobkova, E.; Nagel, D.; Hofmann, A.; Chramko, T.; Smirnova, E.; Kopečná, O.; Pagáčová, E.; Boreyko, A.; et al. Single Molecule Localization Microscopy Analyses of DNA-Repair Foci and Clusters Detected along Particle Damage Tracks. Front. Phys. Sect. Med. Phys. Imaging 2020, 8, 578662. [Google Scholar] [CrossRef]
- Lorat, Y.; Timm, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered Double-Strand Breaks in Heterochromatin Perturb DNA Repair after High Linear Energy Transfer Irradiation. Radiother. Oncol. 2016, 121, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Lorat, Y.; Brunner, C.U.; Schanz, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Nanoscale Analysis of Clustered DNA Damage after High-LET Irradiation by Quantitative Electron Microscopy–The Heavy Burden to Repair. DNA Repair 2015, 28, 93–106. [Google Scholar] [CrossRef]
- Ayache, J.; Beaunier, L.; Boumendil, J.; Ehret, G.; Laub, D. Artifacts in Transmission Electron Microscopy. In Sample Preparation Handbook for Transmission Electron Microscopy; Springer: New York, NY, USA, 2010; pp. 125–170. ISBN 978-0-387-98181-9. [Google Scholar]
- Cremer, C. Optics Far Beyond the Diffraction Limit. In Springer Handbook of Lasers and Optics; Träger, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1359–1397. ISBN 978-3-642-19408-5. [Google Scholar]
- Cremer, C.; Masters, B.R. Resolution Enhancement Techniques in Microscopy. Eur. Phys. J. H 2013, 38, 281–344. [Google Scholar] [CrossRef]
- Hausmann, M.; Ilić, N.; Pilarczyk, G.; Lee, J.-H.; Logeswaran, A.; Borroni, A.; Krufczik, M.; Theda, F.; Waltrich, N.; Bestvater, F.; et al. Challenges for Super-Resolution Localization Microscopy and Biomolecular Fluorescent Nano-Probing in Cancer Research. Int. J. Mol. Sci. 2017, 18, 2066. [Google Scholar] [CrossRef]
- Du, G.; Drexler, G.A.; Friedland, W.; Greubel, C.; Hable, V.; Krücken, R.; Kugler, A.; Tonelli, L.; Friedl, A.A.; Dollinger, G. Spatial Dynamics of DNA Damage Response Protein Foci along the Ion Trajectory of High-LET Particles. Radiat. Res. 2011, 176, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Lopez Perez, R.; Best, G.; Nicolay, N.H.; Greubel, C.; Rossberger, S.; Reindl, J.; Dollinger, G.; Weber, K.-J.; Cremer, C.; Huber, P.E. Superresolution Light Microscopy Shows Nanostructure of Carbon Ion Radiation-Induced DNA Double-Strand Break Repair Foci. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 2767–2776. [Google Scholar] [CrossRef]
- Varga, D.; Majoros, H.; Újfaludi, Z.; Erdélyi, M.; Pankotai, T. Quantification of DNA Damage Induced γH2AX Focus Formation via Super-Resolution dSTORM Localization Microscopy. Nanoscale 2019, 11, 14226–14236. [Google Scholar] [CrossRef]
- Cremer, C.; Edelmann, P.; Esa, A.; Bornfleth, H.; Schneider, B.; BradI, J.; Rinke, B.; Trakhtenbrot, L.; Dietzel, S.; Hausmann, M.; et al. Spektrale Präzisionsdistanzmikroskopie in Der Genomforschung. Z. Med. Phys. 1999, 9, 14–20. [Google Scholar] [CrossRef]
- Esa, A.; Edelmann, P.; Kreth, G.; Trakhtenbrot, L.; Amariglio, N.; Rechavi, B.G.; Hausmann, B.M.; Cremer, C. Three-Dimensional Spectral Precision Distance Microscopy of Chromatin Nanostructures after Triple-Colour DNA Labelling: A Study of the BCR Region on Chromosome 22 and the Philadelphia Chromosome. J. Microsc. 2000, 199, 96–105. [Google Scholar] [CrossRef]
- Rauch, J.; Knoch, T.A.; Solovei, I.; Teller, K.; Stein, S.; Buiting, K.; Horsthemke, B.; Langowski, J.; Cremer, T.; Hausmann, M.; et al. Light Optical Precision Measurements of the Active and Inactive Prader–Willi Syndrome Imprinted Regions in Human Cell Nuclei. Differentiation 2008, 76, 66–82. [Google Scholar] [CrossRef]
- Lemmer, P.; Gunkel, M.; Baddeley, D.; Kaufmann, R.; Urich, A.; Weiland, Y.; Reymann, J.; Müller, P.; Hausmann, M.; Cremer, C. SPDM: Light Microscopy with Single-Molecule Resolution at the Nanoscale. Appl. Phys. B 2008, 93, 1–12. [Google Scholar] [CrossRef]
- Hendrix, J.; Flors, C.; Dedecker, P.; Hofkens, J.; Engelborghs, Y. Dark States in Monomeric Red Fluorescent Proteins Studied by Fluorescence Correlation and Single Molecule Spectroscopy. Biophys. J. 2008, 94, 4103–4113. [Google Scholar] [CrossRef]
- Sinnecker, D.; Voigt, P.; Hellwig, N.; Schaefer, M. Reversible Photobleaching of Enhanced Green Fluorescent Proteins. Biochemistry 2005, 44, 7085–7094. [Google Scholar] [CrossRef]
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef]
- Deschout, H.; Zanacchi, F.C.; Mlodzianoski, M.; Diaspro, A.; Bewersdorf, J.; Hess, S.T.; Braeckmans, K. Precisely and Accurately Localizing Single Emitters in Fluorescence Microscopy. Nat. Methods 2014, 11, 253–266. [Google Scholar] [CrossRef]
- Hofmann, A.; Krufczik, M.; Heermann, D.W.; Hausmann, M. Using Persistent Homology as a New Approach for Super-Resolution Localization Microscopy Data Analysis and Classification of ΓH2AX Foci/Clusters. Int. J. Mol. Sci. 2018, 19, 2263. [Google Scholar] [CrossRef]
- Abbe, E. Beiträge Zur Theorie Des Mikroskops Und Der MikroskopischenWahrnehmung. Arch. F Mikrosk. Anat. 1873, 9, 411–468. [Google Scholar] [CrossRef]
- Lee, J.H.; Tchetgna, F.L.; Krufczik, M.; Schmitt, E.; Cremer, C.; Bestvater, F.; Hausmann, M. COMBO-FISH: A Versatile Tool Beyond Standard FISH to Study Chromatin Organization by Fluorescence Light Microscopy. OBM Genet. 2018, 3, 28. [Google Scholar] [CrossRef]
- Ripley, B.D. Modelling Spatial Patterns. J. R. Stat. Soc. Ser. B Methodol. 1977, 39, 172–192. [Google Scholar] [CrossRef]
- Cremer, M.; Hase, J.V.; Volm, T.; Brero, A.; Kreth, G.; Walter, J.; Fischer, C.; Solovei, I.; Cremer, C.; Cremer, T. Non-Random Radial Higher-Order Chromatin Arrangements in Nuclei of Diploid Human Cells. Chromosome Res. 2001, 9, 541–567. [Google Scholar] [CrossRef] [PubMed]
- Kozubek, S.; Lukásová, E.; Jirsová, P.; Koutná, I.; Kozubek, M.; Ganová, A.; Bártová, E.; Falk, M.; Paseková, R. 3D Structure of the Human Genome: Order in Randomness. Chromosoma 2002, 111, 321–331. [Google Scholar] [CrossRef]
- Falk, M.; Lukásová, E.; Kozubek, S.; Kozubek, M. Topography of Genetic Elements of X-Chromosome Relative to the Cell Nucleus and to the Chromosome X Territory Determined for Human Lymphocytes. Gene 2002, 292, 13–24. [Google Scholar] [CrossRef]
- Huang, K.; Li, Y.; Shim, A.R.; Virk, R.K.A.; Agrawal, V.; Eshein, A.; Nap, R.J.; Almassalha, L.M.; Backman, V.; Szleifer, I. Physical and Data Structure of 3D Genome. Sci. Adv. 2020, 6, eaay4055. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin Structure Influences the Sensitivity of DNA to Gamma-Radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef]
- Radulescu, I.; Elmroth, K.; Stenerlöw, B. Chromatin Organization Contributes to Non-Randomly Distributed Double-Strand Breaks after Exposure to High-LET Radiation. Radiat. Res. 2004, 161, 1–8. [Google Scholar] [CrossRef]
- Lukásová, E.; Kozubek, S.; Kozubek, M.; Falk, M.; Amrichová, J. The 3D Structure of Human Chromosomes in Cell Nuclei. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2002, 10, 535–548. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, M. Chromosome Territories. Cold Spring Harb. Perspect. Biol. 2010, 2, a003889. [Google Scholar] [CrossRef]
- Aleksandrov, R.; Hristova, R.; Stoynov, S.; Gospodinov, A. The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells 2020, 9, 1853. [Google Scholar] [CrossRef]
- Kleppe, A.; Albregtsen, F.; Vlatkovic, L.; Pradhan, M.; Nielsen, B.; Hveem, T.S.; Askautrud, H.A.; Kristensen, G.B.; Nesbakken, A.; Trovik, J.; et al. Chromatin Organisation and Cancer Prognosis: A Pan-Cancer Study. Lancet Oncol. 2018, 19, 356–369. [Google Scholar] [CrossRef]
- Máté, G.; Hofmann, A.; Wenzel, N.; Heermann, D.W. A Topological Similarity Measure for Proteins. Biochim. Biophys. Acta BBA Biomembr. 2014, 1838, 1180–1190. [Google Scholar] [CrossRef]
- Braden, B. The Surveyor’s Area Formula. Coll. Math. J. 1986, 17, 326–337. [Google Scholar] [CrossRef]
- Ghrist, R. Barcodes: The Persistent Topology of Data. Bull. Am. Math. Soc. 2007, 45, 61–76. [Google Scholar] [CrossRef]
- Jaccard, P. Etude Comparative de La Distribution Florale Dans Une Portion Des Alpes et Des Jura. Bull. Soc. Vaud. Sci. Nat. 1901, 37, 547–579. [Google Scholar]
- Rouvreau, V. Cython Interface. In GUDHI Editorial Board, 3.3.0, GUDHI User and Reference Manual. 2020. Available online: https://gudhi.inria.fr/doc/3.3.0/ (accessed on 11 January 2021).
- Máté, G.; Heermann, D.W. Statistical Analysis of Protein Ensembles. Front. Phys. 2014, 2, 20. [Google Scholar] [CrossRef]
- Knoch, T.A. Approaching the Three-Dimensional Organization of the Human Genome. Ph.D. Thesis, Faculty of Physics and Astronomy, Heidelberg University, Heidelberg, Germany, 2003. [Google Scholar] [CrossRef]
- Lukášová, E.; Kozubek, S.; Falk, M.; Kozubek, M.; Žaloudík, J.; Vagunda, V.; Pavlovský, Z. Topography of Genetic Loci in the Nuclei of Cells of Colorectal Carcinoma and Adjacent Tissue of Colonic Epithelium. Chromosoma 2004, 112, 221–230. [Google Scholar] [CrossRef]
- Bach, M.; Savini, C.; Krufczik, M.; Cremer, C.; Rösl, F.; Hausmann, M. Super-Resolution Localization Microscopy of γ-H2AX and Heterochromatin after Folate Deficiency. Int. J. Mol. Sci. 2017, 18, 1726. [Google Scholar] [CrossRef]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA Damage Response: Spatiotemporal Relationships among Checkpoint and Repair Proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef]
- Scherthan, H.; Lee, J.-H.; Maus, E.; Schumann, S.; Muhtadi, R.; Chojowski, R.; Port, M.; Lassmann, M.; Bestvater, F.; Hausmann, M. Nanostructure of Clustered DNA Damage in Leukocytes after In-Solution Irradiation with the Alpha Emitter Ra-223. Cancers 2019, 11, 1877. [Google Scholar] [CrossRef] [PubMed]
- Sevcik, J.; Falk, M.; Kleiblova, P.; Lhota, F.; Stefancikova, L.; Janatova, M.; Weiterova, L.; Lukasova, E.; Kozubek, S.; Pohlreich, P.; et al. The BRCA1 Alternative Splicing Variant Δ14-15 with an in-Frame Deletion of Part of the Regulatory Serine-Containing Domain (SCD) Impairs the DNA Repair Capacity in MCF-7 Cells. Cell. Signal. 2012, 24, 1023–1030. [Google Scholar] [CrossRef]
- Sevcik, J.; Falk, M.; Macurek, L.; Kleiblova, P.; Lhota, F.; Hojny, J.; Stefancikova, L.; Janatova, M.; Bartek, J.; Stribrna, J.; et al. Expression of Human BRCA1Δ17-19 Alternative Splicing Variant with a Truncated BRCT Domain in MCF-7 Cells Results in Impaired Assembly of DNA Repair Complexes and Aberrant DNA Damage Response. Cell. Signal. 2013, 25, 1186–1193. [Google Scholar] [CrossRef]
- Jezkova, L.; Zadneprianetc, M.; Kulikova, E.; Smirnova, E.; Bulanova, T.; Depes, D.; Falkova, I.; Boreyko, A.; Krasavin, E.; Davidkova, M.; et al. Particles with Similar LET Values Generate DNA Breaks of Different Complexity and Reparability: A High-Resolution Microscopy Analysis of Γh2AX/53BP1 Foci. Nanoscale 2018, 10, 1162–1179. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Falk, M.; Komůrková, D.; Falková, I.; Bačíková, A.; Klejdus, B.; Pagáčová, E.; Štefančíková, L.; Weiterová, L.; Angelis, K.J.; et al. Two New Faces of Amifostine: Protector from DNA Damage in Normal Cells and Inhibitor of DNA Repair in Cancer Cells. J. Med. Chem. 2016, 59, 3003–3017. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukášová, E.; Štefančíková, L.; Baranová, E.; Falková, I.; Ježková, L.; Davídková, M.; Bačíková, A.; Vachelová, J.; Michaelidesová, A.; et al. Heterochromatinization Associated with Cell Differentiation as a Model to Study DNA Double Strand Break Induction and Repair in the Context of Higher-Order Chromatin Structure. Appl. Radiat. Isot. Data Instrum. Methods Use Agric. Ind. Med. 2014, 83 Pt B, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lukášová, E.; Kořistek, Z.; Klabusay, M.; Ondřej, V.; Grigoryev, S.; Bačíková, A.; Řezáčová, M.; Falk, M.; Vávrová, J.; Kohútová, V.; et al. Granulocyte Maturation Determines Ability to Release Chromatin NETs and Loss of DNA Damage Response; These Properties Are Absent in Immature AML Granulocytes. Biochim. Biophys. Acta 2013, 1833, 767–779. [Google Scholar] [CrossRef]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Chromatin Dynamics during DSB Repair. Biochim. Biophys. Acta 2007, 1773, 1534–1545. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hausmann, M.; Falk, M.; Neitzel, C.; Hofmann, A.; Biswas, A.; Gier, T.; Falkova, I.; Heermann, D.W.; Hildenbrand, G. Elucidation of the Clustered Nano-Architecture of Radiation-Induced DNA Damage Sites and Surrounding Chromatin in Cancer Cells: A Single Molecule Localization Microscopy Approach. Int. J. Mol. Sci. 2021, 22, 3636. https://doi.org/10.3390/ijms22073636
Hausmann M, Falk M, Neitzel C, Hofmann A, Biswas A, Gier T, Falkova I, Heermann DW, Hildenbrand G. Elucidation of the Clustered Nano-Architecture of Radiation-Induced DNA Damage Sites and Surrounding Chromatin in Cancer Cells: A Single Molecule Localization Microscopy Approach. International Journal of Molecular Sciences. 2021; 22(7):3636. https://doi.org/10.3390/ijms22073636
Chicago/Turabian StyleHausmann, Michael, Martin Falk, Charlotte Neitzel, Andreas Hofmann, Abin Biswas, Theresa Gier, Iva Falkova, Dieter W. Heermann, and Georg Hildenbrand. 2021. "Elucidation of the Clustered Nano-Architecture of Radiation-Induced DNA Damage Sites and Surrounding Chromatin in Cancer Cells: A Single Molecule Localization Microscopy Approach" International Journal of Molecular Sciences 22, no. 7: 3636. https://doi.org/10.3390/ijms22073636
APA StyleHausmann, M., Falk, M., Neitzel, C., Hofmann, A., Biswas, A., Gier, T., Falkova, I., Heermann, D. W., & Hildenbrand, G. (2021). Elucidation of the Clustered Nano-Architecture of Radiation-Induced DNA Damage Sites and Surrounding Chromatin in Cancer Cells: A Single Molecule Localization Microscopy Approach. International Journal of Molecular Sciences, 22(7), 3636. https://doi.org/10.3390/ijms22073636