
Linear B-Cell Epitope Prediction for In Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Suitable Linear B-Cell Epitope Predictors
2.2. Development of the Consensus Method
2.3. Data Sets
2.4. Performance Measures
3. Results and Discussion
3.1. Performance of All Predictors on Consensus_NR
3.2. Overall Method Performance and Comparison with BepiPred-2.0
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BCE | Linear B-cell Epitope |
| HMM | Hidden Markov Model |
| SVM | Support Vector Machine |
| AAP | Amino Acid Pairs |
| ERT | Extremely Randomized Tree |
| GB | Gradient Boosting |
| CLI | Command Line Interface |
| GUI | Graphical User Interface |
| AUC | Area Under Curve |
| ROC | Receiver Operating Characteristic |
| MCC | Matthews Correlation Coefficient |
| IEDB | Immune Epitope Data Base |
| Consensus_R | Consensus_Redundant |
| Consensus_NR | Consensus_Non_Redundant |
| SN | Sensitivity |
| SP | Specificity |
| ACC | Accuracy |
| TP | True Positive |
| TN | True Negative |
| FP | False Positive |
| FN | False Negative |
References
- Regenmortel, M.H. What Is a B-Cell Epitope? Adv. Struct. Saf. Stud. 2009, 524, 3–20. [Google Scholar]
- Boehm, T. Design principles of adaptive immune systems. Nat. Rev. Immunol. 2011, 11, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Dudek, N.L.; Perlmutter, P.; Aguilar, M.-I.; Croft, N.P.; Purcell, A.W. Epitope discovery and their use in peptide-based vaccines. Curr. Pharm. Des. 2010, 16, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Russi, R.C.; Bourdin, E.; García, M.I.; Veaute, C.M.I. In silico prediction of T- and B-cell epitopes in PmpD: First step towards to the design of a Chlamydia trachomatis vaccine. Biomed. J. 2018, 41, 109–117. [Google Scholar] [CrossRef]
- Yadav, M.; Liebau, E.; Haldar, C.; Rathaur, S. Identification of major antigenic peptide of filarial glutathione-S-transferase. Vaccine 2011, 29, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooij, W.J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- Chirino, A.J.; Ary, M.L.; A Marshall, S. Minimizing the immunogenicity of protein therapeutics. Drug Discov. Today 2004, 9, 82–90. [Google Scholar] [CrossRef]
- Shirai, H.; Prades, C.; Vita, R.; Marcatili, P.; Popovic, B.; Xu, J.; Overington, J.P.; Hirayama, K.; Soga, S.; Tsunoyama, K.; et al. Antibody informatics for drug discovery. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 2002–2015. [Google Scholar] [CrossRef]
- Nardin, E.H.; Calvo-Calle, J.M.; Oliveira, G.A.; Nussenzweig, R.S.; Schneider, M.; Tiercy, J.-M.; Loutan, L.; Hochstrasser, D.; Rose, K. A Totally Synthetic Polyoxime Malaria Vaccine ContainingPlasmodium falciparumB Cell and Universal T Cell Epitopes Elicits Immune Responses in Volunteers of Diverse HLA Types. J. Immunol. 2001, 166, 481–489. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Mahajan, S.; Peters, B.; Nielsen, M.; Marcatili, P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information From Antibody-Antigen Protein Complexes. Front. Immunol. 2019, 10, 298. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Nielsen, M.; Padkjær, S.B.; Lund, O. Structural analysis of B-cell epitopes in antibody:protein complexes. Mol. Immunol. 2013, 53, 24–34. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H.V. Immunoinformatics may lead to a reappraisal of the nature of B cell epitopes and of the feasibility of synthetic peptide vaccines. J. Mol. Recognit. 2006, 19, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Reineke, U.; Schutkowski, M. Epitope Mapping Protocols; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Zhang, J.; Zhao, X.; Sun, P.; Gao, B.; Ma, Z. Conformational B-Cell Epitopes Prediction from Sequences Using Cost-Sensitive Ensemble Classifiers and Spatial Clustering. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- El-Manzalawy, Y.; Honavar, V. Recent advances in B-cell epitope prediction methods. Immunome Res. 2010, 6, S2. [Google Scholar] [CrossRef]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Tomar, N.; De, R.K. Immunoinformatics: A Brief Review. Methods Mol. Biol. 2014, 1184, 23–55. [Google Scholar] [CrossRef] [PubMed]
- Flower, D.R. Immunoinformatics. Predicting immunogenicity in silico. Preface. Methods Mol. Biol. 2007, 409, 5–6. [Google Scholar]
- Sweredoski, M.J.; Baldi, P. COBEpro: A novel system for predicting continuous B-cell epitopes. Protein Eng. Des. Sel. 2008, 22, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Hopp, T.P.; Woods, K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3824–3828. [Google Scholar] [CrossRef]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Parker, J.M.R.; Guo, D.; Hodges, R.S. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and x-ray-derived accessible sites. Biochemistry 1986, 25, 5425–5432. [Google Scholar] [CrossRef]
- Kolaskar, A.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef]
- Pellequer, J.-L.; Westhof, E.; Van Regenmortel, M.H. Correlation between the location of antigenic sites and the prediction of turns in proteins. Immunol. Lett. 1993, 36, 83–99. [Google Scholar] [CrossRef]
- Pellequer†, J.; Westhof, E. PREDITOP: A program for antigenicity prediction. J. Mol. Graph. 1993, 11, 204–210. [Google Scholar] [CrossRef]
- Alix, A.J. Predictive estimation of protein linear epitopes by using the program PEOPLE. Vaccine 1999, 18, 311–314. [Google Scholar] [CrossRef]
- Odorico, M.; Pellequer, J.-L. BEPITOPE: Predicting the location of continuous epitopes and patterns in proteins. J. Mol. Recognit. 2003, 16, 20–22. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. BcePred: Prediction of Continuous B-Cell Epitopes in Antigenic Sequences Using Physico-chemical Properties. Lect. Notes Comput. Sci. 2004, 3239, 197–204. [Google Scholar]
- Blythe, M.J.; Flower, D.R. Benchmarking B cell epitope prediction: Underperformance of existing methods. Protein Sci. 2009, 14, 246–248. [Google Scholar] [CrossRef]
- Larsen, J.E.P.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef][Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins: Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Rubinstein, N.D.; Mayrose, I.; Martz, E.; Pupko, T. Epitopia: A web-server for predicting B-cell epitopes. BMC Bioinform. 2009, 10, 287. [Google Scholar] [CrossRef]
- El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 2008, 21, 243–255. [Google Scholar] [CrossRef] [PubMed]
- El-Manzalawy, Y.; Dobbs, D.; Honavar, V.; Markstein, P.; Xu, Y. Predicting flexible length linear b-cell epitopes. Comput. Syst. Bioinform. 2008, 7, 121–132. [Google Scholar] [CrossRef]
- Davydov, Y.I.; Tonevitsky, A.G. Prediction of linear B-cell epitopes. Mol. Biol. 2009, 43, 150–158. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Yang, J.; Chou, K.-C. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 2007, 33, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Wee, L.J.; Simarmata, D.; Kam, Y.-W.; Ng, L.F.; Tong, J.C. SVM-based prediction of linear B-cell epitopes using Bayes Feature Extraction. BMC Genom. 2010, 11, S21. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Lin, Y.-C.; Pai, T.-W.; Chang, H.-T. Prediction of B-cell Linear Epitopes with a Combination of Support Vector Machine Classification and Amino Acid Propensity Identification. J. Biomed. Biotechnol. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, W.; Negre, N.N.; White, K.P.; Li, C.; Shah, P.K. Determinants of antigenicity and specificity in immune response for protein sequences. BMC Bioinform. 2011, 12, 251. [Google Scholar] [CrossRef]
- Lian, Y.; Ge, M.; Pan, X.-M. EPMLR: Sequence-based linear B-cell epitope prediction method using multiple linear regression. BMC Bioinform. 2014, 15, 414. [Google Scholar] [CrossRef]
- Lian, Y.; Huang, Z.C.; Ge, M.; Pan, X.M. An Improved Method for Predicting Linear B-cell Epitope Using Deep Maxout Networks. Biomed. Environ. Sci. 2015, 28, 460–463. [Google Scholar] [PubMed]
- Sher, G.; Zhi, D.; Zhang, S. DRREP: Deep ridge regressed epitope predictor. BMC Genom. 2017, 18, 676. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef]
- Söllner, J.; Mayer, B. Machine learning approaches for prediction of linear B-cell epitopes on proteins. J. Mol. Recognit. 2006, 19, 200–208. [Google Scholar] [CrossRef]
- Gao, J.; Faraggi, E.; Zhou, Y.; Ruan, J.; Kurgan, L. BEST: Improved Prediction of B-Cell Epitopes from Antigen Sequences. PLoS ONE 2012, 7, e40104. [Google Scholar] [CrossRef]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.-H.; Cheng, C.-W.; Su, E.C.-Y. Prediction of B-cell epitopes using evolutionary information and propensity scales. BMC Bioinform. 2013, 14, S10. [Google Scholar] [CrossRef]
- Singh, H.; Ansari, H.R.; Raghava, G.P.S. Improved Method for Linear B-Cell Epitope Prediction Using Antigen’s Primary Sequence. PLoS ONE 2013, 8, e62216. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-H.; Wen, M.; Tang, L.-J.; Xie, H.-L.; Fu, L.; Liang, Y.-Z.; Lu, H.-M. Using random forest to classify linear B-cell epitopes based on amino acid properties and molecular features. Biochimie 2014, 103, 1–6. [Google Scholar] [CrossRef]
- Saravanan, V.; Gautham, N. Harnessing Computational Biology for Exact Linear B-Cell Epitope Prediction: A Novel Amino Acid Composition-Based Feature Descriptor. Omics J. Integr. Biol. 2015, 19, 648–658. [Google Scholar] [CrossRef]
- Shen, W.; Cao, Y.; Cha, L.; Zhang, X.; Ying, X.; Zhang, W.; Ge, K.; Li, W.; Zhong, L. Predicting linear B-cell epitopes using amino acid anchoring pair composition. Biodata Min. 2015, 8, 1–12. [Google Scholar] [CrossRef]
- Tsolis, A.C.; Papandreou, N.C.; Iconomidou, V.A.; Hamodrakas, S.J. A Consensus Method for the Prediction of ‘Aggregation-Prone’ Peptides in Globular Proteins. PLoS ONE 2013, 8, e54175. [Google Scholar] [CrossRef]
- Ji, C.; Ma, S. Combinations of weak classifiers. IEEE Trans. Neural Netw. 1997, 8, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.-H.; Grey, H.M.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef]
- Saha, S.; Bhasin, M.; Raghava, G.P.S. Bcipep: A database of B-cell epitopes. BMC Genom. 2005, 6, 79. [Google Scholar] [CrossRef]
- Toseland, C.P.; Clayton, D.J.; Mcsparron, H.; Hemsley, S.L.; Blythe, M.J.; Paine, K.; A Doytchinova, I.; Guan, P.; Hattotuwagama, C.K.; Flower, D.R. AntiJen: A quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immunome Res. 2005, 1, 4. [Google Scholar] [CrossRef][Green Version]
- Pollastri, G.; Baldi, P.; Fariselli, P.; Casadio, R. Prediction of coordination number and relative solvent accessibility in proteins. Proteins Struct. Funct. Bioinform. 2002, 47, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Zarebski, L.; Greenbaum, J.A.; Emami, H.; Hoof, I.; Salimi, N.; Damle, R.; Sette, A.; Peters, B. The Immune Epitope Database 2.0. Nucleic Acids Res. 2009, 38, D854–D862. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef]
- Frousios, K.K.; Iconomidou, V.; Karletidi, C.-M.; Hamodrakas, S.J. Amyloidogenic determinants are usually not buried. BMC Struct. Biol. 2009, 9, 44. [Google Scholar] [CrossRef]
- Peters, C.; Tsirigos, K.D.; Shu, N.; Elofsson, A. Improved topology prediction using the terminal hydrophobic helices rule. Bioinformatics 2015, 32, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [CrossRef] [PubMed]
- Greenbaum, J.A.; Andersen, P.H.; Blythe, M.; Bui, H.-H.; Cachau, R.E.; Crowe, J.; Davies, M.; Kolaskar, A.S.; Lund, O.; Morrison, S.; et al. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J. Mol. Recognit. 2007, 20, 75–82. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Chicco, D. Ten quick tips for machine learning in computational biology. Biodata Min. 2017, 10, 1–17. [Google Scholar] [CrossRef]
- Boughorbel, S.; Jarray, F.; El-Anbari, M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS ONE 2017, 12, e0177678. [Google Scholar] [CrossRef]
- Konstantinou, G.N. T-Cell Epitope Prediction. Adv. Struct. Saf. Stud. 2017, 1592, 211–222. [Google Scholar] [CrossRef]
- Flower, D.R. Immunoinformatics and the in silico Prediction of Immunogenicity. Adv. Struct. Saf. Stud. 2007, 409, 1–15. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Pandey, R.K.; Ojha, R.; Mishra, A.; Prajapati, V.K. Designing B- and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J. Cell. Biochem. 2018, 119, 7631–7642. [Google Scholar] [CrossRef]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 1–21. [Google Scholar] [CrossRef]
- Yazdani, Z.; Rafiei, A.; Yazdani, M.; Valadan, R. Design an Efficient Multi-Epitope Peptide Vaccine Candidate Against SARS-CoV-2: An in silico Analysis. Infect. Drug Resist. 2020, 13, 3007–3022. [Google Scholar] [CrossRef]


{kind=link}
| Predictor | Description | Status | Institution | Link |
|---|---|---|---|---|
| Antigenic [24] | Physico-chemical propensity scales, occurrence of residues | Not currently available online | Department of Zoology, University of Poona, India | http://www.emboss.bioinformatics.nl/cgi-bin/emboss/antigenic |
| PEOPLE [29] | Physico-chemical propensity scales | Not available online | Laboratoire de Spectroscopies et Structures Biomoléculaire, Université de Reims Champagne Ardenne, France | - |
| BEPITOPE [30] | Physico-chemical propensity scales | Freely available online | Ιnstitute of environmental biology and biotechnology, CEA, France | http://bepitope.ibs.fr/ |
| BcePred [31] | Physico-chemical propensity scales | Freely available online and downloadable | Ιnstitute of Microbial Technology, Chandigarh, Ιndia | http://crdd.osdd.net/raghava/bcepred/index.html |
| BepiPred-1.0 [33] | HMM & Parker hydrophilicity scale | Freely available online and downloadable | Center for Biological Sequence Analysis, Technical University of Denmark | http://www.cbs.dtu.dk/services/BepiPred-1.0/ |
| Söllner [47] | Physicochemical Properties, Molecular Operating Environment, K-Nearest Neighbor, Decision Tree | Not available online | emergentec biodevelopment GmbH, Vienna, Austria | - |
| Chen [39] | SVM & AAP | Not available online | Institute of Image Processing and Pattern Recognition, Shanghai Jiaotong University, Shanghai, China | - |
| ABCpred [34] | Neural networks (feed forward & reccurent) | Freely available online and downloadable | Ιnstitute of Microbial Technology, Chandigarh, Ιndia | http://crdd.osdd.net/raghava/abcpred/index.html |
| BCPREDS [36,37] | SVM | Freely available online and downloadable | Dep. of Computer Science & Dep. of Genetics, Development and Cell Biology, Ιowa State University, USA | http://ailab.ist.psu.edu/bcpred/ |
| AAPPred [38] | SVM & AAP | Not currently available online | Faculty of Biology, Moscow State University Russia | http://www.bioinf.ru/aappred/predict |
| Epitopia [35] | Machine Learning algorithm trained to discern antigenic features | Freely available online and downloadable | Tel Aviv Uni. Ιsrael & Uni. of British Columbia Canada & Uni. of Massachusetts, USA | http://epitopia.tau.ac.il/index.html |
| COBEpro [19] | SVM | Freely available online and downloadable upon request | Dep. of Computer Science and Ιnstitute for Genomics and Bioinformatics, University of California USA | http://scratch.proteomics.ics.uci.edu/ |
| BayesB [40] | SVM | Not currently available online | Singapore Ιmmunology Network & Dep. of Biochemistry, National Uni. of Singapore | http://immunopred.org/bayesb/index.html |
| LEPS [41] | SVM & Physicochemical propensity scales & Amino Acid Segments | Not currently available online | National Taiwan Ocean University Taiwan, China Medical University Taiwan | http://leps.cs.ntou.edu.tw/ |
| BEOracle [42] | SVM | Not available online | Department of Biostatistics and Computational Biology, Dana-Farber Cancer Ιnstitute & Harvard School of Public Health, Boston, USA | - |
| BEST [48] | SVM | Not currently available online | School of Mathematical Sciences and LPMC, Nankai University, Tianjin, People’s Republic of China | http://biomine.ece.ualberta/ |
| SVMTriP [49] | SVM | Freely available online and downloadable | University of Nebraska USA, Osaka Uni. Japan | http://sysbio.unl.edu/SVMTriP/ |
| BEEPro [50] | SVM & Physicochemical propensity scales & Position Specific Scoring Matrix | Not available online | School of Medicine, Taipei Medical University, Taipei, Taiwan | - |
| LBtope [51] | SVM & Physicochemical propensity scales & AAP | Freely available online and downloadable | Ιnstitute of Microbial Technology, Chandigarh, Ιndia | http://crdd.osdd.net/raghava/lbtope/protein.php |
| Random Forest [52] | Amino acid descriptors & Random Forest | Not currently available online | Research Center of Modernization of Traditional Chinese Medicines, Central South University, Changsha, China | http://sysbio.yznu.cn/Research/Epitopesprediction.aspx |
| EPMLR [43] | Multiple Linear Regression | Not currently available online | The Key Laboratory of Bioinformatics, Ministry of Education, School of Life Sciences, Tsinghua University, Beijing, China | http://www.bioinfo.tsinghua.edu.cn/epitope/EPMLR/ |
| DMN-LBE [44] | Deep Maxout Networks | Not currently available online | The Key Laboratory of Bioinformatics, Ministry of Education, School of Life Sciences, Tsinghua University, Beijing, China | http://bioinfo.tsinghua.edu.cn/epitope/DMNLBE/ |
| LBEEP [53] | Deviation from Expected Mean—SVM | Freely available download | Center for Advanced Study in Crystallography and Biophysics, University of Madras, Guindy Campus, Chennai, Tamil Nadu, Ιndia. | https://github.com/brsaran/LBEEP |
| APCpred [54] | Amino acid Anchoring Pair Composition & SVM | Not currently available online | Department of Molecular Biology, Hebei University College of Life Sciences, China | http://ccb.bmi.ac.cn/APCpred/ |
| DRREP [45] | Deep Ridge Neural Network | Not currently available online | Department of Computer Science, University of Central Florida, Orlando, FL, USA | https://github.com/gsher1/DRREP |
| BepiPred-2.0 [20] | Random forest algorithm trained on epitopes derived from crystal structures | Freely available online and downloadable | Department of Bio and Health Informatics, Technical University of Denmark, Denmark | http://www.cbs.dtu.dk/services/BepiPred/ |
| iBCE-EL [46] | Ensemble framework combining ERT & GB | Freely available online | Department of Physiology, Ajou University School of Medicine, Suwon, South Korea | http://thegleelab.org/iBCE-EL/ |
| Predictor | Threshold | Mode | Threshold Type |
|---|---|---|---|
| BcePred | 2 | Combined | Not Default |
| BepiPred-1.0 | 0.35 | BepiPred | Default |
| ABCpred | 0.51 | 20 | Default |
| COBEpro | 4 | - | Not Default |
| SVMTriP | 0.2 | 20 | Default |
| LBtope | 0.6 | LBtop_Confirm | Default |
| LBEEP | 0.6 | Balanced | Default |
| Predictor | Prediction | Window Size |
|---|---|---|
| ABCpred | Per peptide | 10, 12, 14, 16, 18, 20 |
| SVMTriP | Per peptide | 10, 12, 14, 16, 18, 20 |
| LBEEP | Per peptide | 5–15 |
| BcePred | Per residue | - |
| BepiPred-1.0 | Per residue | - |
| COBEpro | Per residue | - |
| LBtope | Per residue | - |
| Predictor | Positive | Negative |
|---|---|---|
| BcePred | BCIPEP (2004) | 1029 random sequences |
| BepiPred-1.0 | HΙV/PELLEQUER/ANTIJEN | Not described in the original publication |
| ABCpred | BCIPEP (2006) | 700 random sequences |
| COBEpro | HΙV/PELLEQUER | HIV/Pellequer non-Epitopes |
| SVMTriP | ΙEDB (2012) | 4925 IEDB non-epitopes |
| LBtope | ΙEDB (2012) | IEDB (2012) non-epitopes |
| LBEEP | ΙEDB (2015) | IEDB (2015) non-epitopes |
| Data Set | Epitopes | Non-Epitopes |
|---|---|---|
| BepiPred-2.0 * | 11,814 | 18,689 |
| Consensus_R | 7675 | 15,617 |
| Consensus_NR | 4286 | 5266 |
| Predictor | SN% | SP% | ACC% | MCC |
|---|---|---|---|---|
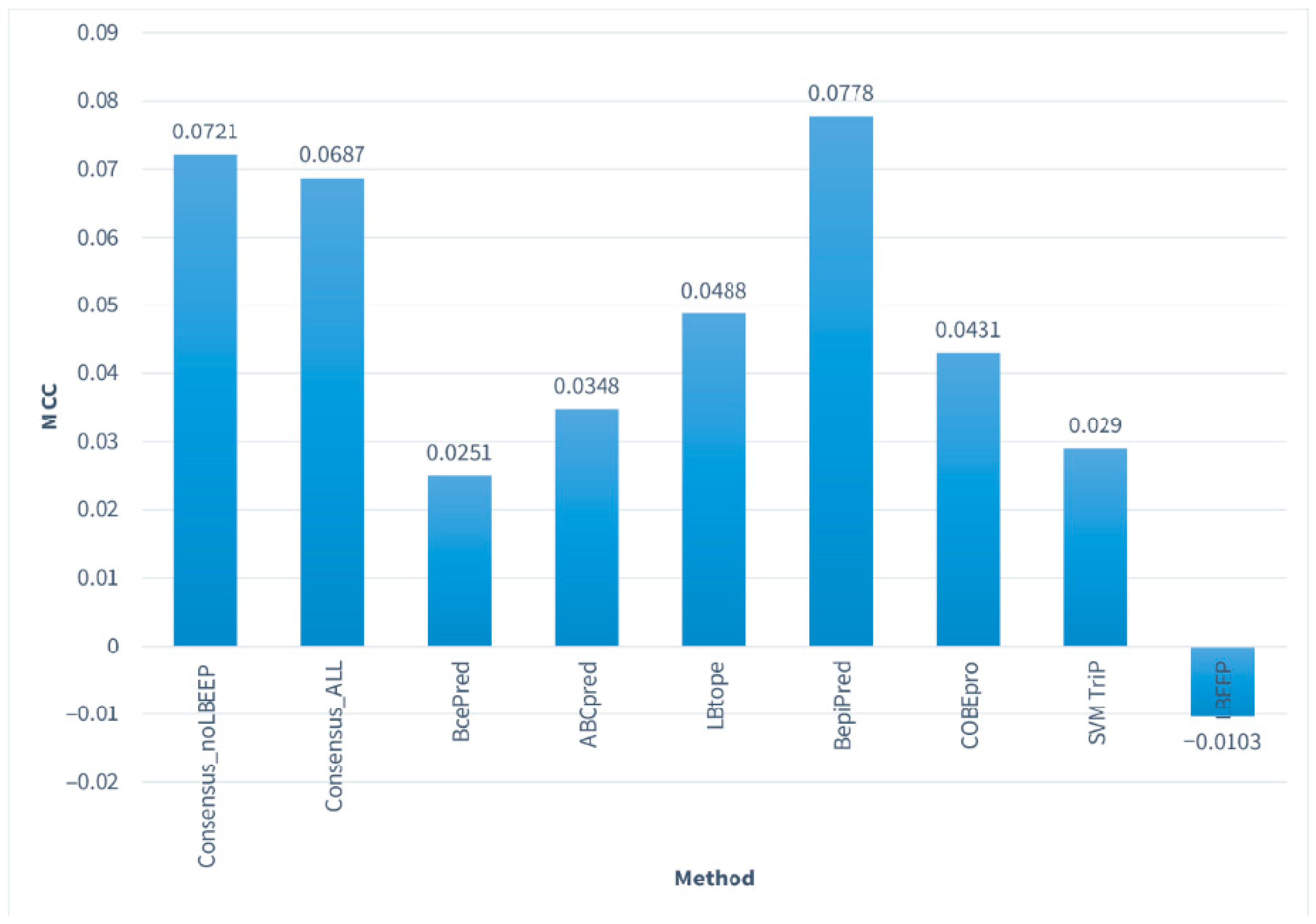
| Consensus_noLBEEP | 48.39 | 58.81 | 54.14 | 0.0721 |
| Consensus_ALL | 27.15 | 78.73 | 55.59 | 0.0687 |
| BcePred | 22.21 | 79.85 | 53.99 | 0.0251 |
| ABCpred | 66.44 | 36.9 | 50.16 | 0.0348 |
| LBtope | 45.91 | 58.94 | 53.1 | 0.0488 |
| BepiPred-1.0 | 49.95 | 57.84 | 54.3 | 0.0778 |
| COBEpro | 58.63 | 45.67 | 51.49 | 0.0431 |
| SVMTriP | 16.21 | 85.87 | 54.62 | 0.0290 |
| LBEEP | 19.06 | 80.12 | 52.72 | −0.0103 |
| Predictor | SN% | SP% | ACC% | MCC |
|---|---|---|---|---|
| Consensus_RES | 46.64 | 58.24 | 53.04 | 0.0489 |
| BcePred | 29.18 | 72.21 | 52.9 | 0.0154 |
| LBtope | 45.56 | 57.47 | 52.13 | 0.0304 |
| BepiPred-1.0 | 48.12 | 56.76 | 52.88 | 0.0488 |
| COBEpro | 49.27 | 52.49 | 51.05 | 0.0175 |
| Predictor | SN% | SP% | ACC% | MCC |
|---|---|---|---|---|
| Consensus_noLBEEP | 50.18 | 58.54 | 54.07 | 0.0873 |
| BepiPred-2.0 | 63.35 | 42.63 | 51.93 | 0.0607 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galanis, K.A.; Nastou, K.C.; Papandreou, N.C.; Petichakis, G.N.; Pigis, D.G.; Iconomidou, V.A. Linear B-Cell Epitope Prediction for In Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface. Int. J. Mol. Sci. 2021, 22, 3210. https://doi.org/10.3390/ijms22063210
Galanis KA, Nastou KC, Papandreou NC, Petichakis GN, Pigis DG, Iconomidou VA. Linear B-Cell Epitope Prediction for In Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface. International Journal of Molecular Sciences. 2021; 22(6):3210. https://doi.org/10.3390/ijms22063210
Chicago/Turabian StyleGalanis, Kosmas A., Katerina C. Nastou, Nikos C. Papandreou, Georgios N. Petichakis, Diomidis G. Pigis, and Vassiliki A. Iconomidou. 2021. "Linear B-Cell Epitope Prediction for In Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface" International Journal of Molecular Sciences 22, no. 6: 3210. https://doi.org/10.3390/ijms22063210
APA StyleGalanis, K. A., Nastou, K. C., Papandreou, N. C., Petichakis, G. N., Pigis, D. G., & Iconomidou, V. A. (2021). Linear B-Cell Epitope Prediction for In Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface. International Journal of Molecular Sciences, 22(6), 3210. https://doi.org/10.3390/ijms22063210