
The Winged Helix Domain of CSB Regulates RNAPII Occupancy at Promoter Proximal Pause Sites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
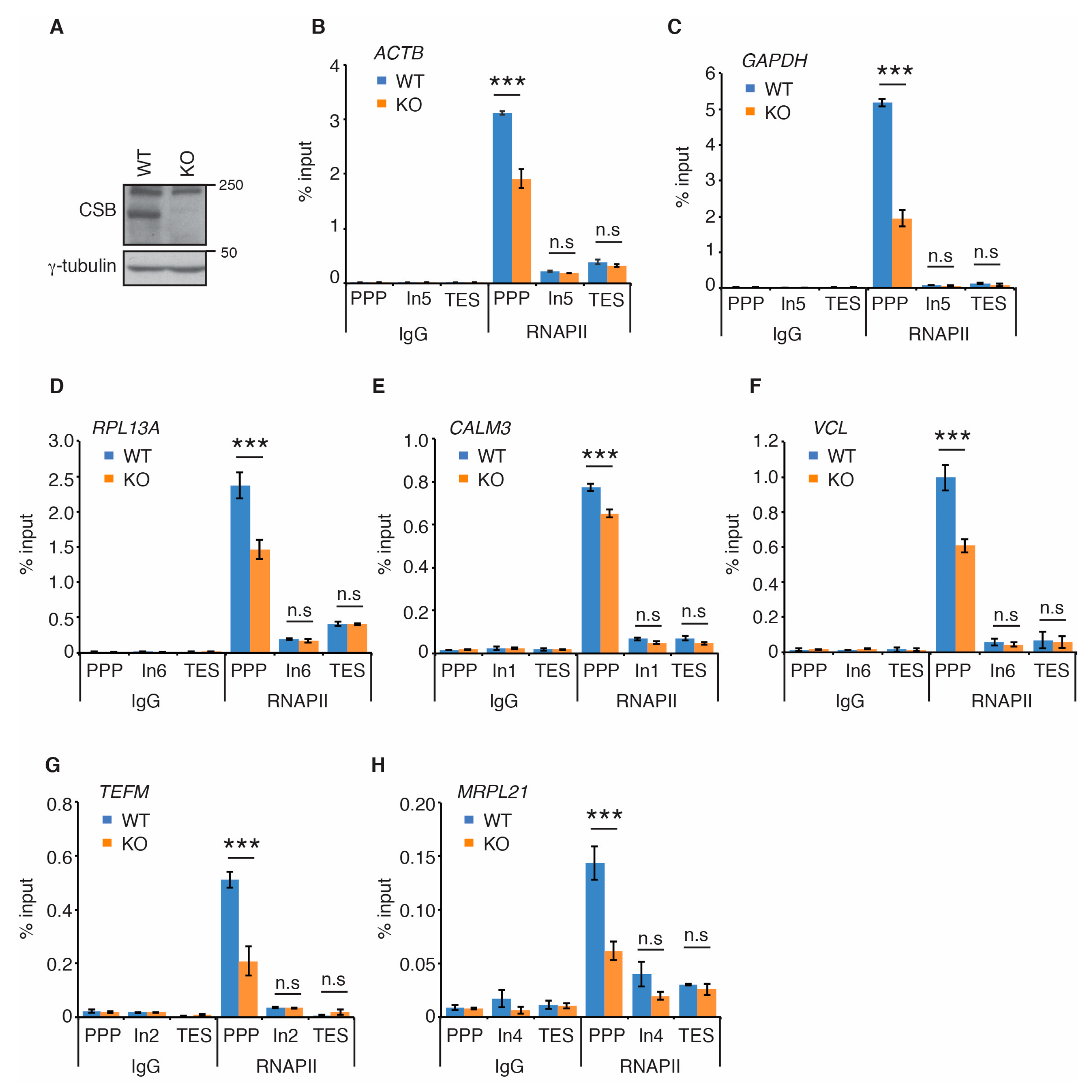
2.1. CSB Promotes RNAPII Accumulation at PPP Sites of Several Actively Transcribed Genes
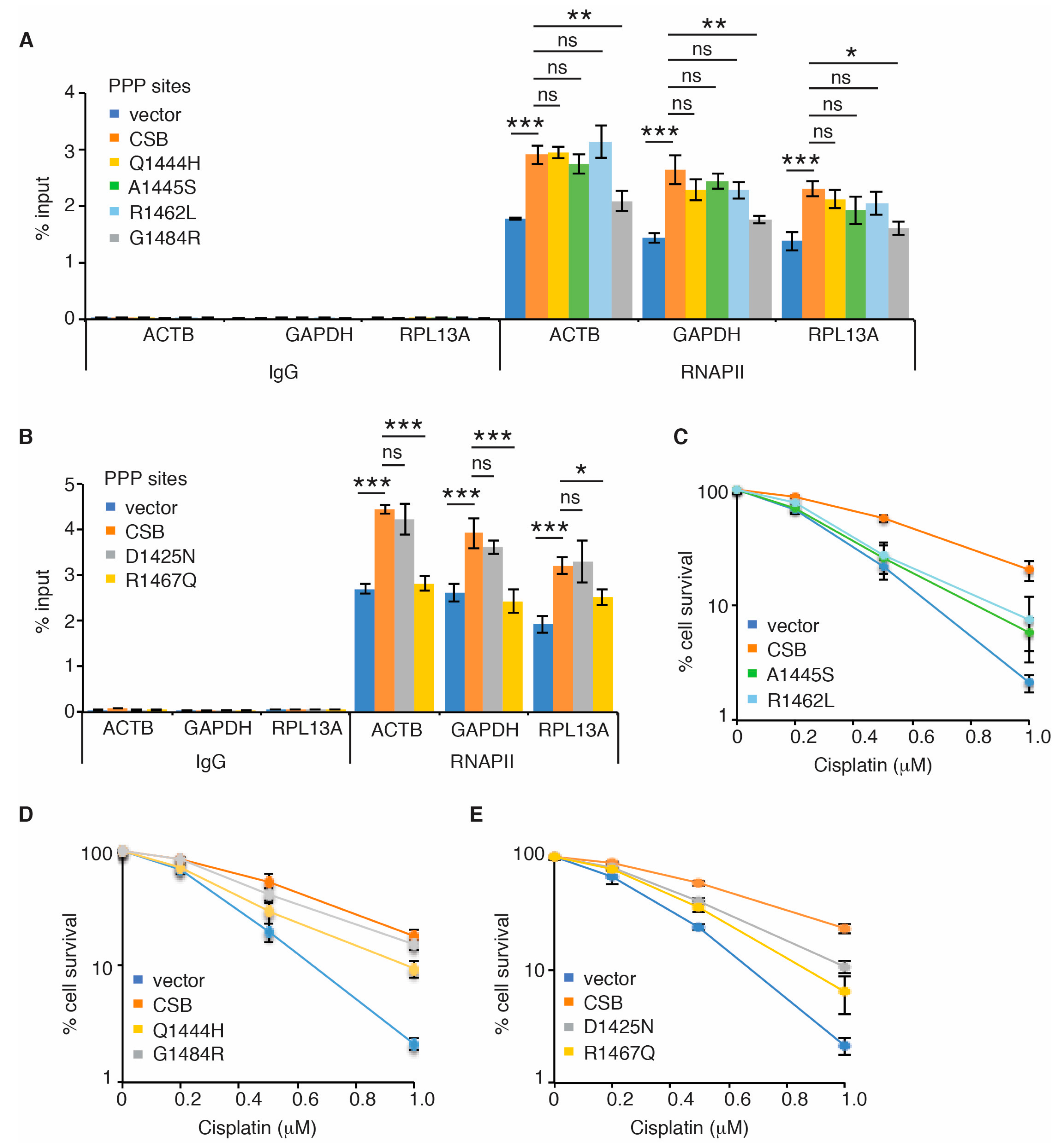
2.2. The WHD of CSB Is Necessary for Its Ability to Promote RNAPII Association with PPP Sites
2.3. Ubiquitin Binding-Defective Mutations of CSB Sensitize Cells to Cisplatin But Have No Effect on RNAPII Occupancy at PPP Sites
2.4. Cancer-Associated R1467Q and G1484R Mutations Impair CSB’s Ability to Promote RNAPII Occupancy at PPP Sites of ACTB, GAPDH and RPL13A Genes
3. Discussion
4. Materials and Methods
4.1. Plasmids and Antibodies
4.2. Cell Culture, Transfection, Retroviral Infection and Treatment
4.3. Chromatin Immunoprecipitation (ChIP)
4.4. Immunoblotting
4.5. Clonogenic Survival Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Troelstra, C.; van Gool, A.; de Wit, J.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 1992, 71, 939–953. [Google Scholar] [CrossRef]
- Van der Horst, G.T.; van Steeg, H.; Berg, R.J.; van Gool, A.J.; de Wit, J.; Weeda, G.; Morreau, H.; Beems, R.B.; van Kreijl, C.F.; de Gruijl, F.R.; et al. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell 1997, 89, 425–435. [Google Scholar] [CrossRef]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef]
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2009, 31, 113–126. [Google Scholar] [CrossRef]
- Calmels, N.; Botta, E.; Jia, N.; Fawcett, H.; Nardo, T.; Nakazawa, Y.; Lanzafame, M.; Moriwaki, S.; Sugita, K.; Kubota, M.; et al. Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J. Med. Genet. 2018, 55, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef]
- Balajee, A.S.; May, A.; Dianov, G.L.; Friedberg, E.C.; Bohr, V.A. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc. Natl. Acad. Sci. USA 1997, 94, 4306–4311. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Houle, J.-F.; Iyer, N.; Bohr, V.A.; Friedberg, E.C. Reduced RNA polymerase II transcription in extracts of cockayne syndrome and xeroderma pigmentosum/Cockayne syndrome cells. Nucleic Acids Res. 1997, 25, 3636–3642. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dianov, G.; Bischoff, C.; Sunesen, M.; Bohr, V.A. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 1999, 27, 1365–1368. [Google Scholar] [CrossRef]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; de Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M., III; Stevnsner, T.; et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010, 24, 2334–2346. [Google Scholar] [CrossRef]
- Berquist, B.R.; Canugovi, C.; Sykora, P.; Wilson, D.M., III; Bohr, V.A. Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic Acids Res. 2012, 40, 8392–8405. [Google Scholar] [CrossRef]
- Paccosi, E.; Costanzo, F.; Costantino, M.; Balzerano, A.; Monteonofrio, L.; Soddu, S.; Prantera, G.; Brancorsini, S.; Egly, J.-M.; Proietti-De-Santis, L. The Cockayne syndrome group A and B proteins are part of a ubiquitin–proteasome degradation complex regulating cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30498–30508. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Mitchell, T.R.H.; Leach, D.M.; Rainbow, A.J.; Zhu, X.-D. Cockayne Syndrome group B protein interacts with TRF2 and regulates telomere length and stability. Nucleic Acids Res. 2012, 40, 9661–9674. [Google Scholar] [CrossRef]
- Feng, E.; Batenburg, N.L.; Walker, J.R.; Ho, A.; Mitchell, T.R.H.; Qin, J.; Zhu, X.-D. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 2020, 133, jcs234914. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Walker, J.R.; Noordermeer, S.M.; Moatti, N.; Durocher, D.; Zhu, X.D. ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat. Commun. 2017, 8, 1921. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Walker, J.R.; Coulombe, Y.; Sherker, A.; Masson, J.Y.; Zhu, X.D. CSB interacts with BRCA1 in late S/G2 to promote MRN- and CtIP-mediated DNA end resection. Nucleic Acids Res. 2019, 47, 10678–10692. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Thompson, E.L.; Hendrickson, E.A.; Zhu, X.D. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015, 34, 1399–1416. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Bailey, A.D.; Weiner, A.M. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc. Natl. Acad. Sci. USA 2006, 103, 9613–9618. [Google Scholar] [CrossRef]
- Lake, R.J.; Boetefuer, E.L.; Tsai, P.-F.; Jeong, J.; Choi, I.; Won, K.-J.; Fan, H.-Y. The Sequence-Specific Transcription Factor c-Jun Targets Cockayne Syndrome Protein B to Regulate Transcription and Chromatin Structure. PLoS Genet. 2014, 10, e1004284. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, U.; Epanchintsev, A.; Rauschendorf, M.A.; Laugel, V.; Stevnsner, T.; Bohr, V.A.; Coin, F.; Egly, J.M. Regulatory interplay of Cockayne syndrome B ATPase and stress-response gene ATF3 following genotoxic stress. Proc. Natl. Acad. Sci. USA 2013, 110, E2261–E2270. [Google Scholar] [CrossRef]
- Epanchintsev, A.; Costanzo, F.; Rauschendorf, M.A.; Caputo, M.; Ye, T.; Donnio, L.M.; de Santis, L.P.; Coin, F.; Laugel, V.; Egly, J.M. Cockayne’s Syndrome A and B Proteins Regulate Transcription Arrest after Genotoxic Stress by Promoting ATF3 Degradation. Mol. Cell 2017, 68, 1054–1066. [Google Scholar] [CrossRef]
- Ciaffardini, F.; Nicolai, S.; Caputo, M.; Canu, G.; Paccosi, E.; Costantino, M.; Frontini, M.; Balajee, A.S.; Proietti-De-Santis, L. The cockayne syndrome B protein is essential for neuronal differentiation and neuritogenesis. Cell Death Dis. 2014, 5, e1268. [Google Scholar] [CrossRef]
- Wang, Y.; Chakravarty, P.; Ranes, M.; Kelly, G.; Brooks, P.J.; Neilan, E.; Stewart, A.; Schiavo, G.; Svejstrup, J.Q. Dysregulation of gene expression as a cause of Cockayne syndrome neurological disease. Proc. Natl. Acad. Sci. USA 2014, 111, 14454–14459. [Google Scholar] [CrossRef]
- Wang, Y.; Jones-Tabah, J.; Chakravarty, P.; Stewart, A.; Muotri, A.; Laposa, R.R.; Svejstrup, J.Q. Pharmacological Bypass of Cockayne Syndrome B Function in Neuronal Differentiation. Cell Rep. 2016, 14, 2554–2561. [Google Scholar] [CrossRef][Green Version]
- Van Gool, A.J.; Citterio, E.; Rademakers, S.; Van Os, R.; Vermeulen, W.; Constantinou, A.; Egly, J.-M.; Bootsma, D.; Hoeijmakers, J.H. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J. 1997, 16, 5955–5965. [Google Scholar] [CrossRef]
- Selby, C.P.; Sancar, A. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. USA 1997, 94, 11205–11209. [Google Scholar] [CrossRef] [PubMed]
- Tantin, D.; Kansal, A.; Carey, M. Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol. Cell. Biol. 1997, 17, 6803–6814. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef]
- Svejstrup, J.Q. Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem. Sci. 2007, 32, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Lainé, J.-P.; Egly, J.-M. Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J. 2006, 25, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Lindsey-Boltz, L.A.; Sancar, A. RNA polymerase: The most specific damage recognition protein in cellular responses to DNA damage? Proc. Natl. Acad. Sci. USA 2007, 104, 13213–13214. [Google Scholar] [CrossRef] [PubMed]
- Saxowsky, T.T.; Doetsch, P.W. RNA Polymerase Encounters with DNA Damage: Transcription-Coupled Repair or Transcriptional Mutagenesis? Chem. Rev. 2006, 106, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lahiri, I.; Wang, W.; Wier, A.; Cianfrocco, M.A.; Chong, J.; Hare, A.A.; Dervan, P.B.; DiMaio, F.; Leschziner, A.E.; et al. Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nat. Cell Biol. 2017, 551, 653–657. [Google Scholar] [CrossRef]
- Adelman, K.; Lis, J.T. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.S.; Sato, Y.; Yamagata, A.; Goto-Ito, S.; Saijo, M.; Fukai, S. Structural basis of ubiquitin recognition by the winged-helix domain of Cockayne syndrome group B protein. Nucleic Acids Res. 2019, 47, 3784–3794. [Google Scholar] [CrossRef]
- Sin, Y.; Tanaka, K.; Saijo, M. The C-terminal Region and SUMOylation of Cockayne Syndrome Group B Protein Play Critical Roles in Transcription-coupled Nucleotide Excision Repair. J. Biol. Chem. 2016, 291, 1387–1397. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Qin, J.; Walker, J.R.; Zhu, X.-D. Efficient UV repair requires disengagement of the CSB winged helix domain from the CSB ATPase domain. DNA Repair. 2018, 68, 58–67. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, G.; Li, W. Elevated Expression of ERCC6 Confers Resistance to 5-Fluorouracil and Is Associated with Poor Patient Survival in Colorectal Cancer. DNA Cell Biol. 2017, 36, 781–786. [Google Scholar] [CrossRef]
- Lu, Y.; Mani, S.; Kandimalla, E.; Yu, N.; Agrawal, S.; States, J.; Bregman, D. The Cockayne syndrome group B DNA repair protein as an anti-cancer target. Int. J. Oncol. 2001, 19, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Caputo, M.; Frontini, M.; Velez-Cruz, R.; Nicolai, S.; Prantera, G.; Proietti-De-Santis, L. The CSB repair factor is overexpressed in cancer cells, increases apoptotic resistance, and promotes tumor growth. DNA Repair. 2013, 12, 293–299. [Google Scholar] [CrossRef]
- Proietti-De-Santis, L.; Balzerano, A.; Prantera, G. CSB: An Emerging Actionable Target for Cancer Therapy. Trends Cancer 2018, 4, 172–175. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Renaudin, X.; Williams, C.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef]
- Geijer, M.E.; Marteijn, J.A. What happens at the lesion does not stay at the lesion: Transcription-coupled nucleotide excision repair and the effects of DNA damage on transcription in cis and trans. DNA Repair. 2018, 71, 56–68. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krishnamurthy, S. Cellular Responses to Cisplatin-Induced DNA Damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef]
- Selzer, R.R.; Nyaga, S.; Tuo, J.; May, A.; Muftuoglu, M.; Christiansen, M.; Citterio, E.; Brosh, R.M., Jr.; Bohr, V.A. Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res. 2002, 30, 782–793. [Google Scholar] [CrossRef]
- Ranes, M.; Boeing, S.; Wang, Y.; Wienholz, F.; Menoni, H.; Walker, J.; Encheva, V.; Chakravarty, P.; Mari, P.-O.; Stewart, A.; et al. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2016, 44, 5246–5255. [Google Scholar] [CrossRef]
- Wilson, F.R.; Ho, A.; Walker, J.R.; Zhu, X.-D. Cdk-dependent phosphorylation regulates TRF1 recruitment to PML bodies and promotes C-circle production in ALT cells. J. Cell Sci. 2016, 129, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xiao, S.; Zhu, X.-D. MRE11–RAD50–NBS1 and ATM function as co-mediators of TRF1 in telomere length control. Nat. Struct. Mol. Biol. 2007, 14, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Mitchell, T.R.; Zhu, X.-D. Human XPF controls TRF2 and telomere length maintenance through distinctive mechanisms. Mech. Ageing Dev. 2008, 129, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Kuster, B.; Mann, M.; Petrini, J.H.; Lange, T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat. Genet. 2000, 25, 347–352. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batenburg, N.L.; Cui, S.; Walker, J.R.; Schellhorn, H.E.; Zhu, X.-D. The Winged Helix Domain of CSB Regulates RNAPII Occupancy at Promoter Proximal Pause Sites. Int. J. Mol. Sci. 2021, 22, 3379. https://doi.org/10.3390/ijms22073379
Batenburg NL, Cui S, Walker JR, Schellhorn HE, Zhu X-D. The Winged Helix Domain of CSB Regulates RNAPII Occupancy at Promoter Proximal Pause Sites. International Journal of Molecular Sciences. 2021; 22(7):3379. https://doi.org/10.3390/ijms22073379
Chicago/Turabian StyleBatenburg, Nicole L., Shixin Cui, John R. Walker, Herb E. Schellhorn, and Xu-Dong Zhu. 2021. "The Winged Helix Domain of CSB Regulates RNAPII Occupancy at Promoter Proximal Pause Sites" International Journal of Molecular Sciences 22, no. 7: 3379. https://doi.org/10.3390/ijms22073379
APA StyleBatenburg, N. L., Cui, S., Walker, J. R., Schellhorn, H. E., & Zhu, X.-D. (2021). The Winged Helix Domain of CSB Regulates RNAPII Occupancy at Promoter Proximal Pause Sites. International Journal of Molecular Sciences, 22(7), 3379. https://doi.org/10.3390/ijms22073379