
Combined Genome, Transcriptome and Metabolome Analysis in the Diagnosis of Childhood Cerebellar Ataxia

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
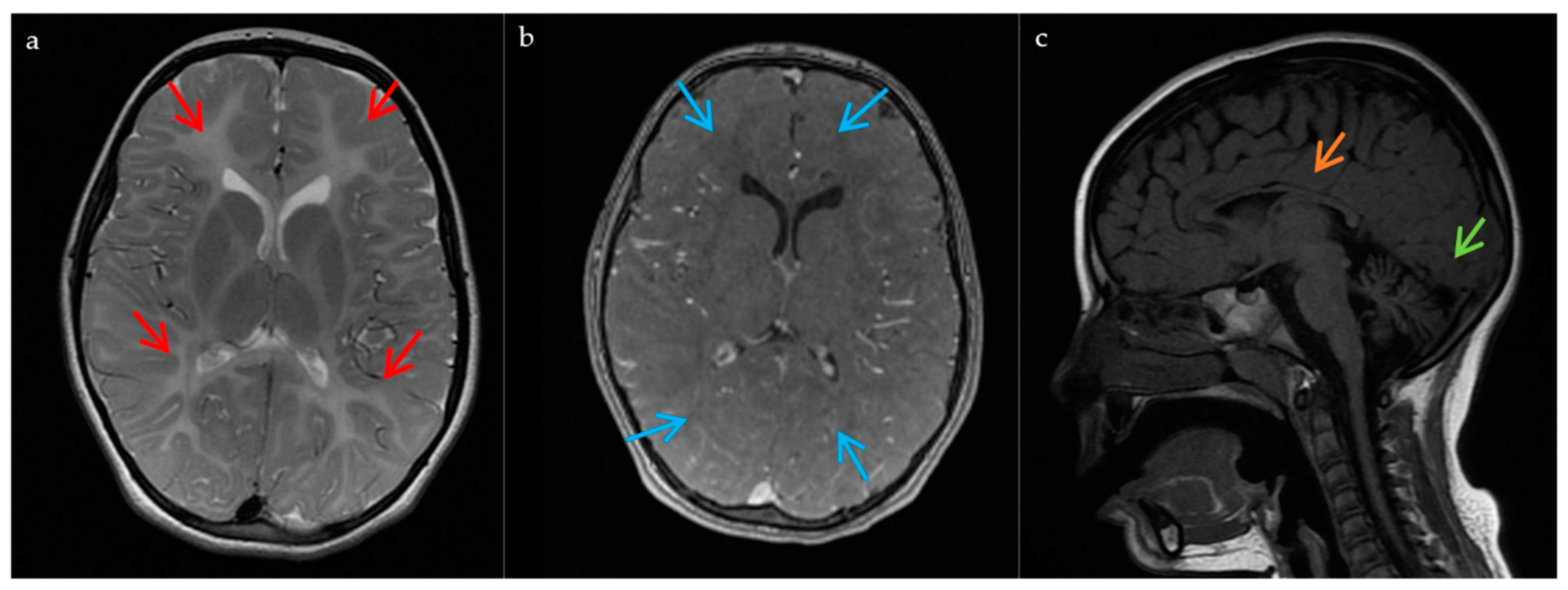
2.1. Case Report
2.2. Whole-Exome Analysis
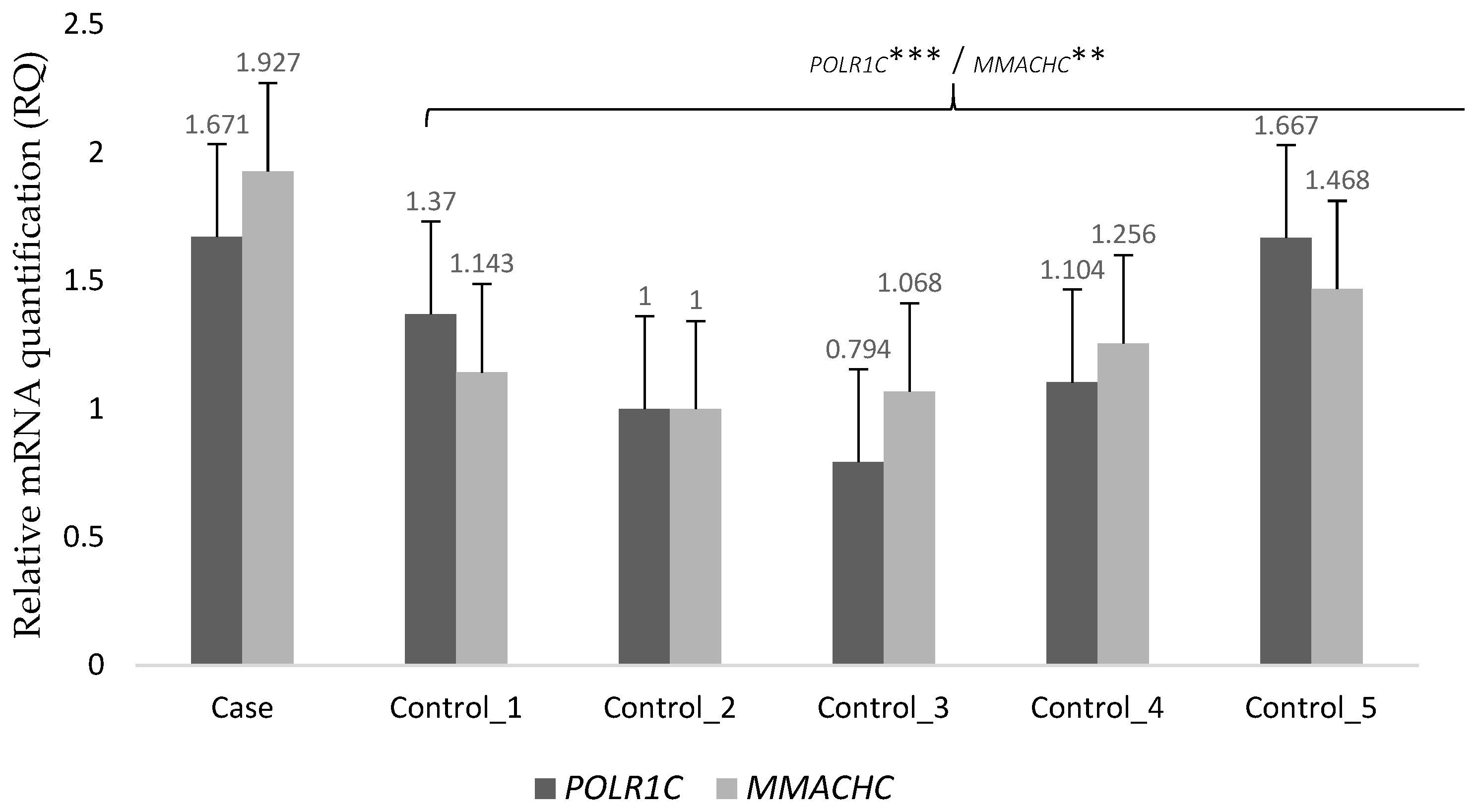
2.3. Transcriptome Analysis
2.4. Metabolome Analysis
3. Discussion
4. Materials and Methods
4.1. Consent and Approval
4.2. WES for Candidate Gene Identification
4.3. RNA-Seq for Gene Expression Analysis
4.4. Validation of Next-Generation Sequencing (NGS) Candidate Variants
4.5. Functional Validation through Metabolomics
4.5.1. Metabolite Extraction and Mass Spectrometry Analysis
4.5.2. Data Treatment
4.5.3. Metabolite Identification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fogel, B.L. Childhood cerebellar ataxia. J. Child Neurol. 2012, 27, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Vedolin, L.; Gonzalez, G.; Souza, C.F.; Lourenço, C.; Barkovich, A.J. Inherited cerebellar ataxia in childhood: A pattern-recognition approach using brain MRI. Am. J. Neuroradiol. 2013, 34, 925–934. [Google Scholar] [CrossRef]
- Konczak, J.; Timmann, D. The effect of damage to the cerebellum on sensorimotor and cognitive function in children and adolescents. Neurosci. Biobehav. Rev. 2007, 31, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Martindale, J.; Shanmugarajah, P.; Grünewald, R.A.; Sarrigiannis, P.G.; Beauchamp, N.; Garrard, K.; Warburton, R.; Sanders, D.S.; Friend, D.; et al. Causes of progressive cerebellar ataxia: Prospective evaluation of 1500 patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 301–309. [Google Scholar] [CrossRef]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Musselman, K.E.; Stoyanov, C.T.; Marasigan, R.; Jenkins, M.E.; Konczak, J.; Morton, S.M.; Bastian, A.J. Prevalence of ataxia in children: A systematic review. Neurology 2014, 82, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Sandford, E.; Burmeister, M. Genes and genetic testing in hereditary ataxias. Genes 2014, 5, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Lee, H.; Deignan, J.L.; Strom, S.P.; Kantarci, S.; Wang, X.; Quintero-Rivera, F.; Vilain, E.; Grody, W.W.; Perlman, S.; et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014, 71, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Subramony, S.H. Chronic childhood ataxia: The cause depends on how you look. Dev. Med. Child Neurol. 2013, 55, 298–299. [Google Scholar] [CrossRef][Green Version]
- Rossi, M.; Anheim, M.; Durr, A.; Klein, C.; Koenig, M.; Synofzik, M.; Marras, C.; van de Warrenburg, B.P. The genetic nomenclature of recessive cerebellar ataxias. Mov. Disord. 2018, 33, 1056–1076. [Google Scholar] [CrossRef]
- Holmboe, E.S.; Durning, S.J. Assessing clinical reasoning: Moving from in vitro to in vivo. Diagnosis 2014, 1, 111–117. [Google Scholar] [CrossRef] [PubMed]
- De Silva, R.N.; Vallortigara, J.; Greenfield, J.; Hunt, B.; Giunti, P.; Hadjivassiliou, M. Diagnosis and management of progressive ataxia in adults. Pract. Neurol. 2019, 19, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Praticò, A.D.; Pavone, V.; Lubrano, R.; Falsaperla, R.; Rizzo, R.; Ruggieri, M. Ataxia in children: Early recognition and clinical evaluation. Ital. J. Pediatr. 2017, 43, 6. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Schwartzentruber, J.; Beaulieu, C.L.; Dyment, D.; Smith, A.; Chardon, J.W.; Yoon, G.; Rouleau, G.A.; Suchowersky, O.; Siu, V.; et al. Exome Sequencing as a Diagnostic Tool for Pediatric-Onset Ataxia. Hum. Mutat. 2014, 35, 45–49. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef]
- Smith, H.S.; Swint, J.M.; Lalani, S.R.; Yamal, J.M.; de Oliveira Otto, M.C.; Castellanos, S.; Taylor, A.; Lee, B.H.; Russell, H.V. Clinical Application of Genome and Exome Sequencing as a Diagnostic Tool for Pediatric Patients: A Scoping Review of the Literature. Genet. Med. 2019, 21, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Green, R.C. Diagnostic Clinical Genome and Exome Sequencing. N. Engl. J. Med. 2014, 370, 2418–2425. [Google Scholar] [CrossRef]
- Sanderson, S.C.; Hill, M.; Patch, C.; Searle, B.; Lewis, C.; Chitty, L.S. Delivering genome sequencing in clinical practice: An interview study with healthcare professionals involved in the 100 000 Genomes Project. BMJ Open 2019, 9, e029699. [Google Scholar] [CrossRef] [PubMed]
- Thiffault, I.; Wolf, N.I.; Forget, D.; Guerrero, K.; Tran, L.T.; Choquet, K.; Lavallée-Adam, M.; Poitras, C.; Brais, B.; Yoon, G.; et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat. Commun. 2015, 6, 7623. [Google Scholar] [CrossRef] [PubMed]
- Kraoua, I.; Karkar, A.; Drissi, C.; Benrhouma, H.; Klaa, H.; Samaan, S.; Renaldo, F.; Elmaleh, M.; Ben Hamouda, M.; Abdelhak, S.; et al. Novel POLR1C mutation in RNA polymerase III-related leukodystrophy with severe myoclonus and dystonia. Mol. Genet. Genom. Med. 2019, 7, e914. [Google Scholar] [CrossRef]
- Han, J.Y.; Kim, S.Y.; Cheon, J.E.; Choi, M.; Lee, J.S.; Chae, J.H. A familial case of childhood ataxia with leukodystrophy due to novel POLR1C mutations. J. Clin. Neurol. 2020, 16, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Gauquelin, L.; Cayami, F.K.; Sztriha, L.; Yoon, G.; Tran, L.T.; Guerrero, K.; Hocke, F.; van Spaendonk, R.M.L.; Fung, E.L.; D’Arrigo, S.; et al. Clinical spectrum of POLR3-related leukodystrophy caused by biallelic POLR1C pathogenic variants. Neurol. Genet. 2019, 5, e369. [Google Scholar] [CrossRef] [PubMed]
- Zwaenepoel, I.; Mustapha, M.; Leibovici, M.; Verpy, E.; Goodyear, R.; Liu, X.Z.; Nouaille, S.; Nance, W.E.; Kanaan, M.; Avraham, K.B.; et al. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc. Natl. Acad. Sci. USA 2002, 99, 6240–6245. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Dauwerse, J.G.; Dixon, J.; Seland, S.; Ruivenkamp, C.A.L.; Van Haeringen, A.; Hoefsloot, L.H.; Peters, D.J.M.; De Boers, A.C.; Daumer-Haas, C.; Maiwald, R.; et al. Mutations in genes encoding subunits of RNA polymerases i and III cause Treacher Collins syndrome. Nat. Genet. 2011, 43, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Ellis, J.P.; Tirone, J.C.; Pawelek, P.D.; Doré, C.; Atkinson, J.L.; Watkins, D.; Morel, C.F.; Fujiwara, T.M.; Moras, E.; Hosack, A.R.; et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet. 2006, 38, 93–100. [Google Scholar] [CrossRef]
- Froese, D.S.; Kopec, J.; Fitzpatrick, F.; Schuller, M.; McCorvie, T.J.; Chalk, R.; Plessl, T.; Fettelschoss, V.; Fowler, B.; Baumgartner, M.R.; et al. Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J. Biol. Chem. 2015, 290, 29167–29177. [Google Scholar] [CrossRef]
- Perrier, S.; Michell-Robinson, M.A.; Bernard, G. POLR3-Related Leukodystrophy: Exploring Potential Therapeutic Approaches. Front. Cell. Neurosci. 2021, 14, 487. [Google Scholar] [CrossRef]
- Bernard, G.; Thiffault, I.; Tetreault, M.; Putorti, M.L.; Bouchard, I.; Sylvain, M.; Melançon, S.; Laframboise, R.; Langevin, P.; Bouchard, J.P.; et al. Tremor-ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3-10q23.31. Neurogenetics 2010, 11, 457–464. [Google Scholar] [CrossRef]
- Tétreault, M.; Choquet, K.; Orcesi, S.; Tonduti, D.; Balottin, U.; Teichmann, M.; Fribourg, S.; Schiffmann, R.; Brais, B.; Vanderver, A.; et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 652–655. [Google Scholar] [CrossRef]
- Daoud, H.; Tétreault, M.; Gibson, W.; Guerrero, K.; Cohen, A.; Gburek-Augustat, J.; Synofzik, M.; Brais, B.; Stevens, C.A.; Sanchez-Carpintero, R.; et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J. Med. Genet. 2013, 50, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Dorboz, I.; Dumay-Odelot, H.; Boussaid, K.; Bouyacoub, Y.; Barreau, P.; Samaan, S.; Jmel, H.; Eymard-Pierre, E.; Cances, C.; Bar, C.; et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol. Genet. 2018, 4, e289. [Google Scholar] [CrossRef] [PubMed]
- Terhal, P.A.; Vlaar, J.M.; Middelkamp, S.; Nievelstein, R.A.J.; Nikkels, P.G.J.; Ross, J.; Créton, M.; Bos, J.W.; Voskuil-Kerkhof, E.S.M.; Cuppen, E.; et al. Biallelic variants in POLR3GL cause endosteal hyperostosis and oligodontia. Eur. J. Hum. Genet. 2020, 28, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, M.; Hernandez, N. RNA polymerase III transcription as a disease factor. Genes Dev. 2020, 34, 865–882. [Google Scholar] [CrossRef]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Koy, A.; Lin, J.P.; Sanger, T.D.; Marks, W.A.; Mink, J.W.; Timmermann, L. Advances in management of movement disorders in children. Lancet Neurol. 2016, 15, 719–735. [Google Scholar] [CrossRef]
- Kashiki, H.; Li, H.; Miyamoto, S.; Ueno, H.; Tsurusaki, Y.; Ikeda, C.; Kurata, H.; Okada, T.; Shimazu, T.; Imamura, H.; et al. POLR1C variants dysregulate splicing and cause hypomyelinating leukodystrophy. Neurol. Genet. 2020, 6, e524. [Google Scholar] [CrossRef] [PubMed]
- Morel, C.F.; Lerner-Ellis, J.P.; Rosenblatt, D.S. Combined methylmalonic aciduria and homocystinuria (cblC): Phenotype-genotype correlations and ethnic-specific observations. Mol. Genet. Metab. 2006, 88, 315–321. [Google Scholar] [CrossRef]
- Nogueira, C.; Aiello, C.; Cerone, R.; Martins, E.; Caruso, U.; Moroni, I.; Rizzo, C.; Diogo, L.; Leão, E.; Kok, F.; et al. Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, cblC type. Mol. Genet. Metab. 2008, 93, 475–480. [Google Scholar] [CrossRef]
- Allan Drummond, D.; Wilke, C.O. The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 2009, 10, 715–724. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.Y.; Mitoma, H.; Fatemi, A. Metabolic ataxias. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 155, pp. 117–127. [Google Scholar]
- Blake, J.A.; Dolan, M.; Drabkin, H.; Hill, D.P.; Ni, L.; Sitnikov, D.; Bridges, S.; Burgess, S.; Buza, T.; McCarthy, F.; et al. Gene ontology annotations and resources. Nucleic Acids Res. 2013, 41, D530–D535. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient | Family | Sex (F/M) | Ethnicity | Consanguinity | Age at Last Assessment (Years) | Age of Onset (Years) | Genetic Characteristics | |
|---|---|---|---|---|---|---|---|---|
| Mutation 1 | Mutation 2 | |||||||
| Index | XXIV | F | Caucasian (Spanish) | No | 14 | 2.5 | c.193A>G; | c.836G>A; |
| p.Met65Val | p.Arg279Gln | |||||||
| 1 [19] | I | M | Libyan | Yes | 8 | 0.5 | c.95A>T; | c.95A>T; |
| p.Asn32Ile | p.Asn32Ile | |||||||
| 2 [19] | II | M | Hungarian | No | 10 | 1 | c.221A>G; | c.221A>G; |
| p.Asn74Ser | p.Asn74Ser | |||||||
| 3 [19] | III | M | Asian (Chinese) | No | 4 | 1 | c.436T>C; | c.883_885delAAG; |
| p.Cys146Arg | p.Lys295del | |||||||
| 4 [19] | IV | F | Caucasian (Armenian/Russian) | No | 6 | 2.5 | c.77C>T; | c.326G>A; |
| p.Thr26Ile | p.Arg109His | |||||||
| 5 [19] | V | F | Caucasian | No | 9 | 1.5 | c.193A>G; | c.572G>A; |
| p.Met65Val | p.Arg191Gln | |||||||
| 6 [19] | VI | F | Caucasian (Turkish) | Suspected | 18 | 4 | c.326G>A; | c.970G>A; |
| p.Arg109His | p.Glu324Lys | |||||||
| 7 [19] | VII | M | Caucasian | No | 33 | 2 | c.395G>A; | c.461_462delAA; |
| p.Gly132Asp | p.Lys154Argfs*4 | |||||||
| 8 [19] | VIII | F | Caucasian | No | 2 | 1 | c.281T>C; | c.785T>C; |
| p.Val94Ala | p.Ile262Thr | |||||||
| 9 [20] | IX | F | Tunisian | Yes | 25 | 5 | c.863T>C; | c.863T>C; |
| p.Phe288Ser | p.Phe288Ser | |||||||
| 10 [22] | X | M | Caucasian (British) | No | NA | 0 | c.69+1G>A; | c.836G>A; |
| p.Asn24Asnfs55*; (prediction) | p.Arg279Gln | |||||||
| 11 [22] | XI | M | Caucasian | No | NA | 4 | c.916_920delTATAT; | c.938C>T; |
| 12 [22] | M | p.Tyr306Leufs*4 | p.Thr313Met | |||||
| 13 [22] | XII | M | Caucasian (Dutch) | No | NA | 2 | c.193A>G; | c.733G>A; |
| p.Met65Val | p.Val245Met | |||||||
| 14 [22] | XIII | F | Caucasian (English) | No | NA | 3 | c.313A>T; | c.916_920delTATAT; |
| p.Ile105Phe | p.Tyr306Leufs*4 | |||||||
| 15 [22] | XIV | F | Caucasian (English) | Yes | NA | 2 | c.836G>A; | c.836G>A; |
| p.Arg279Gln | p.Arg279Gln | |||||||
| 16 [22] | XV | F | Norwegian | No | NA | 0.3 | c.88C>T; | c.916_920delTATAT; |
| p.Pro30Ser | p.Tyr306Leufs*4 | |||||||
| 17 [22] | XVI | F | Caucasian (English) | No | NA | 0 | c.221A>G; | c.502G>A; |
| p.Asn74Ser | p.Val168Met + splicing error | |||||||
| 18 [22] | XVII | F | Caucasian | No | NA | 6 | c.79A>G; | c.349G>C; |
| p.Thr27Ala | p.Ala117Pro | |||||||
| 19 [22] | XVIII | F | Caucasian | No | NA | 0.4 | c.322C>T; | c.325C>T; |
| p.His108Tyr | p.Arg109Cys | |||||||
| 20 [22] | XIX | F | African/American | No | NA | 2 | c.70-1G>A; | c.835C>T; |
| p.Asn24Profs27* (prediction) | p.Arg279Trp | |||||||
| 21 [22] | XX | F | Caucasian | No | NA | 0 | c.699C>G; | c.883_885delAAG; |
| p.Tyr233* | p.Lys295del | |||||||
| 22 [22] | XXI | M | Caucasian | No | NA | 1 | c.88C>T; | c.615delC; |
| 23 [22] | F | 0 | p.Pro30Ser | p.Gln206Lysfs*48 | ||||
| 24 [22] | XXII | F | Asian | No | NA | 3.5 | c.77C>T; | c.77C>T; |
| p.Thr26Ile | p.Thr26Ile | |||||||
| 25 [21] | XXIII | M | Asian (Korean) | NA | NA | 5 | c.698_699insAA; | c.713A>G; |
| 26 [21] | F | p.Tyr233fs | p.Asp238Gly | |||||
| Patient | Symptoms at Onset 1 | Develop-mental Delay 1 | Age at Walking without Support (Months) 1 | Abnormal Cognition 1 | Cerebellar Signs | Tremor 1 | Pyramidal Signs | Dystonia | Myoclonus | Age at Wheelchair (Years) | Myopia | Dental AbN | Hypogonadotropic Hypogonadism |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Index | Delayed motor development, tremor, ataxia, dysmetria | + | 28 | + | + | + | + | + | + | 7 | + | − | − |
| 1 [19] | Delayed motor development | + | 22 | + | + | + | + | − | − | 3 | − | + | Too young |
| 2 [19] | Ataxia, tremor, head titubation | + | 18 | + | + | + | + | − | − | 9 * | − | − | Too young |
| 3 [19] | Delayed motor development and failure to thrive | + | Never autonomously | + | + | + | + | + | − | Always | − | − | Too young |
| 4 [19] | Tremor, dysmetria | + | 15 | − | + | + | + | − | − | − * | − | + | Too young |
| 5 [19] | Delayed motor development | + | 24 | + | + | + | + | − | − | Always * | + | − | Too young |
| 6 [19] | Clumsy gait, frequent falls | − | 18 | + | + | + | + | + | − | 9 (long distances) * | + | − | − |
| 7 [19] | Delayed motor development | + | Never autonomously | + | + | + | − | − | + | Puberty | + | − | − |
| 8 [19] | Delayed motor development | + | 24 (with support) | − | + | + | − | − | − | − | − | + | Too young |
| 9 [20] | Tremor, ataxia | − | 12 | − | + | + | + | + | + | 21 * | − | − | − |
| 10 [22] | NA | NA | NA | NA | + | NA | + | − | NA | NA | − | + | Too young |
| 11 [22] | NA | NA | NA | NA | + | NA | − | − | NA | − | − | + | − |
| 12 [22] | NA | NA | NA | NA | + | NA | − | − | NA | − | + | + | − |
| 13 [22] | NA | NA | NA | NA | + | NA | − | − | NA | − | + | − | − |
| 14 [22] | NA | NA | NA | NA | + | NA | − | − | NA | 12 | + | + | Too young |
| 15 [22] | NA | NA | NA | NA | + | NA | + | + | NA | 7 | − | − | Too young |
| 16 [22] | NA | NA | NA | NA | + | NA | − | + | NA | 0 | + | + | Too young |
| 17 [22] | NA | NA | NA | NA | + | NA | + | + | NA | 0 | + | + | Too young |
| 18 [22] | NA | NA | NA | NA | + | NA | + | − | NA | − | − | + | − |
| 19 [22] | NA | NA | NA | NA | + | NA | + | − | NA | 0 | − | + | − |
| 20 [22] | NA | NA | NA | NA | + | NA | + | − | NA | 11 | − | − | Too young |
| 21 [22] | NA | NA | NA | NA | + | NA | + | + | NA | 3 | + | + | Too young |
| 22 [22] | NA | NA | NA | NA | + | NA | + | + | NA | 4 | +C | + | Too young |
| 23 [22] | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | − | Too young |
| 24 [22] | NA | NA | NA | NA | + | NA | − | − | NA | − | + | + | Too young |
| 25 [21] | Tremor, ataxia | + | NA | + | + | + | + | NA | − | NA | + | − | − |
| 26 [21] | Tremor, ataxia | + | NA | + | + | + | + | NA | + | NA | + | − | − |
| Patient | Diffuse Hypomyelination | Cerebellar Atrophy | Thin Corpus Callosum |
|---|---|---|---|
| Index | + | + | + |
| 1 [19] | + | − | + |
| 2 [19] | + | − | + |
| 3 [19] | + | − | + |
| 4 [19] | + | + | + |
| 5 [19] | + | + | + |
| 6 [19] | + | + | + |
| 7 [19] | + | + | + |
| 8 [19] | + | + | + |
| 9 [20] | + | + | + |
| 10 [22] | + | − | + |
| 11 [22] | + | + | + |
| 12 [22] | + | + | + |
| 13 [22] | + | + | + |
| 14 [22] | + | + | + |
| 15 [22] | + | + | + |
| 16 [22] | + | − | − |
| 17 [22] | + | + | + |
| 18 [22] | + | + | + |
| 19 [22] | + | + | + |
| 20 [22] | + | + | + |
| 21 [22] | + | − | + |
| 22 [22] | + | + | + |
| 23 [22] | NA | NA | NA |
| 24 [22] | + | + | + |
| 25 [21] | + | − | NA |
| 26 [21] | + | − | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ching-López, A.; Martinez-Gonzalez, L.J.; Arrabal, L.; Sáiz, J.; Gavilán, Á.; Barbas, C.; Lorente, J.A.; Roldán, S.; Sánchez, M.J.; Gutierrez-Ríos, P. Combined Genome, Transcriptome and Metabolome Analysis in the Diagnosis of Childhood Cerebellar Ataxia. Int. J. Mol. Sci. 2021, 22, 2990. https://doi.org/10.3390/ijms22062990
Ching-López A, Martinez-Gonzalez LJ, Arrabal L, Sáiz J, Gavilán Á, Barbas C, Lorente JA, Roldán S, Sánchez MJ, Gutierrez-Ríos P. Combined Genome, Transcriptome and Metabolome Analysis in the Diagnosis of Childhood Cerebellar Ataxia. International Journal of Molecular Sciences. 2021; 22(6):2990. https://doi.org/10.3390/ijms22062990
Chicago/Turabian StyleChing-López, Ana, Luis Javier Martinez-Gonzalez, Luisa Arrabal, Jorge Sáiz, Ángela Gavilán, Coral Barbas, Jose Antonio Lorente, Susana Roldán, Maria José Sánchez, and Purificacion Gutierrez-Ríos. 2021. "Combined Genome, Transcriptome and Metabolome Analysis in the Diagnosis of Childhood Cerebellar Ataxia" International Journal of Molecular Sciences 22, no. 6: 2990. https://doi.org/10.3390/ijms22062990
APA StyleChing-López, A., Martinez-Gonzalez, L. J., Arrabal, L., Sáiz, J., Gavilán, Á., Barbas, C., Lorente, J. A., Roldán, S., Sánchez, M. J., & Gutierrez-Ríos, P. (2021). Combined Genome, Transcriptome and Metabolome Analysis in the Diagnosis of Childhood Cerebellar Ataxia. International Journal of Molecular Sciences, 22(6), 2990. https://doi.org/10.3390/ijms22062990