
Cellular and Molecular Mechanisms Mediating Methylmercury Neurotoxicity and Neuroinflammation

 , , ,
, , , 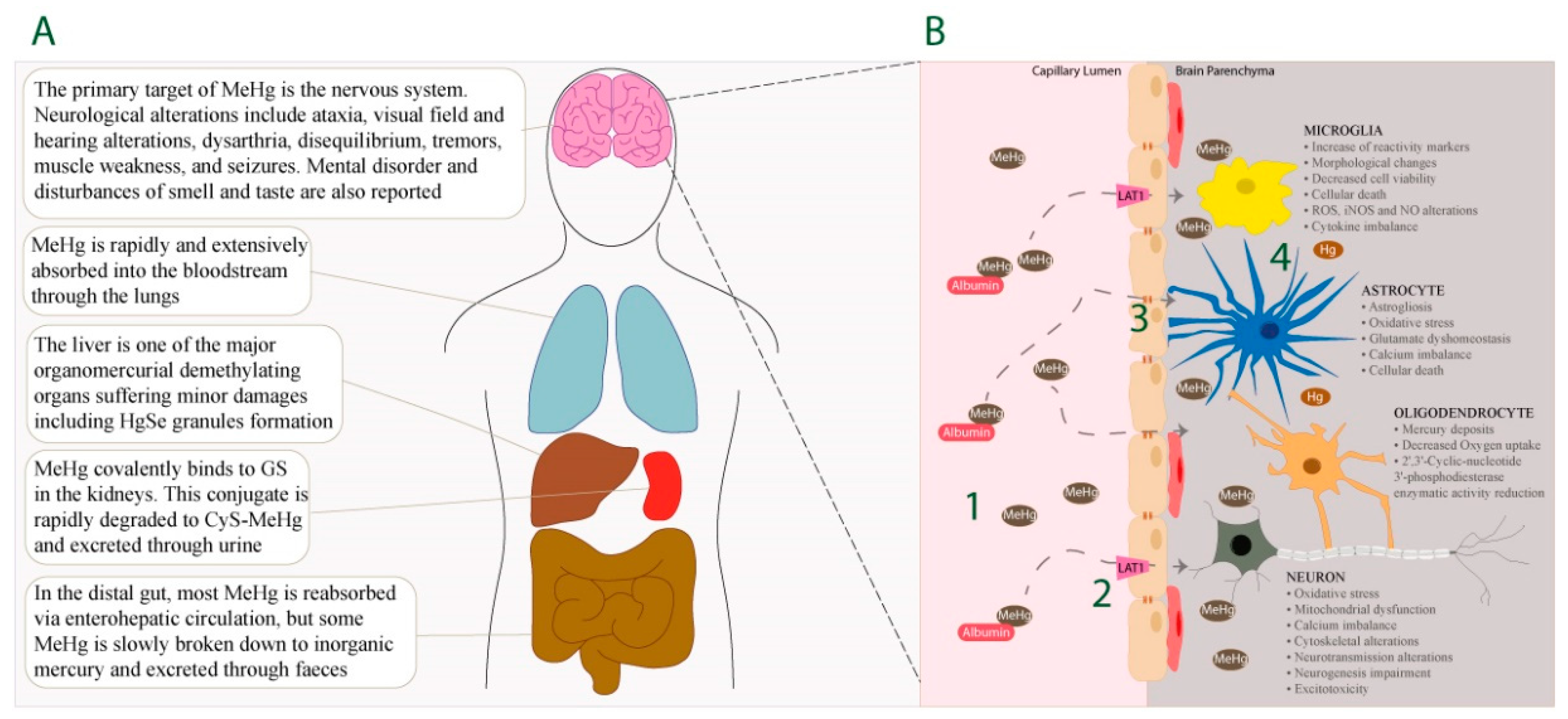{kind=link}
{kind=link}
Abstract
1. Introduction
2. Environmental Impact of Mercury
2.1. Mercury Cycle
2.2. Methylmercury Biomagnification
2.3. The Minamata Disaster
2.4. Additional Impactful Events of MeHg Poisoning
2.5. The Minamata Convention
3. Toxicokinetic Properties of MeHg
3.1. Absorption and Distribution
3.2. Metabolism and Excretion
3.3. Influence of Microbiota
3.4. MeHg Exposure Prevention and Therapeutic Strategies
4. Cellular and Molecular Mechanisms Involved in MeHg Neurotoxicity
4.1. Microglia
4.2. Oligodendrocytes
4.3. Astrocytes
4.4. Neurons
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cesario, R.; Mota, A.M.; Caetano, M.; Nogueira, M.; Canario, J. Mercury and methylmercury transport and fate in the water column of Tagus estuary (Portugal). Mar. Polut. Bull. 2018, 127, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Pacyna, E.G.P.; Pacyna, J.M.; Sundseth, K.; Munthe, J.; Kindbom, K.; Wilson, S.; Steenhuisen, F.; Maxson, P. Global emission of mercury to the atmosphere from anthropogenic sources in 2005 and projections to 2020. Atmos. Environ. 2010, 44, 2487–2499. [Google Scholar] [CrossRef]
- Pinheiro, M.C.; Crespo-Lopez, M.E.; Vieira, J.L.; Oikawa, T.; Guimaraes, G.A.; Araujo, C.C.; Amoras, W.W.; Ribeiro, D.R.; Herculano, A.M.; do Nascimento, J.L.; et al. Mercury pollution and childhood in Amazon riverside villages. Environ. Int. 2007, 33, 56–61. [Google Scholar] [CrossRef]
- Park, J.D.; Zheng, W. Human exposure and health effects of inorganic and elemental mercury. J. Prev. Med. Public Health 2012, 45, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C. Chronic Neurological Disease Due to Methylmercury Poisoning. Can. J. Neurol. Sci. 2018, 45, 620–623. [Google Scholar] [CrossRef]
- Kimakova, T.; Nasser, B.; Issa, M.; Uher, I. Mercury cycling in the terrestrial, aquatic and atmospheric environment of the Slovak Republic—An overview. Ann. Agric. Environ. Med. 2019, 26, 273–279. [Google Scholar] [CrossRef]
- Sakamoto, M.; Tatsuta, N.; Izumo, K.; Phan, P.T.; Vu, L.D.; Yamamoto, M.; Nakamura, M.; Nakai, K.; Murata, K. Health Impacts and Biomarkers of Prenatal Exposure to Methylmercury: Lessons from Minamata, Japan. Toxics 2018, 6, 45. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Wang, F.; Luo, Z.; Guo, S.; Strahle, U. Toxicity of mercury: Molecular evidence. Chemosphere 2020, 245, 125586. [Google Scholar] [CrossRef]
- Rice, K.M.; Walker, E.M., Jr.; Wu, M.; Gillette, C.; Blough, E.R. Environmental mercury and its toxic effects. J. Prev. Med. Public Health 2014, 47, 74–83. [Google Scholar] [CrossRef]
- O’Donoghue, J.L.; Watson, G.E.; Brewer, R.; Zareba, G.; Eto, K.; Takahashi, H.; Marumoto, M.; Love, T.; Harrington, D.; Myers, G.J. Neuropathology associated with exposure to different concentrations and species of mercury: A review of autopsy cases and the literature. Neurotoxicology 2020, 78, 88–98. [Google Scholar] [CrossRef]
- Berzas Nevado, J.J.; Rodriguez Martin-Doimeadios, R.C.; Guzman Bernardo, F.J.; Jimenez Moreno, M.; Herculano, A.M.; do Nascimento, J.L.; Crespo-Lopez, M.E. Mercury in the Tapajos River basin, Brazilian Amazon: A review. Environ. Int. 2010, 36, 593–608. [Google Scholar] [CrossRef]
- Farina, M.; Rocha, J.B.; Aschner, M. Mechanisms of methylmercury-induced neurotoxicity: Evidence from experimental studies. Life Sci. 2011, 89, 555–563. [Google Scholar] [CrossRef]
- Sundseth, K.; Pacyna, J.M.; Pacyna, E.G.; Pirrone, N.; Thorne, R.J. Global Sources and Pathways of Mercury in the Context of Human Health. Int. J. Environ. Res. Public Health 2017, 14, 105. [Google Scholar] [CrossRef] [PubMed]
- Selin, N.E. Global change and mercury cycling: Challenges for implementing a global mercury treaty. Environ. Toxicol. Chem. 2014, 33, 1202–1210. [Google Scholar] [CrossRef]
- Moody, K.H.; Hasan, K.M.; Aljic, S.; Blakeman, V.M.; Hicks, L.P.; Loving, D.C.; Moore, M.E.; Hammett, B.S.; Silva-Gonzalez, M.; Seney, C.S.; et al. Mercury emissions from Peruvian gold shops: Potential ramifications for Minamata compliance in artisanal and small-scale gold mining communities. Environ. Res. 2020, 182, 109042. [Google Scholar] [CrossRef]
- Obrist, D.; Kirk, J.L.; Zhang, L.; Sunderland, E.M.; Jiskra, M.; Selin, N.E. A review of global environmental mercury processes in response to human and natural perturbations: Changes of emissions, climate, and land use. Ambio 2018, 47, 116–140. [Google Scholar] [CrossRef]
- Beckers, F.R.; Rinklebe, J. Cycling of mercury in the environment: Sources, fate, and human health implications: A review. Crit. Rev. Environ. Sci. Technol. 2017, 47, 693–794. [Google Scholar] [CrossRef]
- Grigal, D.F. Mercury sequestration in forests and peatlands: A review. J. Environ. Qual. 2003, 32, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Queipo Abad, S.; Rodriguez-Gonzalez, P.; Davis, W.C.; Garcia Alonso, J.I. Development of a Common Procedure for the Determination of Methylmercury, Ethylmercury, and Inorganic Mercury in Human Whole Blood, Hair, and Urine by Triple Spike Species-Specific Isotope Dilution Mass Spectrometry. Anal. Chem. 2017, 89, 6731–6739. [Google Scholar] [CrossRef]
- Lei, P.; Zhong, H.; Duan, D.; Pan, K. A review on mercury biogeochemistry in mangrove sediments: Hotspots of methylmercury production? Sci. Total Environ. 2019, 680, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Choi, A.L.; Fitzgerald, W.F.; Hammerschmidt, C.R.; Lamborg, C.H.; Soerensen, A.L.; Sunderland, E.M. Mercury biogeochemical cycling in the ocean and policy implications. Environ. Res. 2012, 119, 101–117. [Google Scholar] [CrossRef]
- Leermakers, M.G.S.; De Galan, S.; Brion, N.; Baeyens, W. Mercury in the Southern North Sea and Scheldt estuary. Mar. Chem. 2001, 75, 229–248. [Google Scholar] [CrossRef]
- Harding, G.; Dalziel, J.; Vass, P. Bioaccumulation of methylmercury within the marine food web of the outer Bay of Fundy, Gulf of Maine. PLoS ONE 2018, 13, e0197220. [Google Scholar] [CrossRef]
- Mason, R.P.; Reinfelder, J.R.; Morel, F.M.M. Bioaccumulation of mercury and methylmercury. Water Air Soil Pollut. 1995, 80, 915–921. [Google Scholar] [CrossRef]
- Watras, C.J.; Back, R.C.; Halvorsen, S.; Hudson, R.J.; Morrison, K.A.; Wente, S.P. Bioaccumulation of mercury in pelagic freshwater food webs. Sci. Total Environ. 1998, 219, 183–208. [Google Scholar] [CrossRef]
- Pickhardt, P.C.; Fisher, N.S. Accumulation of inorganic and methylmercury by freshwater phytoplankton in two contrasting water bodies. Environ. Sci. Technol. 2007, 41, 125–131. [Google Scholar] [CrossRef]
- Xu, Z.; Fan, W.; Shi, Z.; Tan, C.; Cui, M.; Tang, S.; Qiu, G.; Feng, X. Mercury and methylmercury bioaccumulation in a contaminated bay. Mar. Polut. Bull. 2019, 143, 134–139. [Google Scholar] [CrossRef]
- Eto, K. Minamata disease. Neuropathology 2000, 20, S14–S19. [Google Scholar] [CrossRef]
- James, A.K.; Nehzati, S.; Dolgova, N.V.; Sokaras, D.; Kroll, T.; Eto, K.; O’Donoghue, J.L.; Watson, G.E.; Myers, G.J.; Krone, P.H.; et al. Rethinking the Minamata Tragedy: What Mercury Species Was Really Responsible? Environ. Sci. Technol. 2020, 54, 2726–2733. [Google Scholar] [CrossRef]
- Yorifuji, T. Lessons From an Early-stage Epidemiological Study of Minamata Disease. J. Epidemiol. 2020, 30, 12–14. [Google Scholar] [CrossRef]
- Yorifuji, T.; Takaoka, S.; Grandjean, P. Accelerated functional losses in ageing congenital Minamata disease patients. Neurotoxicol. Teratol. 2018, 69, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Miyata, C.; Tomita, M.; Date, S.; Kojima, T.; Minamoto, H.; Kurimoto, S.; Noguchi, Y.; Nakagawa, R. A Central Nervous System Disease of Unknown Cause That Occurred in the Minamata Region: Results of an Epidemiological Study. J. Epidemiol. 2020, 30, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, P.J.; Stegeman, J.J.; Fleming, L.E.; Allemand, D.; Anderson, D.M.; Backer, L.C.; Brucker-Davis, F.; Chevalier, N.; Corra, L.; Czerucka, D.; et al. Human Health and Ocean Pollution. Ann. Glob. Health 2020, 86, 1–64. [Google Scholar] [CrossRef]
- Vahter, M.; Akesson, A.; Lind, B.; Bjors, U.; Schutz, A.; Berglund, M. Longitudinal study of methylmercury and inorganic mercury in blood and urine of pregnant and lactating women, as well as in umbilical cord blood. Environ. Res. 2000, 84, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Peng, S.; He, L.; Wang, Y.; Li, Y.; Ma, D.; Wang, Y.; Sun, L.; Zheng, H.; Yang, W.; et al. Methylmercury cytotoxicity and possible mechanisms in human trophoblastic HTR-8/SVneo cells. Ecotoxicol. Environ. Saf. 2021, 207, 111520. [Google Scholar] [CrossRef]
- Bakir, F.; Damluji, S.F.; Amin-Zaki, L.; Murtadha, M.; Khalidi, A.; al-Rawi, N.Y.; Tikriti, S.; Dahahir, H.I.; Clarkson, T.W.; Smith, J.C.; et al. Methylmercury poisoning in Iraq. Science 1973, 181, 230–241. [Google Scholar] [CrossRef]
- Malm, O. Gold mining as a source of mercury exposure in the Brazilian Amazon. Environ. Res. 1998, 77, 73–78. [Google Scholar] [CrossRef]
- Kehrig, H.A.P.; Pinto, F.N.; Moreira, I.; Malm, O. Heavy metals and methylmercury in a tropical coastal estuary and a mangrove in Brazil. Org. Geochem. 2003, 34, 661–669. [Google Scholar] [CrossRef]
- Raposo, R.S.; Pinto, D.V.; Moreira, R.; Dias, R.P.; Ribeiro, C.A.F.; Oriá, R.B.; Malva1, J.O. Methylmercury Impact on Adult Neurogenesis: Is the Worst Yet to Come from Recent Brazilian Environmental Disasters? Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef]
- Crespo-Lopez, M.E.; Augusto-Oliveira, M.; Lopes-Araujo, A.; Santos-Sacramento, L.; Yuki Takeda, P.; Macchi, B.M.; do Nascimento, J.L.M.; Maia, C.S.F.; Lima, R.R.; Arrifano, G.P. Mercury: What can we learn from the Amazon? Environ. Int. 2020, 146, 106223. [Google Scholar] [CrossRef]
- Mambrey, V.; Rakete, S.; Tobollik, M.; Shoko, D.; Moyo, D.; Schutzmeier, P.; Steckling-Muschack, N.; Muteti-Fana, S.; Bose-O’Reilly, S. Artisanal and small-scale gold mining: A cross-sectional assessment of occupational mercury exposure and exposure risk factors in Kadoma and Shurugwi, Zimbabwe. Environ. Res. 2020, 184, 109379. [Google Scholar] [CrossRef]
- Basu, N. The Minamata Convention on Mercury and the role for the environmental sciences community. Environ. Toxicol. Chem. 2018, 37, 2951–2952. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.; Shanley, J.B.; Riscassi, A.; de Wit, H.A.; Eklof, K.; Meng, B.; Mitchell, C.; Osterwalder, S.; Schuster, P.F.; Webster, J.; et al. Recent advances in understanding and measurement of mercury in the environment: Terrestrial Hg cycling. Sci. Total Environ. 2020, 721, 137647. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K. (Ed.) Chapter 9—Principles and basic concepts of toxicokinetics. In Fundamentals of Toxicology; Academic Press: Cambridge, MA, USA, 2016; pp. 87–107. [Google Scholar] [CrossRef]
- Jo, S.; Woo, H.D.; Kwon, H.-J.; Oh, S.-Y.; Park, J.-D.; Hong, Y.-S.; Pyo, H.; Park, K.S.; Ha, M.; Kim, H.; et al. Estimation of the Biological Half-Life of Methylmercury Using a Population Toxicokinetic Model. Int. J. Environ. Res. Public Health 2015, 12, 9054–9067. [Google Scholar] [CrossRef]
- Nogara, P.A.; Oliveira, C.S.; Schmitz, G.L.; Piquini, P.C.; Farina, M.; Aschner, M.; Rocha, J.B.T. Methylmercury’s chemistry: From the environment to the mammalian brain. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129284. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Aschner, M. Glutathione antioxidant system and methylmercury-induced neurotoxicity: An intriguing interplay. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129285. [Google Scholar] [CrossRef]
- Montgomery, K.S.; Mackey, J.; Thuett, K.; Ginestra, S.; Bizon, J.L.; Abbott, L.C. Chronic, low-dose prenatal exposure to methylmercury impairs motor and mnemonic function in adult C57/B6 mice. Behav. Brain Res. 2008, 191, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Lopez, M.E.; Costa-Malaquias, A.; Oliveira, E.H.C.; Miranda, M.S.; Arrifano, G.P.F.; Souza-Monteiro, J.R.; Sagica, F.E.-S.; Fontes-Junior, E.A.; Maia, C.S.F.; Macchi, B.M.; et al. Is Low Non-Lethal Concentration of Methylmercury Really Safe? A Report on Genotoxicity with Delayed Cell Proliferation. PLoS ONE 2016, 11, e0162822. [Google Scholar] [CrossRef]
- Li, P.; Du, B.; Chan, H.M.; Feng, X.; Li, B. Mercury bioaccumulation and its toxic effects in rats fed with methylmercury polluted rice. Sci. Total Environ. 2018, 633, 93–99. [Google Scholar] [CrossRef]
- Antunes Dos Santos, A.; Appel Hort, M.; Culbreth, M.; López-Granero, C.; Farina, M.; Rocha, J.B.; Aschner, M. Methylmercury and brain development: A review of recent literature. J. Trace Elem. Med. Biol. 2016, 38, 99–107. [Google Scholar] [CrossRef]
- Björkman, L.; Lundekvam, B.F.; Laegreid, T.; Bertelsen, B.I.; Morild, I.; Lilleng, P.; Lind, B.; Palm, B.; Vahter, M. Mercury in human brain, blood, muscle and toenails in relation to exposure: An autopsy study. Environ. Health 2007, 6, 30. [Google Scholar] [CrossRef]
- Mergler, D.; Anderson, H.A.; Chan, L.H.; Mahaffey, K.R.; Murray, M.; Sakamoto, M.; Stern, A.H. Methylmercury exposure and health effects in humans: A worldwide concern. Ambio 2007, 36, 3–11. [Google Scholar] [CrossRef]
- Korbas, M.; Lai, B.; Vogt, S.; Gleber, S.C.; Karunakaran, C.; Pickering, I.J.; Krone, P.H.; George, G.N. Methylmercury targets photoreceptor outer segments. ACS Chem. Biol. 2013, 8, 2256–2263. [Google Scholar] [CrossRef]
- Albers, A.; Gies, U.; Raatschen, H.J.; Klintschar, M. Another umbrella murder?—A rare case of Minamata disease. Forensic Sci. Med. Pathol. 2020, 16, 504–509. [Google Scholar] [CrossRef]
- Hong, Y.-S.; Kim, Y.-M.; Lee, K.-E. Methylmercury exposure and health effects. J. Prev. Med. Public Health 2012, 45, 353–363. [Google Scholar] [CrossRef]
- Fang, S.C. Comparative study of uptake and tissue distribution of methylmercury in female rats by inhalation and oral routes of administration. Bull. Environ. Contamin. Toxicol. 1980, 24, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Mechanisms involved in the transport of mercuric ions in target tissues. Arch. Toxicol. 2017, 91, 63–81. [Google Scholar] [CrossRef]
- Zayas, Z.P.; Ouerdane, L.; Mounicou, S.; Lobinski, R.; Monperrus, M.; Amouroux, D. Hemoglobin as a major binding protein for methylmercury in white-sided dolphin liver. Anal. Bioanal. Chem. 2014, 406, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, A.; Hirayama, K.; Inoue, M. Interaction of methylmercury compounds with albumin. Arch. Toxicol. 1990, 64, 639–643. [Google Scholar] [CrossRef]
- Li, Y.; Yan, X.-P.; Chen, C.; Xia, Y.-L.; Jiang, Y. Human Serum Albumin−Mercurial Species Interactions. J. Proteome Res. 2007, 6, 2277–2286. [Google Scholar] [CrossRef]
- Carneiro, M.F.; Grotto, D.; Barbosa, F., Jr. Inorganic and methylmercury levels in plasma are differentially associated with age, gender, and oxidative stress markers in a population exposed to mercury through fish consumption. J. Toxicol. Environ. Health A 2014, 77, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Jiang, H.; Syversen, T.; Rocha, J.B.; Farina, M.; Aschner, M. The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J. Neurochem. 2008, 107, 1083–1090. [Google Scholar] [CrossRef]
- Diez, S. Human health effects of methylmercury exposure. Rev. Environ. Contam. Toxicol. 2009, 198, 111–132. [Google Scholar] [CrossRef]
- Kerper, L.E.; Ballatori, N.; Clarkson, T.W. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am. J. Physiol. 1992, 262, R761–R765. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F.; Sawada, M.; Takashima, A. Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain. Neurotoxicology 2009, 30, 1000–1007. [Google Scholar] [CrossRef]
- Vahter, M.E.; Mottet, N.K.; Friberg, L.T.; Lind, S.B.; Charleston, J.S.; Burbacher, T.M. Demethylation of methyl mercury in different brain sites of Macaca fascicularis monkeys during long-term subclinical methyl mercury exposure. Toxicol. Appl. Pharmacol. 1995, 134, 273–284. [Google Scholar] [CrossRef]
- Uchikawa, T.; Kanno, T.; Maruyama, I.; Mori, N.; Yasutake, A.; Ishii, Y.; Yamada, H. Demethylation of methylmercury and the enhanced production of formaldehyde in mouse liver. J. Toxicol. Sci. 2016, 41, 479–487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Palmisano, F.; Cardellicchio, N.; Zambonin, P.G. Speciation of mercury in dolphin liver: A two-stage mechanism for the demethylation accumulation process and role of selenium. Mar. Environ. Res. 1995, 40, 109–121. [Google Scholar] [CrossRef]
- Khan, M.A.K.; Wang, F. Chemical Demethylation of Methylmercury by Selenoamino Acids. Chem. Res. Toxicol. 2010, 23, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Friberg, L.; Mottet, N.K. Accumulation of methylmercury and inorganic mercury in the brain. Biol. Trace Elem. Res. 1989, 21, 201–206. [Google Scholar] [CrossRef]
- Vázquez, M.; Vélez, D.; Devesa, V. In vitro evaluation of inorganic mercury and methylmercury effects on the intestinal epithelium permeability. Food Chem. Toxicol. 2014, 74, 349–359. [Google Scholar] [CrossRef]
- Seko, Y.; Takahashi, M.; Hasegawa, T.; Miura, T. Intestinal Absorption of Mercury in Vitro from Intestinal Contents of Methylmercury Administered Mice. J. Health Sci. 2001, 47, 508–511. [Google Scholar] [CrossRef]
- Nuttall, K.L. Interpreting mercury in blood and urine of individual patients. Ann. Clin. Lab. Sci. 2004, 34, 235–250. [Google Scholar]
- Yasutake, A.; Hirayama, K.; Inoue, M. Mechanism of urinary excretion of methylmercury in mice. Arch. Toxicol. 1989, 63, 479–483. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F.; Unoki, T. Decreased plasma thiol antioxidant capacity precedes neurological signs in a rat methylmercury intoxication model. Food Chem. Toxicol. 2020, 146, 111810. [Google Scholar] [CrossRef]
- Rothenberg, S.E.; Keiser, S.; Ajami, N.J.; Wong, M.C.; Gesell, J.; Petrosino, J.F.; Johs, A. The role of gut microbiota in fetal methylmercury exposure: Insights from a pilot study. Toxicol. Lett. 2016, 242, 60–67. [Google Scholar] [CrossRef]
- Rowland, I.R.; Mallett, A.K.; Flynn, J.; Hargreaves, R.J. The effect of various dietary fibres on tissue concentration and chemical form of mercury after methylmercury exposure in mice. Arch. Toxicol. 1986, 59, 94–98. [Google Scholar] [CrossRef]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Digest. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef]
- Schaefer, J.K.; Yagi, J.; Reinfelder, J.R.; Cardona, T.; Ellickson, K.M.; Tel-Or, S.; Barkay, T. Role of the bacterial organomercury lyase (MerB) in controlling methylmercury accumulation in mercury-contaminated natural waters. Environ. Sci. Technol. 2004, 38, 4304–4311. [Google Scholar] [CrossRef]
- Liu, Y.; Ji, J.; Zhang, W.; Suo, Y.; Zhao, J.; Lin, X.; Cui, L.; Li, B.; Hu, H.; Chen, C.; et al. Selenium modulated gut flora and promoted decomposition of methylmercury in methylmercury-poisoned rats. Exotoxicol. Environ. Saf. 2019, 185, 109720. [Google Scholar] [CrossRef]
- Myers, G.J.; Davidson, P.W.; Watson, G.E.; van Wijngaarden, E.; Thurston, S.W.; Strain, J.J.; Shamlaye, C.F.; Bovet, P. Methylmercury exposure and developmental neurotoxicity. Bull. World Health Organ. 2015, 93, 132. [Google Scholar] [CrossRef]
- Ye, B.J.; Kim, B.G.; Jeon, M.J.; Kim, S.Y.; Kim, H.C.; Jang, T.W.; Chae, H.J.; Choi, W.J.; Ha, M.N.; Hong, Y.S. Evaluation of mercury exposure level, clinical diagnosis and treatment for mercury intoxication. Ann. Occup. Environ. Med. 2016, 28, 5. [Google Scholar] [CrossRef]
- Bridges, C.C.; Joshee, L.; Zalups, R.K. Effect of DMPS and DMSA on the placental and fetal disposition of methylmercury. Placenta 2009, 30, 800–805. [Google Scholar] [CrossRef]
- Dawn, L.; Whited, L. Dimercaprol; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Flora, S.J.; Pachauri, V. Chelation in metal intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [Google Scholar] [CrossRef]
- Beyrouty, P.; Chan, H.M. Co-consumption of selenium and vitamin E altered the reproductive and developmental toxicity of methylmercury in rats. Neurotoxicol. Teratol. 2006, 28, 49–58. [Google Scholar] [CrossRef]
- Nobre, P.; Cabral, M.F.; Costa, J.; Castro-Caldas, M.; Carvalho, C.; Branco, V. In Vitro Assessment of the Efficacy of a Macrocyclic Chelator in Reversing Methylmercury Toxicity. Int. J. Environ. Res. Public Health 2019, 16, 4817. [Google Scholar] [CrossRef]
- Rafati-Rahimzadeh, M.; Rafati-Rahimzadeh, M.; Kazemi, S.; Moghadamnia, A.A. Current approaches of the management of mercury poisoning: Need of the hour. Daru 2014, 22, 46. [Google Scholar] [CrossRef]
- Bertossi, M.; Girolamo, F.; Errede, M.; Virgintino, D.; Elia, G.; Ambrosi, L.; Roncali, L. Effects of methylmercury on the microvasculature of the developing brain. Neurotoxicology 2004, 25, 849–857. [Google Scholar] [CrossRef]
- Noguchi, Y.; Shinozaki, Y.; Fujishita, K.; Shibata, K.; Imura, Y.; Morizawa, Y.; Gachet, C.; Koizumi, S. Astrocytes protect neurons against methylmercury via ATP/P2Y(1) receptor-mediated pathways in astrocytes. PLoS ONE 2013, 8, e57898. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Nomura, M.; Iwatsuki, K.; Moriyama, Y.; Gachet, C.; Koizumi, S. Microglia trigger astrocyte-mediated neuroprotection via purinergic gliotransmission. Sci. Rep. 2014, 4, 4329. [Google Scholar] [CrossRef]
- Takahashi, T.; Shimohata, T. Vascular Dysfunction Induced by Mercury Exposure. Int. J. Mol. Sci. 2019, 20, 2435. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Fujimura, M.; Koyama, M.; Kanazawa, M.; Usuki, F.; Nishizawa, M.; Shimohata, T. Methylmercury Causes Blood-Brain Barrier Damage in Rats via Upregulation of Vascular Endothelial Growth Factor Expression. PLoS ONE 2017, 12, e0170623. [Google Scholar] [CrossRef] [PubMed]
- Bradford, A.B.; Mancini, J.D.; Atchison, W.D. Methylmercury-Dependent Increases in Fluo4 Fluorescence in Neonatal Rat Cerebellar Slices Depend on Granule Cell Migrational Stage and GABAA Receptor Modulation. J. Pharmacol. Exp. Ther. 2016, 356, 2–12. [Google Scholar] [CrossRef]
- Mancini, J.D.; Autio, D.M.; Atchison, W.D. Continuous exposure to low concentrations of methylmercury impairs cerebellar granule cell migration in organotypic slice culture. Neurotoxicology 2009, 30, 203–208. [Google Scholar] [CrossRef]
- Heimfarth, L.; Delgado, J.; Mingori, M.R.; Moresco, K.S.; Pureur, R.P.; Gelain, D.P.; Moreira, J.C.F. Delayed neurochemical effects of prenatal exposure to MeHg in the cerebellum of developing rats. Toxicol. Lett. 2018, 284, 161–169. [Google Scholar] [CrossRef]
- Obiorah, M.; McCandlish, E.; Buckley, B.; DiCicco-Bloom, E. Hippocampal developmental vulnerability to methylmercury extends into prepubescence. Front. Neurosci. 2015, 9, 150. [Google Scholar] [CrossRef]
- Falluel-Morel, A.; Sokolowski, K.; Sisti, H.M.; Zhou, X.; Shors, T.J.; Dicicco-Bloom, E. Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J. Neurochem. 2007, 103, 1968–1981. [Google Scholar] [CrossRef]
- Aragão, W.A.B.; Teixeira, F.B.; Fagundes, N.C.F.; Fernandes, R.M.; Fernandes, L.M.P.; da Silva, M.C.F.; Amado, L.L.; Sagica, F.E.S.; Oliveira, E.H.C.; Crespo-Lopez, M.E.; et al. Hippocampal Dysfunction Provoked by Mercury Chloride Exposure: Evaluation of Cognitive Impairment, Oxidative Stress, Tissue Injury and Nature of Cell Death. Oxid. Med. Cell Longev. 2018, 2018, 7878050. [Google Scholar] [CrossRef]
- Miura, K.; Imura, N. Mechanism of methylmercury cytotoxicity. Crit. Rev. Toxicol. 1987, 18, 161–188. [Google Scholar] [CrossRef]
- Peraza, M.A.; Ayala-Fierro, F.; Barber, D.S.; Casarez, E.; Rael, L.T. Effects of micronutrients on metal toxicity. Environ. Health Perspect. 1998, 106, 203–216. [Google Scholar] [CrossRef]
- Charleston, J.S.; Bolender, R.P.; Mottet, N.K.; Body, R.L.; Vahter, M.E.; Burbacher, T.M. Increases in the number of reactive glia in the visual cortex of Macaca fascicularis following subclinical long-term methyl mercury exposure. Toxicol. Appl. Pharmacol. 1994, 129, 196–206. [Google Scholar] [CrossRef]
- Falluel-Morel, A.; Lin, L.; Sokolowski, K.; McCandlish, E.; Buckley, B.; DiCicco-Bloom, E. N-acetyl cysteine treatment reduces mercury-induced neurotoxicity in the developing rat hippocampus. J. Neurosci. Res. 2012, 90, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F. Methylmercury-Mediated Oxidative Stress and Activation of the Cellular Protective System. Antioxidants 2020, 9, 1004. [Google Scholar] [CrossRef]
- Toyama, T.; Shinkai, Y.; Yasutake, A.; Uchida, K.; Yamamoto, M.; Kumagai, Y. Isothiocyanates reduce mercury accumulation via an Nrf2-dependent mechanism during exposure of mice to methylmercury. Environ. Health Perspect. 2011, 119, 1117–1122. [Google Scholar] [CrossRef]
- Krishna Chandran, A.M.; Christina, H.; Das, S.; Mumbrekar, K.D.; Satish Rao, B.S. Neuroprotective role of naringenin against methylmercury induced cognitive impairment and mitochondrial damage in a mouse model. Environ. Toxicol. Pharmacol. 2019, 71, 103224. [Google Scholar] [CrossRef]
- Olguín, N.; Müller, M.L.; Rodríguez-Farré, E.; Suñol, C. Neurotransmitter amines and antioxidant agents in neuronal protection against methylmercury-induced cytotoxicity in primary cultures of mice cortical neurons. Neurotoxicology 2018, 69, 278–287. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Unoki, T.; Akiyama, M.; Kumagai, Y.; Gonçalves, F.M.; Farina, M.; da Rocha, J.B.T.; Aschner, M. Molecular Pathways Associated with Methylmercury-Induced Nrf2 Modulation. Front. Genet. 2018, 9, 373. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells as Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Ginhoux, F.; Greter, M.; Bruttger, J. Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol. 2015, 36, 625–636. [Google Scholar] [CrossRef]
- Hanisch, U.K. Functional diversity of microglia—How heterogeneous are they to begin with? Front. Cell Neurosci. 2013, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNgamma+TNFalpha) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia actively regulate the number of functional synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Valero, J.; Bernardino, L.; Cardoso, F.L.; Silva, A.P.; Fontes-Ribeiro, C.; Ambrosio, A.F.; Malva, J.O. Impact of Neuroinflammation on Hippocampal Neurogenesis: Relevance to Aging and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, S161–S168. [Google Scholar] [CrossRef]
- Nishioku, T.; Takai, N.; Miyamoto, K.; Murao, K.; Hara, C.; Yamamoto, K.; Nakanishi, H. Involvement of caspase 3-like protease in methylmercury-induced apoptosis of primary cultured rat cerebral microglia. Brain Res. 2000, 871, 160–164. [Google Scholar] [CrossRef]
- Eskes, C.; Honegger, P.; Juillerat-Jeanneret, L.; Monnet-Tschudi, F. Microglial reaction induced by noncytotoxic methylmercury treatment leads to neuroprotection via interactions with astrocytes and IL-6 release. Glia 2002, 37, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Garg, T.K.; Chang, J.Y. Methylmercury causes oxidative stress and cytotoxicity in microglia: Attenuation by 15-deoxy-delta 12, 14-prostaglandin J2. J. Neuroimmunol. 2006, 171, 17–28. [Google Scholar] [CrossRef]
- Tan, Q.; Zhang, M.; Geng, L.; Xia, Z.; Li, C.; Usman, M.; Du, Y.; Wei, L.; Bi, H. Hormesis of methylmercury-human serum albumin conjugate on N9 microglia via ERK/MAPKs and STAT3 signaling pathways. Toxicol. Appl. Pharmacol. 2019, 362, 59–66. [Google Scholar] [CrossRef]
- Bassett, T.; Bach, P.; Chan, H.M. Effects of methylmercury on the secretion of pro-inflammatory cytokines from primary microglial cells and astrocytes. Neurotoxicology 2012, 33, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Li, X.; Yin, Z.; Sidoryk-Wegrzynowicz, M.; Jiang, H.; Farina, M.; Rocha, J.B.; Syversen, T.; Aschner, M. Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia 2011, 59, 810–820. [Google Scholar] [CrossRef]
- Ni, M.; Li, X.; Rocha, J.B.; Farina, M.; Aschner, M. Glia and methylmercury neurotoxicity. J. Toxicol. Environ. Health A 2012, 75, 1091–1101. [Google Scholar] [CrossRef]
- Ni, M.; Li, X.; Yin, Z.; Jiang, H.; Sidoryk-Wegrzynowicz, M.; Milatovic, D.; Cai, J.; Aschner, M. Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicol. Sci. 2010, 116, 590–603. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F. Induction of apoptosis by mercury compounds depends on maturation and is not associated with microglial activation. J. Neurosci. Res. 1998, 53, 361–367. [Google Scholar] [CrossRef]
- Chang, J.Y. Methylmercury causes glial IL-6 release. Neurosci. Lett. 2007, 416, 217–220. [Google Scholar] [CrossRef]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; De Groef, L. Characterizing microglia activation: A spatial statistics approach to maximize information extraction. Sci. Rep. 2017, 7, 1576. [Google Scholar] [CrossRef]
- Fernandez-Arjona, M.D.M.; Grondona, J.M.; Granados-Duran, P.; Fernandez-Llebrez, P.; Lopez-Avalos, M.D. Microglia Morphological Categorization in a Rat Model of Neuroinflammation by Hierarchical Cluster and Principal Components Analysis. Front. Cell Neurosci. 2017, 11, 235. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Danjo, Y.; Koizumi, S. Microglial ROCK is essential for chronic methylmercury-induced neurodegeneration. J. Neurochem. 2019, 151, 64–78. [Google Scholar] [CrossRef]
- Hoshi, T.; Toyama, T.; Shinozaki, Y.; Koizumi, S.; Lee, J.Y.; Naganuma, A.; Hwang, G.W. Evaluation of M1-microglial activation by neurotoxic metals using optimized organotypic cerebral slice cultures. J. Toxicol. Sci. 2019, 44, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Mariani, A.; Fanelli, R.; Re Depaolini, A.; De Paola, M. Decabrominated diphenyl ether and methylmercury impair fetal nervous system development in mice at documented human exposure levels. Dev. Neurobiol. 2015, 75, 23–38. [Google Scholar] [CrossRef]
- Sakamoto, M.; Miyamoto, K.; Wu, Z.; Nakanishi, H. Possible involvement of cathepsin B released by microglia in methylmercury-induced cerebellar pathological changes in the adult rat. Neurosci. Lett. 2008, 442, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F.; Nakamura, A. Fasudil, a Rho-Associated Coiled Coil-Forming Protein Kinase Inhibitor, Recovers Methylmercury-Induced Axonal Degeneration by Changing Microglial Phenotype in Rats. Toxicol. Sci. 2019, 168, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Charleston, J.S.; Body, R.L.; Mottet, N.K.; Vahter, M.E.; Burbacher, T.M. Autometallographic determination of inorganic mercury distribution in the cortex of the calcarine sulcus of the monkey Macaca fascicularis following long-term subclinical exposure to methylmercury and mercuric chloride. Toxicol. Appl. Pharmacol. 1995, 132, 325–333. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F.; Zurich, M.G.; Honegger, P. Comparison of the developmental effects of two mercury compounds on glial cells and neurons in aggregate cultures of rat telencephalon. Brain Res. 1996, 741, 52–59. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takeya, M.; Ikeshima-Kataoka, H.; Yasui, M.; Kawasaki, Y.; Shiraishi, M.; Majima, E.; Shiraishi, S.; Uezono, Y.; Sasaki, M.; et al. Increased expression of aquaporin-4 with methylmercury exposure in the brain of the common marmoset. J. Toxicol. Sci. 2012, 37, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K. A review of experimental methylmercury toxicity in rats: Neuropathology and evidence for apoptosis. Toxicol. Pathol. 1997, 25, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, Y.; Ehara, S.; Tatsumi, S.; Yoshida, E.; Takahashi, T.; Eto, K.; Kaji, T.; Fujiwara, Y. Methylmercury-induced neural degeneration in rat dorsal root ganglion is associated with the accumulation of microglia/macrophages and the proliferation of Schwann cells. J. Toxicol. Sci. 2019, 44, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Iwai-Shimada, M.; Takahashi, T.; Kim, M.S.; Fujimura, M.; Ito, H.; Toyama, T.; Naganuma, A.; Hwang, G.W. Methylmercury induces the expression of TNF-alpha selectively in the brain of mice. Sci. Rep. 2016, 6, 38294. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.; Choi, B.H. Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology 1996, 17, 17–26. [Google Scholar]
- Issa, Y.; Brunton, P.; Waters, C.M.; Watts, D.C. Cytotoxicity of metal ions to human oligodendroglial cells and human gingival fibroblasts assessed by mitochondrial dehydrogenase activity. Dent. Mater. 2008, 24, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Kum Jew, S.; Doble, P.A.; Bishop, D.P. Elemental imaging shows mercury in cells of the human lateral and medial geniculate nuclei. PLoS ONE 2020, 15, e0231870. [Google Scholar] [CrossRef]
- Pamphlett, R.; Kum Jew, S. Inorganic mercury in human astrocytes, oligodendrocytes, corticomotoneurons and the locus ceruleus: Implications for multiple sclerosis, neurodegenerative disorders and gliomas. Biometals 2018, 31, 807–819. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Belanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [PubMed]
- Freire, M.A.M.; Lima, R.R.; Nascimento, P.C.; Gomes-Leal, W.; Pereira, A., Jr. Effects of methylmercury on the pattern of NADPH diaphorase expression and astrocytic activation in the rat. Ecotoxicol. Environ. Saf. 2020, 201, 110799. [Google Scholar] [CrossRef]
- Ishihara, Y.; Itoh, K.; Oguro, A.; Chiba, Y.; Ueno, M.; Tsuji, M.; Vogel, C.F.A.; Yamazaki, T. Neuroprotective activation of astrocytes by methylmercury exposure in the inferior colliculus. Sci. Rep. 2019, 9, 13899. [Google Scholar] [CrossRef]
- Farina, M.; Cereser, V.; Portela, L.V.; Mendez, A.; Porciúncula, L.O.; Fornaguera, J.; Gonçalves, C.A.; Wofchuk, S.T.; Rocha, J.B.; Souza, D.O. Methylmercury increases S100B content in rat cerebrospinal fluid. Environ. Toxicol. Pharmacol. 2005, 19, 249–253. [Google Scholar] [CrossRef] [PubMed]
- De Simone, U.; Caloni, F.; Gribaldo, L.; Coccini, T. Human Co-culture Model of Neurons and Astrocytes to Test Acute Cytotoxicity of Neurotoxic Compounds. Int. J. Toxicol. 2017, 36, 463–477. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, X.; Zhang, R.; Zhou, Y. Effects of postnatal exposure to methylmercury on spatial learning and memory and brain NMDA receptor mRNA expression in rats. Toxicol. Lett. 2009, 188, 230–235. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Shanker, G.; Mutkus, L.A.; Walker, S.J.; Aschner, M. Methylmercury enhances arachidonic acid release and cytosolic phospholipase A2 expression in primary cultures of neonatal astrocytes. Brain Res. Mol. Brain Res. 2002, 106, 1–11. [Google Scholar] [CrossRef]
- Aschner, M. Astrocytic swelling, phospholipase A2, glutathione and glutamate: Interactions in methylmercury-induced neurotoxicity. Cell. Mol. Biol. 2000, 46, 843–854. [Google Scholar] [PubMed]
- Liu, W.; Xu, Z.; Yang, T.; Deng, Y.; Xu, B.; Feng, S. Tea Polyphenols Protect Against Methylmercury-Induced Cell Injury in Rat Primary Cultured Astrocytes, Involvement of Oxidative Stress and Glutamate Uptake/Metabolism Disorders. Mol. Neurobiol. 2016, 53, 2995–3009. [Google Scholar] [CrossRef] [PubMed]
- Mutkus, L.; Aschner, J.L.; Syversen, T.; Aschner, M. Methylmercury alters the in vitro uptake of glutamate in GLAST- and GLT-1-transfected mutant CHO-K1 cells. Biol. Trace Elem. Res. 2005, 107, 231–245. [Google Scholar] [CrossRef]
- Hertz, L.; Rothman, D.L. Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology 2017, 6, 17. [Google Scholar] [CrossRef]
- Yin, Z.; Milatovic, D.; Aschner, J.L.; Syversen, T.; Rocha, J.B.; Souza, D.O.; Sidoryk, M.; Albrecht, J.; Aschner, M. Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res. 2007, 1131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Lee, E.; Ni, M.; Jiang, H.; Milatovic, D.; Rongzhu, L.; Farina, M.; Rocha, J.B.; Aschner, M. Methylmercury-induced alterations in astrocyte functions are attenuated by ebselen. Neurotoxicology 2011, 32, 291–299. [Google Scholar] [CrossRef][Green Version]
- Aschner, M.; Syversen, T.; Souza, D.O.; Rocha, J.B.; Farina, M. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz. J. Med. Biol. Res. 2007, 40, 285–291. [Google Scholar] [CrossRef]
- Culbreth, M.; Aschner, M. Dysregulation of Glutamate Cycling Mediates Methylmercury-Induced Neurotoxicity. Adv. Neurobiol. 2016, 13, 295–305. [Google Scholar] [CrossRef]
- Juarez, B.I.; Portillo-Salazar, H.; Gonzalez-Amaro, R.; Mandeville, P.; Aguirre, J.R.; Jimenez, M.E. Participation of N-methyl-D-aspartate receptors on methylmercury-induced DNA damage in rat frontal cortex. Toxicology 2005, 207, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Juarez, B.I.; Martinez, M.L.; Montante, M.; Dufour, L.; Garcia, E.; Jimenez-Capdeville, M.E. Methylmercury increases glutamate extracellular levels in frontal cortex of awake rats. Neurotoxicol. Teratol. 2002, 24, 767–771. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. Methylmercury inhibits cysteine uptake in cultured primary astrocytes, but not in neurons. Brain Res. 2001, 914, 159–165. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, L.; Langlois, P.; Mironov, G.; Chan, H.M. Proteome changes in methylmercury-exposed mouse primary cerebellar granule neurons and astrocytes. Toxicol. In Vitro 2019, 57, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Yang, B.; Zhou, Y.; Yin, C.; Liu, T.; Qian, H.; Xing, G.; Wang, S.; Li, F.; Zhang, Y.; et al. Acute Methylmercury Exposure and the Hypoxia-Inducible Factor-1alpha Signaling Pathway under Normoxic Conditions in the Rat Brain and Astrocytes in Vitro. Environ. Health Perspect. 2019, 127, 127006. [Google Scholar] [CrossRef]
- Yang, B.; Yin, C.; Zhou, Y.; Wang, Q.; Jiang, Y.; Bai, Y.; Qian, H.; Xing, G.; Wang, S.; Li, F.; et al. Curcumin protects against methylmercury-induced cytotoxicity in primary rat astrocytes by activating the Nrf2/ARE pathway independently of PKCdelta. Toxicology 2019, 425, 152248. [Google Scholar] [CrossRef]
- Kaur, P.; Aschner, M.; Syversen, T. Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology 2006, 27, 492–500. [Google Scholar] [CrossRef]
- Pierozan, P.; Biasibetti, H.; Schmitz, F.; Avila, H.; Fernandes, C.G.; Pessoa-Pureur, R.; Wyse, A.T.S. Neurotoxicity of Methylmercury in Isolated Astrocytes and Neurons: The Cytoskeleton as a Main Target. Mol. Neurobiol. 2017, 54, 5752–5767. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, H.; Yin, Z.; Aschner, M.; Cai, J. Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicol. Sci. 2009, 107, 135–143. [Google Scholar] [CrossRef]
- Yang, J.; Vitery, M.D.C.; Chen, J.; Osei-Owusu, J.; Chu, J.; Qiu, Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 2019, 102, 813–827.e6. [Google Scholar] [CrossRef]
- Robitaille, S.; Mailloux, R.J.; Chan, H.M. Methylmercury alters glutathione homeostasis by inhibiting glutaredoxin 1 and enhancing glutathione biosynthesis in cultured human astrocytoma cells. Toxicol. Lett. 2016, 256, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Seeger, R.; Puntel, R.; Vargas Barbosa, N. Role of calcium and mitochondria in MeHg-mediated cytotoxicity. J. Biomed. Biotechnol. 2012, 2012, 248764. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.; Mutkus, L.A.; Aschner, M. Methylmercury-mediated inhibition of 3H-D-aspartate transport in cultured astrocytes is reversed by the antioxidant catalase. Brain Res. 2001, 902, 92–100. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, A.A.; López-Granero, C.; Farina, M.; Rocha, J.B.T.; Bowman, A.B.; Aschner, M. Oxidative stress, caspase-3 activation and cleavage of ROCK-1 play an essential role in MeHg-induced cell death in primary astroglial cells. Food Chem. Toxicol. 2018, 113, 328–336. [Google Scholar] [CrossRef]
- Sebbagh, M.; Renvoizé, C.; Hamelin, J.; Riché, N.; Bertoglio, J.; Bréard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001, 3, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Cuello, S.; Goya, L.; Madrid, Y.; Campuzano, S.; Pedrero, M.; Bravo, L.; Cámara, C.; Ramos, S. Molecular mechanisms of methylmercury-induced cell death in human HepG2 cells. Food Chem. Toxicol. 2010, 48, 1405–1411. [Google Scholar] [CrossRef]
- Chen, P.; Miah, M.R.; Aschner, M. Metals and Neurodegeneration. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Faustman, E.M.; Ponce, R.A.; Ou, Y.C.; Mendoza, M.A.; Lewandowski, T.; Kavanagh, T. Investigations of methylmercury-induced alterations in neurogenesis. Environ. Health Perspect. 2002, 110, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Limke, T.L.; Bearss, J.J.; Atchison, W.D. Acute exposure to methylmercury causes Ca2+ dysregulation and neuronal death in rat cerebellar granule cells through an M3 muscarinic receptor-linked pathway. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 80, 60–68. [Google Scholar] [CrossRef]
- Antunes Dos Santos, A.; Ferrer, B.; Marques Gonçalves, F.; Tsatsakis, A.M.; Renieri, E.A.; Skalny, A.V.; Farina, M.; Rocha, J.B.T.; Aschner, M. Oxidative Stress in Methylmercury-Induced Cell Toxicity. Toxics 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, C.; Guo, N.; Sahay, A. Communication, Cross Talk, and Signal Integration in the Adult Hippocampal Neurogenic Niche. Neuron 2020, 105, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.; Cheng, Y.; Li, B.; Petrov, A.; Joshi, P.; Berman, R.F.; Reuhl, K.R.; DiCicco-Bloom, E. Methylmercury elicits rapid inhibition of cell proliferation in the developing brain and decreases cell cycle regulator, cyclin E. Neurotoxicology 2006, 27, 970–981. [Google Scholar] [CrossRef]
- Bose, R.; Onishchenko, N.; Edoff, K.; Janson Lang, A.M.; Ceccatelli, S. Inherited effects of low-dose exposure to methylmercury in neural stem cells. Toxicol. Sci. Off. J. Soc. Toxicol. 2012, 130, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, K.; Obiorah, M.; Robinson, K.; McCandlish, E.; Buckley, B.; DiCicco-Bloom, E. Neural stem cell apoptosis after low-methylmercury exposures in postnatal hippocampus produce persistent cell loss and adolescent memory deficits. Dev. Neurobiol. 2013, 73, 936–949. [Google Scholar] [CrossRef]
- Tian, J.; Luo, Y.; Chen, W.; Yang, S.; Wang, H.; Cui, J.; Lu, Z.; Lin, Y.; Bi, Y. MeHg Suppressed Neuronal Potency of Hippocampal NSCs Contributing to the Puberal Spatial Memory Deficits. Biol. Trace Elem. Res. 2016, 172, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Jebbett, N.J.; Hamilton, J.W.; Rand, M.D.; Eckenstein, F. Low level methylmercury enhances CNTF-evoked STAT3 signaling and glial differentiation in cultured cortical progenitor cells. Neurotoxicology 2013, 38, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, L.O.; Dionizio, A.; Nascimento, P.C.; Puty, B.; Leão, L.K.R.; Luz, D.A.; Silva, M.C.F.; Amado, L.L.; Leite, A.; Buzalaf, M.R.; et al. Proteomic approach underlying the hippocampal neurodegeneration caused by low doses of methylmercury after long-term exposure in adult rats. Metallomics 2019, 11, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Freire, M.A.M.; Santana, L.N.S.; Bittencourt, L.O.; Nascimento, P.C.; Fernandes, R.M.; Leão, L.K.R.; Fernandes, L.M.P.; Silva, M.C.F.; Amado, L.L.; Gomes-Leal, W.; et al. Methylmercury intoxication and cortical ischemia: Pre-clinical study of their comorbidity. Ecotoxicol. Environ. Saf. 2019, 174, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Radio, N.M.; Freudenrich, T.M.; Robinette, B.L.; Crofton, K.M.; Mundy, W.R. Comparison of PC12 and cerebellar granule cell cultures for evaluating neurite outgrowth using high content analysis. Neurotoxicol. Teratol. 2010, 32, 25–35. [Google Scholar] [CrossRef]
- Ferraro, L.; Tomasini, M.C.; Tanganelli, S.; Mazza, R.; Coluccia, A.; Carratù, M.R.; Gaetani, S.; Cuomo, V.; Antonelli, T. Developmental exposure to methylmercury elicits early cell death in the cerebral cortex and long-term memory deficits in the rat. Int. J. Dev. Neurosci. 2009, 27, 165–174. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F. Methylmercury causes neuronal cell death through the suppression of the TrkA pathway: In vitro and in vivo effects of TrkA pathway activators. Toxicol. Appl. Pharmacol. 2015, 282, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F.; Cheng, J.; Zhao, W. Prenatal low-dose methylmercury exposure impairs neurite outgrowth and synaptic protein expression and suppresses TrkA pathway activity and eEF1A1 expression in the rat cerebellum. Toxicol. Appl. Pharmacol. 2016, 298, 1–8. [Google Scholar] [CrossRef]
- Jacob, S.; Sumathi, T. Extenuation of in utero toxic effects of MeHg in the developing neurons by Fisetin via modulating the expression of synaptic transmission and plasticity regulators in hippocampus of the rat offspring. Chem. Biol. Interact. 2019, 305, 3–10. [Google Scholar] [CrossRef]
- Colón-Rodríguez, A.; Colón-Carrión, N.M.; Atchison, W.D. AMPA receptor contribution to methylmercury-mediated alteration of intracellular Ca(2+) concentration in human induced pluripotent stem cell motor neurons. Neurotoxicology 2020, 81, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Takanezawa, Y.; Nakamura, R.; Sone, Y.; Uraguchi, S.; Kiyono, M. An autophagy deficiency promotes methylmercury-induced multinuclear cell formation. Biochem. Biophys. Res. Commun. 2019, 511, 460–467. [Google Scholar] [CrossRef]
- Rolls, M.M.; Thyagarajan, P.; Feng, C. Microtubule dynamics in healthy and injured neurons. Dev. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Hajela, R.K.; Atchison, W.D. Effects of methylmercury on human neuronal L-type calcium channels transiently expressed in human embryonic kidney cells (HEK-293). J. Pharmacol. Exp. Ther. 2002, 302, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.D.S.; Falcão, K.V.G.; Amaral, I.P.G.; Leite, A.C.R.; Bezerra, R.S. Mitochondria as targets for toxicity and metabolism research using zebrafish. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129634. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.; Zhao, L.; Wu, Q.; Wu, C.; Chang, X.; Zhou, Z. Low-Dose Methylmercury-Induced Apoptosis and Mitochondrial DNA Mutation in Human Embryonic Neural Progenitor Cells. Oxid. Med. Cell Longev. 2016, 2016, 5137042. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Yasutake, A.; Marumoto, M.; Hirayama, K. Methylmercury inhibits electron transport chain activity and induces cytochrome c release in cerebellum mitochondria. J. Toxicol. Sci. 2011, 36, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Dreiem, A.; Seegal, R.F. Methylmercury-induced changes in mitochondrial function in striatal synaptosomes are calcium-dependent and ROS-independent. Neurotoxicology 2007, 28, 720–726. [Google Scholar] [CrossRef]
- Chung, Y.P.; Yen, C.C.; Tang, F.C.; Lee, K.I.; Liu, S.H.; Wu, C.C.; Hsieh, S.S.; Su, C.C.; Kuo, C.Y.; Chen, Y.W. Methylmercury exposure induces ROS/Akt inactivation-triggered endoplasmic reticulum stress-regulated neuronal cell apoptosis. Toxicology 2019, 425, 152245. [Google Scholar] [CrossRef]
- Farina, M.; Aschner, M.; da Rocha, J.B.T. The catecholaminergic neurotransmitter system in methylmercury-induced neurotoxicity. Adv. Neurotoxicol. 2017, 1, 47–81. [Google Scholar] [CrossRef]
- Ke, T.; Tsatsakis, A.; Santamaría, A.; Antunes Soare, F.A.; Tinkov, A.A.; Docea, A.O.; Skalny, A.; Bowman, A.B.; Aschner, M. Chronic exposure to methylmercury induces puncta formation in cephalic dopaminergic neurons in Caenorhabditis elegans. Neurotoxicology 2020, 77, 105–113. [Google Scholar] [CrossRef]
- Petroni, D.; Tsai, J.; Mondal, D.; George, W. Attenuation of low dose methylmercury and glutamate induced-cytotoxicity and tau phosphorylation by an N-methyl-D-aspartate antagonist in human neuroblastoma (SHSY5Y) cells. Environ. Toxicol. 2013, 28, 700–706. [Google Scholar] [CrossRef]
- Sceniak, M.P.; Spitsbergen, J.B.; Sabo, S.L.; Yuan, Y.; Atchison, W.D. Acute neurotoxicant exposure induces hyperexcitability in mouse lumbar spinal motor neurons. J. Neurophysiol. 2020, 123, 1448–1459. [Google Scholar] [CrossRef]
- Yuan, Y. Methylmercury: A potential environmental risk factor contributing to epileptogenesis. Neurotoxicology 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Brookes, N. In vitro evidence for the role of glutamate in the CNS toxicity of mercury. Toxicology 1992, 76, 245–256. [Google Scholar] [CrossRef]
- Sakaue, M.; Maki, T.; Kaneko, T.; Hemmi, N.; Sekiguchi, H.; Horio, T.; Kadowaki, E.; Ozawa, A.; Yamamoto, M. Potentiation of Methylmercury-Induced Death in Rat Cerebellar Granular Neurons Occurs by Further Decrease of Total Intracellular GSH with BDNF via TrkB in Vitro. Biol. Pharm. Bull. 2016, 39, 1047–1054. [Google Scholar] [CrossRef]
- Algarve, T.D.; Assmann, C.E.; Cadoná, F.C.; Machado, A.K.; Manica-Cattani, M.F.; Sato-Miyata, Y.; Asano, T.; Duarte, M.; Ribeiro, E.E.; Aigaki, T.; et al. Guarana improves behavior and inflammatory alterations triggered by methylmercury exposure: An in vivo fruit fly and in vitro neural cells study. Environ. Sci. Pollut. Res. Int. 2019, 26, 15069–15083. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F. Methylmercury induces oxidative stress and subsequent neural hyperactivity leading to cell death through the p38 MAPK-CREB pathway in differentiated SH-SY5Y cells. Neurotoxicology 2018, 67, 226–233. [Google Scholar] [CrossRef]
- Santana, L.; Bittencourt, L.O.; Nascimento, P.C.; Fernandes, R.M.; Teixeira, F.B.; Fernandes, L.M.P.; Freitas Silva, M.C.; Nogueira, L.S.; Amado, L.L.; Crespo-Lopez, M.E.; et al. Low doses of methylmercury exposure during adulthood in rats display oxidative stress, neurodegeneration in the motor cortex and lead to impairment of motor skills. J. Trace Elem. Med. Biol. 2019, 51, 19–27. [Google Scholar] [CrossRef]
- Tamm, C.; Duckworth, J.; Hermanson, O.; Ceccatelli, S. High susceptibility of neural stem cells to methylmercury toxicity: Effects on cell survival and neuronal differentiation. J. Neurochem. 2006, 97, 69–78. [Google Scholar] [CrossRef]
- Liu, W.; Yang, T.; Xu, Z.; Xu, B.; Deng, Y. Methyl-mercury induces apoptosis through ROS-mediated endoplasmic reticulum stress and mitochondrial apoptosis pathways activation in rat cortical neurons. Free Radic. Res. 2019, 53, 26–44. [Google Scholar] [CrossRef]
- Castoldi, A.F.; Barni, S.; Turin, I.; Gandini, C.; Manzo, L. Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J. Neurosci. Res. 2000, 59, 775–787. [Google Scholar] [CrossRef]
- Ceccatelli, S.; Bose, R.; Edoff, K.; Onishchenko, N.; Spulber, S. Long-lasting neurotoxic effects of exposure to methylmercury during development. J. Intern. Med. 2013, 273, 490–497. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novo, J.P.; Martins, B.; Raposo, R.S.; Pereira, F.C.; Oriá, R.B.; Malva, J.O.; Fontes-Ribeiro, C. Cellular and Molecular Mechanisms Mediating Methylmercury Neurotoxicity and Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 3101. https://doi.org/10.3390/ijms22063101
Novo JP, Martins B, Raposo RS, Pereira FC, Oriá RB, Malva JO, Fontes-Ribeiro C. Cellular and Molecular Mechanisms Mediating Methylmercury Neurotoxicity and Neuroinflammation. International Journal of Molecular Sciences. 2021; 22(6):3101. https://doi.org/10.3390/ijms22063101
Chicago/Turabian StyleNovo, João P., Beatriz Martins, Ramon S. Raposo, Frederico C. Pereira, Reinaldo B. Oriá, João O. Malva, and Carlos Fontes-Ribeiro. 2021. "Cellular and Molecular Mechanisms Mediating Methylmercury Neurotoxicity and Neuroinflammation" International Journal of Molecular Sciences 22, no. 6: 3101. https://doi.org/10.3390/ijms22063101
APA StyleNovo, J. P., Martins, B., Raposo, R. S., Pereira, F. C., Oriá, R. B., Malva, J. O., & Fontes-Ribeiro, C. (2021). Cellular and Molecular Mechanisms Mediating Methylmercury Neurotoxicity and Neuroinflammation. International Journal of Molecular Sciences, 22(6), 3101. https://doi.org/10.3390/ijms22063101