
Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cyclin-Dependent Kinases (CDKs)
2.1. Cyclin-Dependent Kinase 1 (CDK1)
2.2. Cyclin-Dependent Kinase 2 (CDK2)
2.3. Cyclin-Dependent Kinases 4 and 6 (CDK4/6)
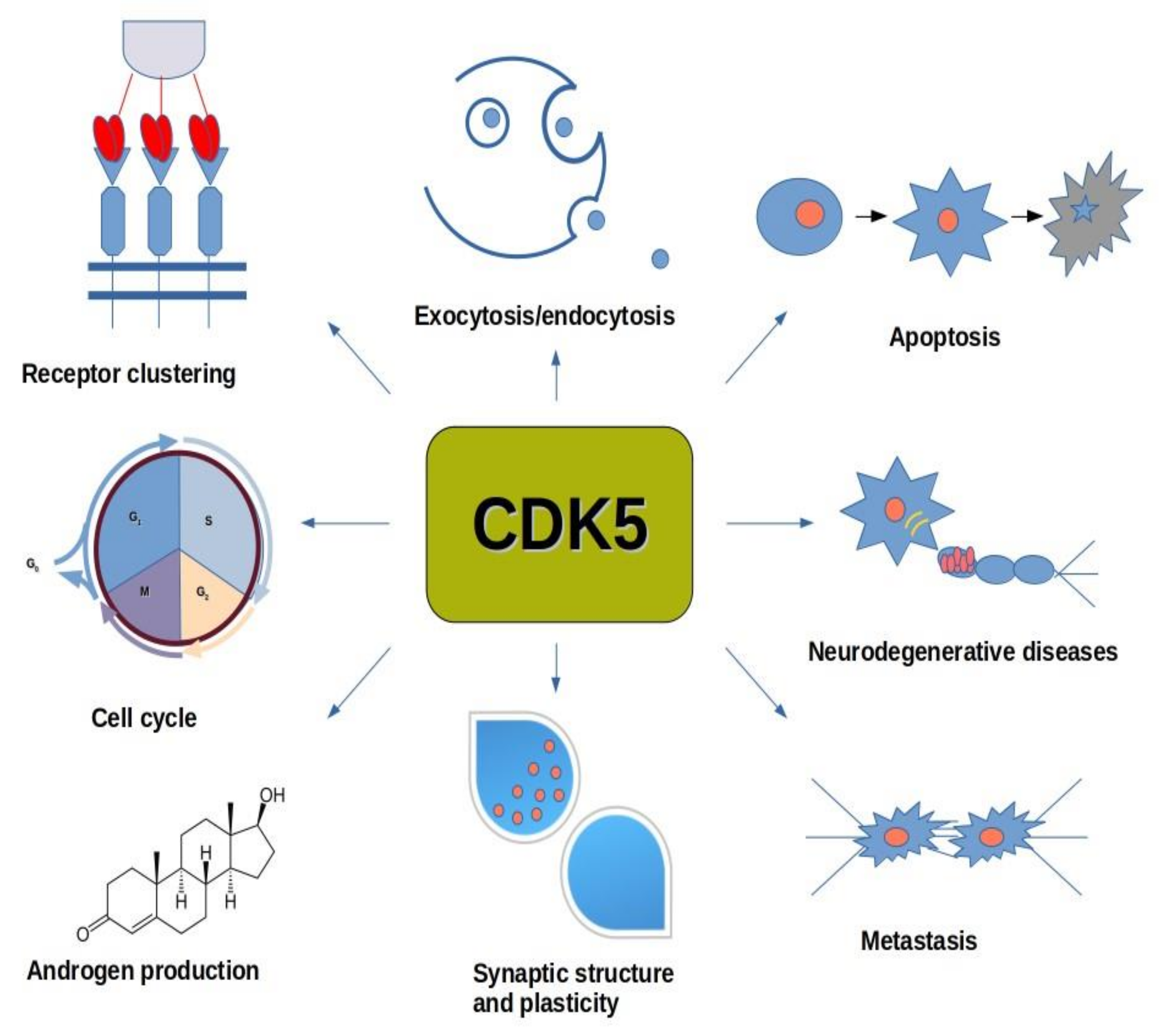
2.4. Cyclin-Dependent Kinase 5 (CDK5)
2.5. Cyclin-Dependent Kinase 7 (CDK7)
2.6. Cyclin-Dependent Kinases 8 and 19 (CDK8, CDK19)
2.7. Cyclin-Dependent Kinase 9 (CDK9)
2.8. Cyclin-Dependent Kinase 10 (CDK10)
2.9. Cyclin-Dependent Kinase 11 (CDK11)
2.10. Cyclin-Dependent Kinases 12 and 13 (CDK12, CDK13)
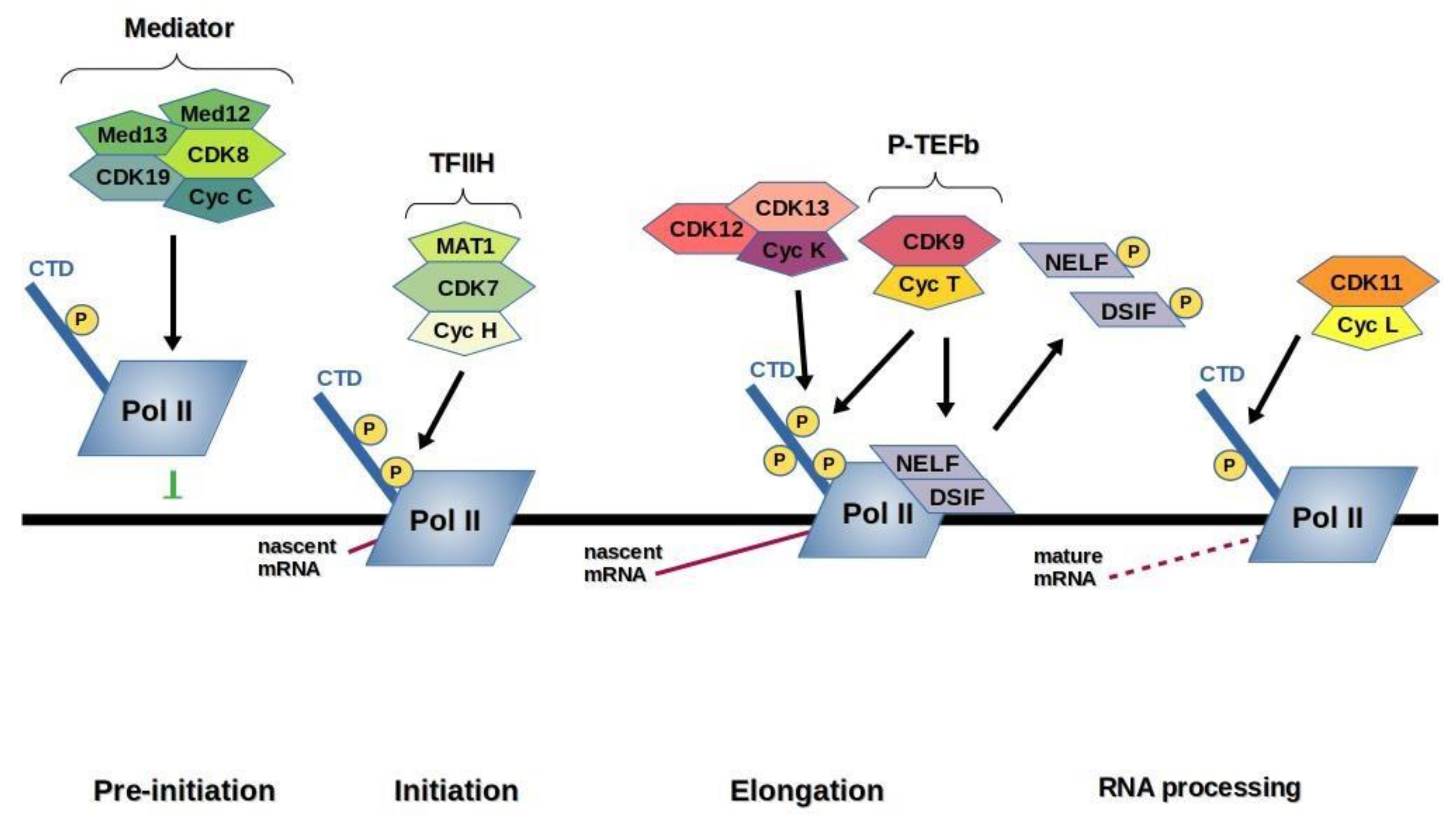
3. Transcription CDKs
4. Cell Cycle CDKs
5. Cell Cycle and Tumor Development
6. Role of CDKs in Cancer Development
6.1. CDK1
6.2. CDK2
6.3. CDK4/6
6.4. CDK5
6.5. CDK7
6.6. CDK9
6.7. CDK11
6.8. CDK12/CDK13
7. Role of CDKs in Rare Developmental Disorders
7.1. CDK4/6
7.2. CDK5
7.3. CDK8/CDK19
7.4. CDK10
7.5. CDK12/CDK13
8. Role of CDKs in Other Disorders
8.1. CDK5 and Neurological Diseases
8.2. CDK9 and HIV
8.3. CDK9 and Cardiac Disorders
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethical Approval
References
- The Nobel Prize in Physiology or Medicine 2001. 2001. Available online: http://nobelprize.org/nobel_prizes/medicine/laureates/2001/ (accessed on 20 November 2020).
- Pavletich, N.P. Mechanisms of cyclin-dependent kinase regulation: Structures of cdks, their cyclin activators, and cip and INK4 inhibitors. J. Mol. Biol. 1999, 287, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Osuga, H.; Osuga, S.; Wang, F.; Fetni, R.; Hogan, M.J.; Slack, R.S.; Hakim, A.M.; Ikeda, J.-E.; Park, D.S. Cyclin-dependent kinases as a therapeutic target for stroke. Proc. Natl. Acad. Sci. USA 2000, 97, 10254–10259. [Google Scholar] [CrossRef]
- Pines, J. Cyclins, CDKs and cancer. Semin. Cancer Biol. 1995, 6, 63–72. [Google Scholar] [CrossRef]
- Wang, S.; Fischer, P.M. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef]
- Arellano, M.; Moreno, S. Regulation of CDK/cyclin Complexes during the Cell Cycle. Int. J. Biochem. Cell Biol. 1997, 29, 559–573. [Google Scholar] [CrossRef]
- Ekholm, S.V.; Reed, S.I. Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 2000, 12, 676–684. [Google Scholar] [CrossRef]
- Senderowicz, A.M. Novel direct and indirect cyclin dependent kinase modulators for the prevention and treatment of human neoplasms. Cancer Chemother. Pharmacol. 2003, 52, S61–S73. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Knockaert, M.; Greengard, P.; Meijer, L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol. Sci. 2002, 23, 417–425. [Google Scholar] [CrossRef]
- Fung, T.K.; Poon, R.Y.C. A roller coaster ride with the mitotic cyclins. Semin. Cell Dev. Biol. 2005, 16, 335–342. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massagué, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef] [PubMed]
- De Bondt, H.L.; Rosenblatt, J.; Jancarik, J.; Jones, H.D.; Morgant, D.O.; Kim, S.H. Crystal structure of cyclin dependent kinase 2. Nature 1993, 363, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Franco, G.; Guzzi, P.; Manca, V.; Mazza, T. Mitotic Oscillators as MP Graphs. In Membrane Computing; Springer: Berlin/Heidelberg, Germany, 2006; pp. 382–394. [Google Scholar]
- Gould, K.L.; Moreno, S.; Owen, D.J.; Sazer, S.; Nurse, P. Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J. 1991, 10, 3297–3309. [Google Scholar] [CrossRef] [PubMed]
- Coleman, T.R.; Dunphy, W.G. Cdc2 regulatory factors. Curr. Opin. Cell Biol. 1994, 6, 877–882. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- London, N.; Biggins, S. Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell Biol. 2014, 15, 736–747. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872. [Google Scholar] [CrossRef]
- Ziebold, U.; Bartsch, O.; Marais, R.; Ferrari, S.; Klempnauer, K.-H. Phosphorylation and activation of B-Myb by cyclin A. Curr. Biol. 1997, 7, 253–260. [Google Scholar] [CrossRef]
- Hydbring, P.; Bahram, F.; Su, Y.; Tronnersjö, S.; Högstrand, K.; von der Lehr, N.; Sharifi, H.R.; Lilischkis, R.; Hein, N.; Wu, S.; et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl. Acad. Sci. USA 2010, 107, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef]
- Hara, E.; Hall, M.; Peters, G. Cdk2-dependent phosphorylation of Id2 modulates activity of E2A-related transcription factors. EMBO J. 1997, 16, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Regan, K.M.; Lou, Z.; Chen, J.; Tindall, D.J. CDK2-Dependent Phosphorylation of FOXO1 as an Apoptotic Response to DNA Damage. Science 2006, 314, 294. [Google Scholar] [CrossRef] [PubMed]
- Major, M.L.; Lepe, R.; Costa, R.H. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol. 2004, 24, 2649–2661. [Google Scholar] [CrossRef]
- Yun, J.; Chae, H.-D.; Choi, T.-S.; Kim, E.-H.; Bang, Y.-J.; Chung, J.; Choi, K.-S.; Mantovani, R.; Shin, D.Y. Cdk2-dependent Phosphorylation of the NF-Y Transcription Factor and Its Involvement in the p53-p21 Signaling Pathway. J. Biol. Chem. 2003, 278, 36966–36972. [Google Scholar] [CrossRef]
- Ying, M.; Shao, X.; Jing, H.; Liu, Y.; Qi, X.; Cao, J.; Chen, Y.; Xiang, S.; Song, H.; Hu, R.; et al. Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood 2018, 131, 2698–2711. [Google Scholar] [CrossRef] [PubMed]
- Grishina, I.; Lattes, B. A Novel Cdk2 Interactor is Phosphorylated by Cdc7 and Associates with Components of the Replication Complexes. Cell Cycle 2005, 4, 4120–4126. [Google Scholar] [CrossRef]
- Saurus, P.; Kuusela, S.; Dumont, V.; Lehtonen, E.; Fogarty, C.L.; Lassenius, M.I.; Forsblom, C.; Lehto, M.; Saleem, M.A.; Groop, P.-H.; et al. Cyclin-dependent kinase 2 protects podocytes from apoptosis. Sci. Rep. 2016, 6, 21664. [Google Scholar] [CrossRef]
- Chunder, N.; Wang, L.; Chen, C.; Hancock, W.W.; Wells, A.D. Cyclin-dependent kinase 2 controls peripheral immune tolerance. J. Immunol. 2012, 189, 5659–5666. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Weintraub, S.J.; Chow, K.N.B.; Luo, R.X.; Zhang, S.H.; He, S.; Dean, D.C. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature 1995, 375, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Luo, R.X.; Santi, A.D.; Postigo, A.A.; Dean, D.C. Cdk Phosphorylation Triggers Sequential Intramolecular Interactions that Progressively Block Rb Functions as Cells Move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef]
- Dhavan, R.; Tsai, L.-H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Kelz, M.B.; Steffen, C.; Ang, E.S.; Zeng, L.; Nestler, E.J. Induction of cyclin-dependent kinase 5 in the hippocampus by chronic electroconvulsive seizures: Role of [Delta]FosB. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 8965–8971. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, K.-T. Induction of cyclin-dependent kinase 5 and its activator p35 through the extracellular-signal-regulated kinase and protein kinase A pathways during retinoic-acid mediated neuronal differentiation in human neuroblastoma SK-N-BE(2)C cells. J. Neurochem. 2004, 91, 634–647. [Google Scholar] [CrossRef]
- Ikiz, B.; Przedborski, S. A Sequel to the Tale of p25/Cdk5 in Neurodegeneration. Neuron 2008, 60, 731–732. [Google Scholar] [CrossRef][Green Version]
- Tang, J.; Ip, J.P.K.; Ye, T.; Ng, Y.-P.; Yung, W.-H.; Wu, Z.; Fang, W.; Fu, A.K.Y.; Ip, N.Y. Cdk5-dependent Mst3 phosphorylation and activity regulate neuronal migration through RhoA inhibition. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 7425–7436. [Google Scholar] [CrossRef]
- Cheung, Z.H.; Ip, N.Y. Cdk5: A multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012, 22, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Arif, A. Extraneuronal activities and regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5. Biochem. Pharmacol. 2012, 84, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, N.; Shibasaki, T.; Kashima, Y.; Miki, T.; Takahashi, K.; Ueno, H.; Sunaga, Y.; Yano, H.; Matsuura, Y.; Iwanaga, T.; et al. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat. Cell Biol. 2000, 2, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.-Y.; Nagashima, K.; Ohshima, T.; Saheki, Y.; Lu, Y.-F.; Matsushita, M.; Yamada, Y.; Mikoshiba, K.; Seino, Y.; Matsui, H.; et al. Cdk5-dependent regulation of glucose-stimulated insulin secretion. Nat. Med. 2005, 11, 1104–1108. [Google Scholar] [CrossRef]
- Su, S.C.; Tsai, L.-H. Cyclin-Dependent Kinases in Brain Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 465–491. [Google Scholar] [CrossRef] [PubMed]
- Chae, T.; Kwon, Y.T.; Bronson, R.; Dikkes, P.; Li, E.; Tsai, L.-H. Mice Lacking p35, a Neuronal Specific Activator of Cdk5, Display Cortical Lamination Defects, Seizures, and Adult Lethality. Neuron 1997, 18, 29–42. [Google Scholar] [CrossRef]
- Balastik, M.; Zhou, X.Z.; Alberich-Jorda, M.; Weissova, R.; Žiak, J.; Pazyra-Murphy, M.F.; Cosker, K.E.; Machonova, O.; Kozmikova, I.; Chen, C.-H.; et al. Prolyl Isomerase Pin1 Regulates Axon Guidance by Stabilizing CRMP2A Selectively in Distal Axons. Cell Rep. 2015, 13, 812–828. [Google Scholar] [CrossRef]
- Grant, N.J.; Coates, P.J.; Woods, Y.L.; Bray, S.E.; Morrice, N.A.; Hastie, C.J.; Lamont, D.J.; Carey, F.A.; Sutherland, C. Phosphorylation of a splice variant of collapsin response mediator protein 2 in the nucleus of tumour cells links cyclin dependent kinase-5 to oncogenesis. BMC Cancer 2015, 15, 885. [Google Scholar] [CrossRef]
- Fang, W.-Q.; Ip, J.P.K.; Li, R.; Ng, Y.P.; Lin, S.-C.; Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Cdk5-mediated phosphorylation of Axin directs axon formation during cerebral cortex development. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 13613–13624. [Google Scholar] [CrossRef]
- Brown, M.; Jacobs, T.; Eickholt, B.; Ferrari, G.; Teo, M.; Monfries, C.; Qi, R.Z.; Leung, T.; Lim, L.; Hall, C. Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 8994–9004. [Google Scholar] [CrossRef]
- Kamei, H.; Saito, T.; Ozawa, M.; Fujita, Y.; Asada, A.; Bibb, J.A.; Saido, T.C.; Sorimachi, H.; Hisanaga, S.-I. Suppression of Calpain-dependent Cleavage of the CDK5 Activator p35 to p25 by Site-specific Phosphorylation. J. Biol. Chem. 2007, 282, 1687–1694. [Google Scholar] [CrossRef]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine Targets, Protein Kinases and Pyridoxal Kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fisher, R.P.; Bailey, P.; Levine, A.J. The CDK7-cycH-p36 complex of transcription factor IIH phosphorylates p53, enhancing its sequence-specific DNA binding activity in vitro. Mol. Cell. Biol. 1997, 17, 5923–5934. [Google Scholar] [CrossRef]
- Fisher, R.P.; Jin, P.; Chamberlin, H.M.; Morgan, D.O. Alternative mechanisms of CAK assembly require an assembly factor or an activating kinase. Cell 1995, 83, 47–57. [Google Scholar] [CrossRef]
- Martinez, A.-M.; Afshar, M.; Martin, F.; Cavadore, J.-C.; Labbé, J.-C.; Dorée, M. Dual phosphorylation of the T-loop in CDK7: Its role in controlling cyclin H binding and CAK activity. EMBO J. 1997, 16, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Robert, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef] [PubMed]
- Frouin, I.; Montecucco, A.; Biamonti, G.; Hübscher, U.; Spadari, S.; Maga, G. Cell cycle-dependant dynamic association of cyclin/CDK complexes with human DNA replication proteins. EMBO J. 2002, 21, 2485–2495. [Google Scholar] [CrossRef]
- Lolli, G.; Johnson, L.N. CAK—Cyclin dependant activating kinase—A key kinase in cell cycle control and a target for drugs. Cell Cycle 2005, 4, 572–577. [Google Scholar] [CrossRef]
- Wade-Harper, J.; Elledge, S.J. The role of Cdk7 in CAK function, a retro-retrospective. Genes Dev. 1998, 12, 285–289. [Google Scholar] [CrossRef]
- Shilatifard, A.; Conaway, R.C.; Conaway, J.W. The RNA polymerase II elongation complex. Annu. Rev. Biochem. 2003, 72, 693–715. [Google Scholar] [CrossRef]
- Goodrich, J.A.; Tjian, R. Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell 1994, 77, 145–156. [Google Scholar] [CrossRef]
- Lu, H.; Zawel, L.; Fisher, L.; Egly, J.M.; Reinberg, D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature 1992, 358, 641–645. [Google Scholar] [CrossRef]
- Oelgeschlager, T. Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J. Cell. Physiol. 2002, 190, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Palancade, B.; Bensaude, O. Investigating RNA polymer ase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem. 2003, 270, 3859–3870. [Google Scholar] [CrossRef]
- Takagi, Y.; Komori, H.; Chang, W.-H.; Hudmon, A.; Erdjument-Bromage, H.; Tempst, P.; Kornberg, R.D. Revised Subunit Structure of Yeast Transcription Factor IIH (TFIIH) and Reconciliation with Human TFIIH. J. Biol. Chem. 2003, 278, 43897–43900. [Google Scholar] [CrossRef]
- Dahmus, M.E. Reversible phosphorylation of the C-terminal domain of RNA polymerase II. J. Biol. Chem. 1996, 271, 19009–19012. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P.; Morgan, D.O. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell Cycle 1994, 78, 713–724. [Google Scholar]
- Tassan, J.; Schultz, S.J.; Bartek, J.; Nigg, E.A. Cell cycle analysis of the activity, subcellular localisation, and subunit composition of human CAK. J. Cell Biol. 1994, 127, 467–478. [Google Scholar] [CrossRef]
- Larochelle, S.; Chen, J.; Knights, R.; Pandur, J.; Morcillo, P.; Erdjument-Bromage, H.; Tempst, P.; Suter, B.; Fisher, R.P. T-loop phosphorylation stabilizes the CDK7-cyclin H-MAT1 complex in vivo and regulates its CTD kinase activity. EMBO J. 2001, 20, 3749–3759. [Google Scholar] [CrossRef]
- Feaver, W.J.; Svejstrup, J.Q.; Henry, N.L.; Kornberg, R.D. Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell 1994, 79, 1103–1109. [Google Scholar] [CrossRef]
- Moncollin, V.V.; Vichi, P.; Egly, J.-M. TFIIH:A transcription factor involved in DNA repair and cell-cycle regulation. Contemp. Cancer Res. 1998, 2, 143–159. [Google Scholar]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef]
- Tassan, J.P.; Jaquenoud, M.; Leopold, P.; Schultz, S.J.; Nigg, E.A. Identification of human cyclin-dependent kinase 8, a putative protein kinase partner for cyclin C. Proc. Natl. Acad. Sci. USA 1995, 92, 8871–8875. [Google Scholar] [CrossRef]
- Boube, M.; Joulia, L.; Cribbs, D.L.; Bourbon, H.M. Evidence for a mediator of RNA polymerase II transcriptional regulation conserved from yeast to man. Cell 2002, 110, 143–151. [Google Scholar] [CrossRef]
- Hengartner, C.J.; Myer, V.E.; Liao, S.-M.; Wilson, C.J.; Koh, S.S.; Young, R.A. Temporal Regulation of RNA Polymerase II by Srb10 and Kin28 Cyclin-Dependent Kinases. Mol. Cell 1998, 2, 43–53. [Google Scholar] [CrossRef]
- Akoulitchev, S.; Chuikov, S.; Reinberg, D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 2000, 407, 102–106. [Google Scholar] [CrossRef]
- Elmlund, H.; Baraznenok, V.; Lindahl, M.; Samuelsen, C.O.; Koeck, P.J.B.; Holmberg, S.; Hebert, H.; Gustafsson, C.M. The cyclin-dependent kinase 8 module sterically blocks Mediator interactions with RNA polymerase II. Proc. Natl. Acad. Sci. USA 2006, 103, 15788. [Google Scholar] [CrossRef] [PubMed]
- Knuesel, M.T.; Meyer, K.D.; Bernecky, C.; Taatjes, D.J. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009, 23, 439–451. [Google Scholar] [CrossRef]
- Tsai, K.-L.; Sato, S.; Tomomori-Sato, C.; Conaway, R.C.; Conaway, J.W.; Asturias, F.J. A conserved Mediator-CDK8 kinase module association regulates Mediator-RNA polymerase II interaction. Nat. Struct. Mol. Biol. 2013, 20, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A Employs CDK8-Mediator to Stimulate RNAPII Elongation in Response to Hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016, 16, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Sessa, M.; Infante, T.; Casamassimi, A. Unraveling framework of the ancestral Mediator complex in human diseases. Biochimie 2012, 94, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Schiano, C.; Casamassimi, A.; Rienzo, M.; de Nigris, F.; Sommese, L.; Napoli, C. Involvement of Mediator complex in malignancy. Biochim. Biophys. Acta BBA Rev. Cancer 2014, 1845, 66–83. [Google Scholar] [CrossRef]
- Syring, I.; Klümper, N.; Offermann, A.; Braun, M.; Deng, M.; Boehm, D.; Queisser, A.; von Mässenhausen, A.; Brägelmann, J.; Vogel, W.; et al. Comprehensive analysis of the transcriptional profile of the Mediator complex across human cancer types. Oncotarget 2016, 7, 23043–23055. [Google Scholar] [CrossRef]
- Zhu, Y.; Qi, C.; Jain, S.; Le Beau, M.M.; Espinosa, R., 3rd; Atkins, G.B.; Lazar, M.A.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 10848–10853. [Google Scholar] [CrossRef]
- Kuuselo, R.; Savinainen, K.; Azorsa, D.O.; Basu, G.D.; Karhu, R.; Tuzmen, S.; Mousses, S.; Kallioniemi, A. Intersex-like (IXL) is a Cell Survival Regulator in Pancreatic Cancer with 19q13 Amplification. Cancer Res. 2007, 67, 1943. [Google Scholar] [CrossRef]
- Bader, S.; Walker, M.; Hendrich, B.; Bird, A.; Bird, C.; Hooper, M.; Wyllie, A. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene 1999, 18, 8044–8047. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Jo, Y.S.; Kim, M.S.; Lee, S.H.; Yoo, N.J. Mutational Heterogeneity of MED23 Gene in Colorectal Cancers. Pathol. Oncol. Res. 2015, 21, 1281–1282. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.F.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Broude, E.V.; Győrffy, B.; Chumanevich, A.A.; Chen, M.; McDermott, M.S.J.; Shtutman, M.; Catroppo, J.F.; Roninson, I.B. Expression of CDK8 and CDK8-interacting Genes as Potential Biomarkers in Breast Cancer. Curr. Cancer Drug Targets 2015, 15, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B.; Győrffy, B.; Mack, Z.T.; Shtil, A.A.; Shtutman, M.S.; Chen, M.; Broude, E.V. Identifying Cancers Impacted by CDK8/19. Cells 2019, 8, 821. [Google Scholar] [CrossRef] [PubMed]
- Becker, F.; Joerg, V.; Hupe, M.C.; Roth, D.; Krupar, R.; Lubczyk, V.; Kuefer, R.; Sailer, V.; Duensing, S.; Kirfel, J.; et al. Increased mediator complex subunit CDK19 expression associates with aggressive prostate cancer. Int. J. Cancer 2019, 146, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Audetat, K.A.; Galbraith, M.D.; Odell, A.T.; Lee, T.; Pandey, A.; Espinosa, J.M.; Dowell, R.D.; Taatjes, D.J. A Kinase-Independent Role for Cyclin-Dependent Kinase 19 in p53 Response. Mol. Cell. Biol. 2017, 37, e00626-16. [Google Scholar] [CrossRef]
- Mallinger, A.; Schiemann, K.; Rink, C.; Sejberg, J.; Honey, M.A.; Czodrowski, P.; Stubbs, M.; Poeschke, O.; Busch, M.; Schneider, R.; et al. 2,8-Disubstituted-1,6-Naphthyridines and 4,6-Disubstituted-Isoquinolines with Potent, Selective Affinity for CDK8/19. ACS Med. Chem. Lett. 2016, 7, 573–578. [Google Scholar] [CrossRef]
- Bergeron, P.; Koehler, M.F.T.; Blackwood, E.M.; Bowman, K.; Clark, K.; Firestein, R.; Kiefer, J.R.; Maskos, K.; McCleland, M.L.; Orren, L.; et al. Design and Development of a Series of Potent and Selective Type II Inhibitors of CDK8. ACS Med. Chem. Lett. 2016, 7, 595–600. [Google Scholar] [CrossRef]
- Rzymski, T.; Mikula, M.; Żyłkiewicz, E.; Dreas, A.; Wiklik, K.; Gołas, A.; Wójcik, K.; Masiejczyk, M.; Wróbel, A.; Dolata, I.; et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget 2017, 8, 33779–33795. [Google Scholar] [CrossRef]
- Marshall, R.M.; Grana, X. Mechanisms controlling CDK9 activity. Front. Biosci. 2006, 11, 2598–2613. [Google Scholar] [CrossRef]
- Fu, T.J.; Peng, J.; Lee, G.; Price, D.H.; Flores, O. Cyclin K Functions as a CDK9 Regulatory Subunit and Participates in RNA Polymerase II Transcription. J. Biol. Chem. 1999, 274, 34527–34530. [Google Scholar] [CrossRef] [PubMed]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.E.; Knapp, S.; Johnson, L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Price, D. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef]
- He, N.; Pezda, A.C.; Zhou, Q. Modulation of a P-TEFb Functional Equilibrium for the Global Control of Cell Growth and Differentiation. Mol. Cell. Biol. 2006, 26, 7068–7076. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.; Ozato, K. The Bromodomain Protein Brd4 Stimulates G1 Gene Transcription and Promotes Progression to S Phase. J. Biol. Chem. 2008, 283, 9040–9048. [Google Scholar] [CrossRef]
- Ping, Y.-H.; Rana, T.M. DSIF and NELF Interact with RNA Polymerase II Elongation Complex and HIV-1 Tat Stimulates P-TEFb-mediated Phosphorylation of RNA Polymerase II and DSIF during Transcription Elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef]
- Kim, Y.K.; Bourgeois, C.F.; Isel, C.; Churcher, M.J.; Karn, J. Phosphorylation of the RNA Polymerase II Carboxyl-Terminal Domain by CDK9 Is Directly Responsible for Human Immunodeficiency Virus Type 1 Tat-Activated Transcriptional Elongation. Mol. Cell. Biol. 2002, 22, 4622–4637. [Google Scholar] [CrossRef] [PubMed]
- Graña, X.; Claudio, P.P.; De Luca, A.; Sang, N.; Giordano, A. PISSLRE, a human novel CDC2-related protein kinase. Oncogene 1994, 9, 2097–2103. [Google Scholar]
- Iorns, E.; Turner, N.C.; Elliott, R.; Syed, N.; Garrone, O.; Gasco, M.; Tutt, A.N.J.; Crook, T.; Lord, C.J.; Ashworth, A. Identification of CDK10 as an Important Determinant of Resistance to Endocrine Therapy for Breast Cancer. Cancer Cell 2008, 13, 91–104. [Google Scholar] [CrossRef]
- Daub, H.; Olsen, J.V.; Bairlein, M.; Gnad, F.; Oppermann, F.S.; Körner, R.; Greff, Z.; Kéri, G.; Stemmann, O.; Mann, M. Kinase-Selective Enrichment Enables Quantitative Phosphoproteomics of the Kinome across the Cell Cycle. Mol. Cell 2008, 31, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Guen, V.J.; Gamble, C.; Lees, J.A.; Colas, P. The awakening of the CDK10/Cyclin M protein kinase. Oncotarget 2017, 8, 50174–50186. [Google Scholar] [CrossRef]
- Guen, V.J.; Gamble, C.; Flajolet, M.; Unger, S.; Thollet, A.; Ferandin, Y.; Superti-Furga, A.; Cohen, P.A.; Meijer, L.; Colas, P. CDK10/cyclin M is a protein kinase that controls ETS2 degradation and is deficient in STAR syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, 19525–19530. [Google Scholar] [CrossRef] [PubMed]
- Boczek, N.J.; Kruisselbrink, T.; Cousin, M.A.; Blackburn, P.R.; Klee, E.W.; Gavrilova, R.H.; Lanpher, B.C. Multigenerational pedigree with STAR syndrome: A novel FAM58A variant and expansion of the phenotype. Am. J. Med. Genet. Part A 2017, 173, 1328–1333. [Google Scholar] [CrossRef]
- Guen, V.J.; Gamble, C.; Perez, D.E.; Bourassa, S.; Zappel, H.; Gärtner, J.; Lees, J.A.; Colas, P. STAR syndrome-associated CDK10/Cyclin M regulates actin network architecture and ciliogenesis. Cell Cycle 2016, 15, 678–688. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Liao, W.-L.; Wang, C.-H.; Tsai, L.-P.; Tang, C.-H.; Chen, C.-H.; Wu, J.-Y.; Liang, W.-M.; Hsieh, A.-R.; Cheng, C.-F.; et al. Association of human height-related genetic variants with familial short stature in Han Chinese in Taiwan. Sci. Rep. 2017, 7, 6372. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Loyer, P.; Hu, D.; Li, T.; Grenet, J.; Lahti, J.M.; Kidd, V.J.; Kivie, M. Cyclin Dependent Kinase 11 in RNA Transcription and Splicing. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2004; pp. 263–288. [Google Scholar]
- Loyer, P.; Trembley, J.H.; Katona, R.; Kidd, V.J.; Lahti, J.M. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell. Signal. 2005, 17, 1033–1051. [Google Scholar] [CrossRef]
- Trembley, J.H.; Hu, D.; Hsu, L.-C.; Yeung, C.-Y.; Slaughter, C.; Lahti, J.M.; Kidd, V.J. PITSLRE p110 Protein Kinases Associate with Transcription Complexes and Affect Their Activity. J. Biol. Chem. 2002, 277, 2589–2596. [Google Scholar] [CrossRef]
- Kobor, M.S.; Greenblatt, J. Regulation of transcription elongation by phosphorylation. Biochim. Biophys. Acta BBA Gene Struct. Expr. 2002, 1577, 261–275. [Google Scholar] [CrossRef]
- Kamada, K.; Roeder, R.G.; Burley, S.K. Molecular mechanism of recruitment of TFIIF- associating RNA polymerase C-terminal domain phosphatase (FCP1) by transcription factor IIF. Proc. Natl. Acad. Sci. USA 2003, 100, 2296–2299. [Google Scholar] [CrossRef]
- Friedl, E.M.; Lane, W.S.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. The C-terminal domain phosphatase and transcription elongation activities of FCP1 are regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 2003, 100, 2328–2333. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-W.G.; Kuzyk, M.A.; Moradian, A.; Ichu, T.-A.; Chang, V.C.-D.; Tien, J.F.; Vollett, S.E.; Griffith, M.; Marra, M.A.; Morin, G.B.J.M.; et al. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol. Cell. Biol. 2012, 32, 4691–4704. [Google Scholar] [CrossRef]
- Buratowski, S. Progression through the RNA polymerase II CTD cycle. Mol. Cell 2009, 36, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.J.C. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; Schultz, N.; Ku, G.Y.; Imtiaz, T.; Kelsen, D.P.; Ilson, D.H.; Solit, D.B.; Berger, M.F.; Ladanyi, M.; Arcila, M.E. Genomic Profiling of Esophagogastric (EG) Tumors in Clinical Practice; American Society of Clinical Oncology: Alexandria, VA, USA, 2015. [Google Scholar]
- Ji, J.; Zhou, C.; Wu, J.; Cai, Q.; Shi, M.; Zhang, H.; Yu, Y.; Zhu, Z.; Zhang, J. Expression pattern of CDK12 protein in gastric cancer and its positive correlation with CD8(+) cell density and CCL12 expression. Int. J. Med. Sci. 2019, 16, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609. [Google Scholar] [CrossRef]
- AACR Project Genie Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, M.; Zhu, J.; Hong, W. Screening of gene mutations associated with bone metastasis in nonsmall cell lung cancer. J. Cancer Res. Ther. 2016, 12, 186. [Google Scholar]
- Biswas, R.; Gao, S.; Cultraro, C.M.; Maity, T.K.; Venugopalan, A.; Abdullaev, Z.; Shaytan, A.K.; Carter, C.A.; Thomas, A.; Rajan, A.; et al. Genomic profiling of multiple sequentially acquired tumor metastatic sites from an “exceptional responder” lung adenocarcinoma patient reveals extensive genomic heterogeneity and novel somatic variants driving treatment response. Mol. Case Stud. 2016, 2, a001263. [Google Scholar] [CrossRef] [PubMed]
- Geyer, J.T.; Subramaniyam, S.; Jiang, Y.; Elemento, O.; Ferry, J.A.; de Leval, L.; Nakashima, M.O.; Liu, Y.-C.; Martin, P.; Mathew, S.; et al. Lymphoblastic transformation of follicular lymphoma: A clinicopathologic and molecular analysis of 7 patients. Hum. Pathol. 2015, 46, 260–271. [Google Scholar] [CrossRef]
- Lui, G.Y.L.; Grandori, C.; Kemp, C.J. CDK12: An emerging therapeutic target for cancer. J. Clin. Pathol. 2018, 71, 957. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Kim, M.; Buratowski, S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol. Cell 2004, 13, 67–76. [Google Scholar] [CrossRef]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.H.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [PubMed]
- Will, C.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef]
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef]
- Cáceres, J.F.; Kornblihtt, A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Tien, J.F.; Mazloomian, A.; Cheng, S.-W.G.; Hughes, C.S.; Chow, C.C.; Canapi, L.T.; Oloumi, A.; Trigo-Gonzalez, G.; Bashashati, A.; Xu, J. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res. 2017, 45, 6698–6716. [Google Scholar] [CrossRef]
- Lei, T.; Zhang, P.; Zhang, X.; Xiao, X.; Zhang, J.; Qiu, T.; Dai, Q.; Zhang, Y.; Min, L.; Li, Q.; et al. Cyclin K regulates prereplicative complex assembly to promote mammalian cell proliferation. Nat. Commun. 2018, 9, 1876. [Google Scholar] [CrossRef]
- Sentenac, A. Eukaryotic RNA Polymerase. Crit. Rev. Biochem. Mol. Biol. 1985, 18, 31–90. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Grummt, I. Regulation of Mammalian Ribosomal Gene Transcription by RNA Polymerase I. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 1998; pp. 109–154. [Google Scholar]
- Willis, I.M. RNA polymerase III. Eur. J. Biochem. 1993, 212, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bregman, D.B.; Pestell, R.G.; Kidd, V.J. Cell cycle regulation and RNA polymerase II. Front. Biosci. 2000, 5, D244–D257. [Google Scholar] [CrossRef] [PubMed]
- Dynlacht, B.D. Regulation of transcription by proteins that control the cell cycle. Nature 1997, 389, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.W. Recycling the cell cycle: Cyclins revisited. Cell 2004, 116, 221–234. [Google Scholar] [CrossRef]
- Lu, H.; Flores, O.; Weinmann, R.; Reinberg, D. The nonphosphorylated form of RNA polymerase II preferentially associates with the preinitiation complex. Proc. Natl. Acad. Sci. USA 1991, 88, 10004–10008. [Google Scholar] [CrossRef]
- Laybourn, P.J.; Dahmus, M.E. Phosphorylation of RNA polymerase IIA occurs subsequent to interaction with the promoter and before the initiation of transcription. J. Biol. Chem. 1990, 265, 13165–13173. [Google Scholar] [CrossRef]
- Kang, M.E.; Dahmus, M.E. RNA polymerases IIA and IIO have distinct roles during transcription from the TATA-less murine dihydrofolate reductase promoter. J. Biol. Chem. 1993, 268, 25033–25040. [Google Scholar] [CrossRef]
- Nogales, E. Recent structural insights into transcription preinitiation complexes. J. Cell Sci. 2000, 113, 4391–4397. [Google Scholar] [PubMed]
- Cadena, D.L.; Dahmus, M.E. Messenger RNA synthesis in mammalian cells is catalyzed by the phosphorylated form of RNA polymerase II. J. Biol. Chem. 1987, 262, 12468–12474. [Google Scholar] [CrossRef]
- Chambers, R.S.; Wang, B.Q.; Burton, Z.F.; Dahmus, M.E. The Activity of COOH-terminal Domain Phosphatase Is Regulated by a Docking Site on RNA Polymerase II and by the General Transcription Factors IIF and IIB. J. Biol. Chem. 1995, 270, 14962–14969. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S. Protein kinases controlling the onset of mitosis. Cell. Mol. Life Sci. 2006, 63, 781–795. [Google Scholar] [CrossRef]
- Morgan, D.O. CYCLIN-DEPENDENT KINASES: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Ren, S.; Rollins, B.J. Cyclin C/Cdk3 Promotes Rb-Dependent G0 Exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Lundberg, A.S.; Weinberg, R.A. Functional Inactivation of the Retinoblastoma Protein Requires Sequential Modification by at Least Two Distinct Cyclin-cdk Complexes. Mol. Cell. Biol. 1998, 18, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Coverley, D.; Pelizon, C.; Trewick, S.; Laskey, R.A. Chromatin-bound Cdc6 persists in S and G2 phases in human cells, while soluble Cdc6 is destroyed in a cyclin A-cdk2 dependent process. J. Cell Sci. 2000, 113, 1929–1938. [Google Scholar]
- Petersen, B.O.; Lukas, J.; Sorensen, C.S.; Bartek, J.; Helin, K. Phosphorylation of mammalian CDC6 by Cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999, 18, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.A.; Tan, T.-T.; Rabson, A.B.; Anderson, D.; Degenhardt, K.; White, E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. 2004, 18, 2095–2107. [Google Scholar] [CrossRef]
- Croce, C.M. Oncogenes and Cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J. Biochemistry, 3rd ed.; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Furuno, N.; den Elzen, N.; Pines, J. Human Cyclin a Is Required for Mitosis until Mid Prophase. J. Cell Biol. 1999, 147, 295–306. [Google Scholar] [CrossRef]
- Riabowol, K.; Draetta, G.; Brizuela, L.; Vandre, D.; Beach, D. The cdc2 kinase is a nuclear protein that is essential for mitosis in mammalian cells. Cell 1989, 57, 393–401. [Google Scholar] [CrossRef]
- Gille, H.; Downward, J. Multiple Ras Effector Pathways Contribute to G1 Cell Cycle Progression. J. Biol. Chem. 1999, 274, 22033–22040. [Google Scholar] [CrossRef]
- Toyoshima, H.; Hunter, T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef]
- Aprelikova, O.; Xiong, Y.; Liu, E.T. Both p16 and p21 Families of Cyclin-dependent Kinase (CDK) Inhibitors Block the Phosphorylation of Cyclin-dependent Kinases by the CDK-activating Kinase. J. Biol. Chem. 1995, 270, 18195–18197. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef]
- Varley, J.M. Germline TP53 mutations and Li-Fraumeni syndrome. Hum. Mutat. 2003, 21, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Sanjeev, G.; George, E.N.K.; Eva, S.; Bertrand, J. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar]
- Pérez de Castro, I.; de Cárcer, G.; Malumbres, M. A census of mitotic cancer genes: New insights into tumor cell biology and cancer therapy. Carcinogenesis 2007, 28, 899–912. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Cai, D.; Kennedy, R.D.; Pathania, S.; Arora, M.; Li, Y.-C.; D’Andrea, A.D.; Parvin, J.D.; Shapiro, G.I. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol. Cell 2009, 35, 327–339. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.-C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- Costa-Cabral, S.; Brough, R.; Konde, A.; Aarts, M.; Campbell, J.; Marinari, E.; Riffell, J.; Bardelli, A.; Torrance, C.; Lord, C.J.; et al. CDK1 Is a Synthetic Lethal Target for KRAS Mutant Tumours. PLoS ONE 2016, 11, e0149099. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer Cell Cycles. Science 1996, 274, 1672. [Google Scholar] [CrossRef]
- Keyomarsi, K.; Leary, N.; Molnar, G.; Lees, E.; Fingert, H.J.; Pardee, A.B. Cyclin E, a Potential Prognostic Marker for Breast Cancer. Cancer Res. 1994, 54, 380. [Google Scholar]
- Keyomarsi, K.; Tucker, S.L.; Buchholz, T.A.; Callister, M.; Ding, Y.; Hortobagyi, G.N.; Bedrosian, I.; Knickerbocker, C.; Toyofuku, W.; Lowe, M.; et al. Cyclin E and Survival in Patients with Breast Cancer. N. Engl. J. Med. 2002, 347, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fang, D.; Chen, H.; Lu, Y.; Dong, Z.; Ding, H.-F.; Jing, Q.; Su, S.-B.; Huang, S. Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget 2015, 6, 20801–20812. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- Barrière, C.; Santamaría, D.; Cerqueira, A.; Galán, J.; Martín, A.; Ortega, S.; Malumbres, M.; Dubus, P.; Barbacid, M. Mice thrive without Cdk4 and Cdk2. Mol. Oncol. 2007, 1, 72–83. [Google Scholar] [CrossRef]
- Lee, Y.-m.; Sicinski, P. Targeting Cyclins and Cyclin-Dependent Kinases in Cancer: Lessons from Mice, Hopes for Therapeutic Applications in Humans. Cell Cycle 2006, 5, 2110–2114. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Fang, D.; Huang, S.; Su, S.-B. Cyclin E1-CDK 2, a potential anticancer target. Aging 2016, 8, 571–572. [Google Scholar] [CrossRef]
- Yin, X.; Yu, J.; Zhou, Y.; Wang, C.; Jiao, Z.; Qian, Z.; Sun, H.; Chen, B. Identification of CDK2 as a novel target in treatment of prostate cancer. Future Oncol. 2018, 14, 709–718. [Google Scholar] [CrossRef]
- Rogatsky, I.; Trowbridge, J.M.; Garabedian, M.J. Potentiation of Human Estrogen Receptor α Transcriptional Activation through Phosphorylation of Serines 104 and 106 by the Cyclin A-CDK2 Complex. J. Biol. Chem. 1999, 274, 22296–22302. [Google Scholar] [CrossRef]
- Pierson-Mullany, L.K.; Lange, C.A. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol. Cell. Biol. 2004, 24, 10542–10557. [Google Scholar] [CrossRef]
- Hu, S.; Lu, Y.; Orr, B.; Godek, K.; Mustachio, L.M.; Kawakami, M.; Sekula, D.; Compton, D.A.; Freemantle, S.; Dmitrovsky, E. Specific CP110 Phosphorylation Sites Mediate Anaphase Catastrophe after CDK2 Inhibition: Evidence for Cooperation with USP33 Knockdown. Mol. Cancer Ther. 2015, 14, 2576–2585. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Ebus, M.E.; Geerts, D.; Koster, J.; Lamers, F.; Valentijn, L.J.; Westerhout, E.M.; Versteeg, R.; Caron, H.N. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12968–12973. [Google Scholar] [CrossRef]
- Faber, A.C.; Chiles, T.C. Inhibition of Cyclin-dependent Kinase-2 Induces Apoptosis in Human Diffuse Large B-cell Lymphomas. Cell Cycle 2007, 6, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Beale, G.; Haagensen, E.J.; Thomas, H.D.; Wang, L.-Z.; Revill, C.H.; Payne, S.L.; Golding, B.T.; Hardcastle, I.R.; Newell, D.R.; Griffin, R.J.; et al. Combined PI3K and CDK2 inhibition induces cell death and enhances in vivo antitumour activity in colorectal cancer. Br. J. Cancer 2016, 115, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, R.; Giebeler, N.; Nevzorova, Y.A.; Bangen, J.-M.; Fahrenkamp, D.; Lambertz, D.; Haas, U.; Hu, W.; Gassler, N.; Cubero, F.J.; et al. Cyclin E1 and cyclin-dependent kinase 2 are critical for initiation, but not for progression of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, 9282–9287. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Widlund, H.R.; Horstmann, M.A.; Ramaswamy, S.; Ross, K.; Huber, W.E.; Nishimura, E.K.; Golub, T.R.; Fisher, D.E. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell 2004, 6, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.-J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.R.; Barlow, C.; Martin, M.P.; Mancusi, C.; Wagner, S.; Self, A.; Barrie, E.; Te Poele, R.; Sharp, S.; Brown, N.; et al. Molecular profiling and combinatorial activity of CCT068127: A potent CDK2 and CDK9 inhibitor. Mol. Oncol. 2018, 12, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Azimi, A.; Caramuta, S.; Seashore-Ludlow, B.; Boström, J.; Robinson, J.L.; Edfors, F.; Tuominen, R.; Kemper, K.; Krijgsman, O.; Peeper, D.S.; et al. Targeting CDK2 overcomes melanoma resistance against BRAF and Hsp90 inhibitors. Mol. Syst. Biol. 2018, 14, e7858. [Google Scholar] [CrossRef]
- Rao, S.S.; Stoehr, J.; Dokic, D.; Wan, L.; Decker, J.T.; Konopka, K.; Thomas, A.L.; Wu, J.; Kaklamani, V.G.; Shea, L.D.; et al. Synergistic effect of eribulin and CDK inhibition for the treatment of triple negative breast cancer. Oncotarget 2017, 8, 83925–83939. [Google Scholar] [CrossRef]
- Deans, A.J.; Khanna, K.K.; McNees, C.J.; Mercurio, C.; Heierhorst, J.; McArthur, G.A. Cyclin-Dependent Kinase 2 Functions in Normal DNA Repair and Is a Therapeutic Target in BRCA1-Deficient Cancers. Cancer Res. 2006, 66, 8219. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Bentley, J.; Wang, L.Z.; Newell, D.R.; Robson, C.N.; Shapiro, G.I.; Curtin, N.J. Pre-clinical evaluation of cyclin-dependent kinase 2 and 1 inhibition in anti-estrogen-sensitive and resistant breast cancer cells. Br. J. Cancer 2010, 102, 342–350. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 11. [Google Scholar] [CrossRef]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.-C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776.e6. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 Inhibitors Expand the Therapeutic Options in Breast Cancer: Palbociclib, Ribociclib and Abemaciclib. BioDrugs 2019, 33, 125–135. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Anwar, F.; Al-Abbasi, F.A.; Zamzami, M.A.; Al-Talhi, H.A.; Kamal, M.A. Neuroprotective Mechanisms Mediated by CDK5 Inhibition. Curr. Pharm. Des. 2016, 22, 527–534. [Google Scholar] [CrossRef]
- Meyer, D.A.; Torres-Altoro, M.I.; Tan, Z.; Tozzi, A.; Di Filippo, M.; DiNapoli, V.; Plattner, F.; Kansy, J.W.; Benkovic, S.A.; Huber, J.D.; et al. Ischemic stroke injury is mediated by aberrant Cdk5. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 8259–8267. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-C.; Leuckx, G.; Sakano, D.; Seymour, P.A.; Mattsson, C.L.; Rautio, L.; Staels, W.; Verdonck, Y.; Serup, P.; Kume, S.; et al. Inhibition of Cdk5 Promotes β-Cell Differentiation From Ductal Progenitors. Diabetes 2018, 67, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Herzog, J.; Ehrlich, S.M.; Pfitzer, L.; Liebl, J.; Fröhlich, T.; Arnold, G.J.; Mikulits, W.; Haider, C.; Vollmar, A.M.; Zahler, S. Cyclin-dependent kinase 5 stabilizes hypoxia-inducible factor-1α: A novel approach for inhibiting angiogenesis in hepatocellular carcinoma. Oncotarget 2016, 7, 27108–27121. [Google Scholar] [CrossRef]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.; Meyer, D.; Guo, A.; Gundara, J.; Mettlach, G.; et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; Garcίa-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis. Front. Oncol. 2014, 4, 96. [Google Scholar] [CrossRef]
- Ianes, C.; Xu, P.; Werz, N.; Meng, Z.; Henne-Bruns, D.; Bischof, J.; Knippschild, U. CK1δ activity is modulated by CDK2/E- and CDK5/p35-mediated phosphorylation. Amino Acids 2016, 48, 579–592. [Google Scholar] [CrossRef]
- Pozo, K.; Hillmann, A.; Augustyn, A.; Plattner, F.; Hai, T.; Singh, T.; Ramezani, S.; Sun, X.; Pfragner, R.; Minna, J.D.; et al. Differential expression of cell cycle regulators in CDK5-dependent medullary thyroid carcinoma tumorigenesis. Oncotarget 2015, 6, 12080–12093. [Google Scholar] [CrossRef]
- Kim, J.W.; Eder, J.P. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology 2014, 28 (Suppl. S3), 15–28. [Google Scholar] [PubMed]
- Liebl, J.; Zhang, S.; Moser, M.; Agalarov, Y.; Demir, C.S.; Hager, B.; Bibb, J.A.; Adams, R.H.; Kiefer, F.; Miura, N.; et al. Cdk5 controls lymphatic vessel development and function by phosphorylation of Foxc2. Nat. Commun. 2015, 6, 8274. [Google Scholar] [CrossRef]
- Kawauchi, T. Cdk5 regulates multiple cellular events in neural development, function and disease. Dev. Growth Differ. 2014, 56, 335–348. [Google Scholar] [CrossRef]
- Pozo, K.; Bibb, J.A. The Emerging Role of Cdk5 in Cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef]
- Bhandari, D.; Lopez-Sanchez, I.; To, A.; Lo, I.C.; Aznar, N.; Leyme, A.; Gupta, V.; Niesman, I.; Maddox, A.L.; Garcia-Marcos, M.; et al. Cyclin-dependent kinase 5 activates guanine nucleotide exchange factor GIV/Girdin to orchestrate migration–proliferation dichotomy. Proc. Natl. Acad. Sci. USA 2015, 112, E4874. [Google Scholar] [CrossRef]
- Wang, H.; Misaki, T.; Taupin, V.; Eguchi, A.; Ghosh, P.; Farquhar, M.G. GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. J. Am. Soc. Nephrol. JASN 2015, 26, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, S.; Pandur, J.; Fisher, R.P.; Salz, H.K.; Suter, B. Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes Dev. 1998, 12, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Saiz, J.E.; Fisher, R.P. A CDK-activating kinase network is required in cell cycle control and transcription in fission yeast. Curr. Biol. 2002, 12, 1100–1105. [Google Scholar] [CrossRef]
- Rossignol, M.; Kolb-Cheynel, I.; Egly, J.-M. Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J. 1997, 16, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Adamczewski, J.P.; Rossignol, M.; Tassan, J.-P.; Nigg, E.A.; Moncollin, V.; Egly, J.-M. MAT1, cdk7 and cyclin H form a kinase complex which is UV light-sensitive upon association with TFIIH. EMBO J. 1996, 15, 1877–1884. [Google Scholar] [CrossRef]
- Lolli, G.; Lowe, E.D.; Brown, N.R.; Johnson, L.L. The Crystal Structure of Human CDK7 and Its Protein Recognition Properties. Structure 2004, 12, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Larochelle, S.; Li, X.; Suter, B. Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature 2003, 424, 228–232. [Google Scholar] [CrossRef]
- Lee, K.M.; Miklos, I.; Du, H.; Watt, S.; Szilagyi, Z.; Saiz, J.E.; Madabhushi, R.; Penkett, C.J.; Sipiczki, M.; Bähler, J.; et al. Impairment of the TFIIH-associated CDK-activating kinase selectively affects cell cycle-regulated gene expression in fission yeast. Mol. Biol. Cell 2005, 16, 2734–2745. [Google Scholar] [CrossRef]
- Wang, B.-Y.; Liu, Q.-Y.; Cao, J.; Chen, J.-W.; Liu, Z.-S. Selective CDK7 inhibition with BS-181 suppresses cell proliferation and induces cell cycle arrest and apoptosis in gastric cancer. Drug Des. Dev. Ther. 2016, 10, 1181–1189. [Google Scholar] [CrossRef]
- Li, B.; Ni Chonghaile, T.; Fan, Y.; Madden, S.F.; Klinger, R.; Connor, A.E.; Walsh, L.; Hurley, G.; Mallya Udupi, G.; Joseph, J.; et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3834. [Google Scholar] [CrossRef]
- Collavin, L.; Lunardi, A.; Del Sal, G. p53-family proteins and their regulators: Hubs and spokes in tumor suppression. Cell Death Differ. 2010, 17, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wierda, W.G.; Chubb, S.; Hawtin, R.E.; Fox, J.A.; Keating, M.J.; Gandhi, V.; Plunkett, W. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 2009, 113, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Pellecchia, M. Apoptosis-based therapies for hematologic malignancies. Blood 2005, 106, 408–418. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chun, H.J.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Golks, A.; Krammer, P.H. Caspases: Pharmacological manipulation of cell death. J. Clin. Investig. 2005, 115, 2665–2672. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Robertson, J.D.; Orrenius, S.; Zhivotovsky, B. Review: Nuclear Events in Apoptosis. J. Struct. Biol. 2000, 129, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Giaccia, A. Transformed cells require continuous activity of RNA polymerase II to resist oncogene-induced apoptosis. Mol. Cell. Biol. 1997, 17, 7306–7316. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, J.; Choy, E.; Harmon, D.; Liu, X.; Nielsen, P.; Mankin, H.; Gray, N.S.; Hornicek, F.J. Systematic Kinome shRNA Screening Identifies CDK11 (PITSLRE) Kinase Expression Is Critical for Osteosarcoma Cell Growth and Proliferation. Clin. Cancer Res. 2012, 18, 4580. [Google Scholar] [CrossRef]
- Cornelis, S.; Bruynooghe, Y.; Denecker, G.; Van Huffel, S.; Tinton, S.; Beyaert, R. Identification and Characterization of a Novel Cell Cycle–Regulated Internal Ribosome Entry Site. Mol. Cell 2000, 5, 597–605. [Google Scholar] [CrossRef]
- Zong, H.; Chi, Y.; Wang, Y.; Yang, Y.; Zhang, L.; Chen, H.; Jiang, J.; Li, Z.; Hong, Y.; Wang, H.; et al. Cyclin D3/CDK11p58 complex is involved in the repression of androgen receptor. Mol. Cell. Biol. 2007, 27, 7125–7142. [Google Scholar] [CrossRef]
- Petretti, C.; Savoian, M.; Montembault, E.; Glover, D.M.; Prigent, C.; Giet, R. The PITSLRE/CDK11p58 protein kinase promotes centrosome maturation and bipolar spindle formation. EMBO Rep. 2006, 7, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Lahti, J.M.; Xiang, J.; Heath, L.S.; Campana, D.; Kidd, V.J. PITSLRE protein kinase activity is associated with apoptosis. Mol. Cell. Biol. 1995, 15, 1–11. [Google Scholar] [CrossRef]
- Chi, Y.; Huang, S.; Wang, L.; Zhou, R.; Wang, L.; Xiao, X.; Li, D.; Cai, Y.; Zhou, X.; Wu, J. CDK11p58 inhibits ERα-positive breast cancer invasion by targeting integrin β3 via the repression of ERα signaling. BMC Cancer 2014, 14, 577. [Google Scholar] [CrossRef]
- Wilker, E.W.; van Vugt, M.A.T.M.; Artim, S.C.; Huang, P.H.; Petersen, C.P.; Reinhardt, H.C.; Feng, Y.; Sharp, P.A.; Sonenberg, N.; White, F.M.; et al. 14-3-3σ controls mitotic translation to facilitate cytokinesis. Nature 2007, 446, 329–332. [Google Scholar] [CrossRef]
- Jia, B.; Choy, E.; Cote, G.; Harmon, D.; Ye, S.; Kan, Q.; Mankin, H.; Hornicek, F.; Duan, Z. Cyclin-dependent kinase 11 (CDK11) is crucial in the growth of liposarcoma cells. Cancer Lett. 2014, 342, 104–112. [Google Scholar] [CrossRef]
- Zhou, Y.; Han, C.; Li, D.; Yu, Z.; Li, F.; Li, F.; An, Q.; Bai, H.; Zhang, X.; Duan, Z.; et al. Cyclin-dependent kinase 11(p110) (CDK11(p110)) is crucial for human breast cancer cell proliferation and growth. Sci. Rep. 2015, 5, 10433. [Google Scholar]
- Chen, H.-R.; Juan, H.-C.; Wong, Y.-H.; Tsai, J.-W.; Fann, M.-J. Cdk12 regulates neurogenesis and late-arising neuronal migration in the developing cerebral cortex. Cereb. Cortex 2016, 27, 2289–2302. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, K.; Wai, P.T.; Maguire, S.L.; Daley, F.; Haider, S.; Kriplani, D.; Campbell, J.; Mirza, H.; Grigoriadis, A.; Tutt, A.; et al. Evaluation of CDK12 Protein Expression as a Potential Novel Biomarker for DNA Damage Response-Targeted Therapies in Breast Cancer. Mol. Cancer Ther. 2018, 17, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, R.; Wilkerson, P.M.; Marchiò, C.; Piscuoglio, S.; Ng, C.K.Y.; Wai, P.; Lambros, M.B.; Samartzis, E.P.; Dedes, K.J.; Frankum, J.; et al. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J. Pathol. 2014, 232, 553–565. [Google Scholar] [CrossRef]
- Zang, Z.J.; Ong, C.K.; Cutcutache, I.; Yu, W.; Zhang, S.L.; Huang, D.; Ler, L.D.; Dykema, K.; Gan, A.; Tao, J.; et al. Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res. 2011, 71, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Tansey, W.P. Mammalian MYC Proteins and Cancer. J. New J. Sci. 2014, 2014, 27. [Google Scholar] [CrossRef]
- Cermelli, S.; Jang, I.S.; Bernard, B.; Grandori, C. Synthetic Lethal Screens as a Means to Understand and Treat MYC-Driven Cancers. Cold Spring Harb. Perspect. Med. 2014, 4, a014209. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef]
- Robinson, K.; Asawachaicharn, N.; Galloway, D.A.; Grandori, C. c-Myc Accelerates S-Phase and Requires WRN to Avoid Replication Stress. PLoS ONE 2009, 4, e5951. [Google Scholar] [CrossRef] [PubMed]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Iniguez, A.B.; Stolte, B.; Wang, E.J.; Conway, A.S.; Alexe, G.; Dharia, N.V.; Kwiatkowski, N.; Zhang, T.; Abraham, B.J.; Mora, J.; et al. EWS/FLI confers tumor cell synthetic lethality to CDK12 inhibition in Ewing sarcoma. Cancer Cell 2018, 33, 202–216.e6. [Google Scholar] [CrossRef]
- Lang, L.; Chansky, H.A.; Hickstein, D.D. EWS·Fli-1 Fusion Protein Interacts with Hyperphosphorylated RNA Polymerase II and Interferes with Serine-Arginine Protein-mediated RNA Splicing. J. Biol. Chem. 2000, 275, 37612–37618. [Google Scholar]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.G.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef] [PubMed]
- Paculová, H.; Kramara, J.; Šimečková, Š.; Fedr, R.; Souček, K.; Hylse, O.; Paruch, K.; Svoboda, M.; Mistrίk, M.; Kohoutek, J. BRCA1 or CDK12 loss sensitizes cells to CHK1 inhibitors. Tumor Biol. 2017, 39, 1010428317727479. [Google Scholar] [CrossRef]
- Cueto-González, A.M.; Fernández-Cancio, M.; Fernández-Alvarez, P.; García-Arumí, E.; Tizzano, E.F. Unusual context of CENPJ variants and primary microcephaly: Compound heterozygosity and nonconsanguinity in an Argentinian patient. Hum. Genome Var. 2020, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Peche, V.S.; Szczepanski, S.; Nürnberg, G.; Tariq, M.; Jameel, M.; Khan, T.N.; Fatima, A.; et al. CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum. Mol. Genet. 2013, 22, 5199–5214. [Google Scholar] [CrossRef]
- Mirzaa, G.; Parry, D.A.; Fry, A.E.; Giamanco, K.A.; Schwartzentruber, J.; Vanstone, M.; Logan, C.V.; Roberts, N.; Johnson, C.A.; Singh, S.; et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat. Genet. 2014, 46, 510–515. [Google Scholar] [CrossRef]
- Sameshima, T.; Morisada, N.; Egawa, T.; Kugo, M.; Iijima, K. MPPH syndrome with aortic coarctation and macrosomia due to CCND2 mutations. Pediatr. Int. 2020, 62, 115–117. [Google Scholar] [CrossRef]
- Kida, A.; Kakihana, K.; Kotani, S.; Kurosu, T.; Miura, O. Glycogen synthase kinase-3β and p38 phosphorylate cyclin D2 on Thr280 to trigger its ubiquitin/proteasome-dependent degradation in hematopoietic cells. Oncogene 2007, 26, 6630–6640. [Google Scholar] [CrossRef] [PubMed]
- Glickstein, S.B.; Alexander, S.; Ross, M.E. Differences in Cyclin D2 and D1 Protein Expression Distinguish Forebrain Progenitor Subsets. Cereb. Cortex 2007, 17, 632–642. [Google Scholar] [CrossRef][Green Version]
- Colas, P. Cyclin-dependent kinases and rare developmental disorders. Orphanet J. Rare Dis. 2020, 15, 203. [Google Scholar] [CrossRef]
- Magen, D.; Ofir, A.; Berger, L.; Goldsher, D.; Eran, A.; Katib, N.; Nijem, Y.; Vlodavsky, E.; Zur, S.; Behar, D.M.; et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with a loss-of-function mutation in CDK5. Hum. Genet. 2015, 134, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Lahiri, D.K. Cdk5 activity in the brain—Multiple paths of regulation. J. Cell Sci. 2014, 127 Pt. 11, 2391–2400. [Google Scholar] [CrossRef]
- Moncini, S.; Castronovo, P.; Murgia, A.; Russo, S.; Bedeschi, M.F.; Lunghi, M.; Selicorni, A.; Bonati, M.T.; Riva, P.; Venturin, M. Functional characterization of CDK5 and CDK5R1 mutations identified in patients with non-syndromic intellectual disability. J. Hum. Genet. 2016, 61, 283–293. [Google Scholar] [CrossRef]
- Harper, T.M.; Taatjes, D.J. The complex structure and function of Mediator. J. Biol. Chem. 2018, 293, 13778–13785. [Google Scholar] [CrossRef]
- Dannappel, M.V.; Sooraj, D.; Loh, J.J.; Firestein, R. Molecular and in vivo Functions of the CDK8 and CDK19 Kinase Modules. Front. Cell Dev. Biol. 2019, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Kramer, J.M.; Merkx, G.; Lugtenberg, D.; Smeets, D.F.; Oortveld, M.A.W.; Blokland, E.A.W.; Agrawal, J.; Schenck, A.; van Bokhoven, H.; et al. CDK19 is disrupted in a female patient with bilateral congenital retinal folds, microcephaly and mild mental retardation. Hum. Genet. 2010, 128, 281–291. [Google Scholar] [CrossRef]
- Simpson, S.; Woodworth, C.D.; Di Paolo, J.A. Altered expression of Erg and Ets-2 transcription factors is associated with genetic changes at 21q22.2-22.3 in immortal and cervical carcinoma cell lines. Oncogene 1997, 14, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Sergère, J.-C.; Thuret, J.-Y.; Le, G.; Edgardo, R.; Carosella, D.; Leteurtre, F. Human CDK10 Gene Isoforms. Biochem. Biophys. Res. Commun. 2000, 276, 271–277. [Google Scholar] [CrossRef]
- Bedeschi, M.F.; Giangiobbe, S.; Paganini, L.; Tabano, S.; Silipigni, R.; Colombo, L.; Crippa, B.L.; Lalatta, F.; Guerneri, S.; Miozzo, M. STAR syndrome plus: The first description of a female patient with the lethal form. Am. J. Med. Genet. Part A 2017, 173, 3226–3230. [Google Scholar] [CrossRef] [PubMed]
- Orge, F.H.; Dar, S.A.; Blackburn, C.N.; Grimes-Hodges, S.J.; Mitchell, A.L. Ocular manifestations of X-linked dominant FAM58A mutation in toe syndactyly, telecanthus, anogenital, and renal malformations (‘STAR’) syndrome. Ophthalmic Genet. 2016, 37, 323–327. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Farrell, J.M.; Alfaro, M.P.; Elhassan, N.O. STAR syndrome is part of the differential diagnosis of females with anorectal malformations. Am. J. Med. Genet. Part A 2015, 167, 1940–1943. [Google Scholar] [CrossRef]
- Boone, P.M.; Bacino, C.A.; Shaw, C.A.; Eng, P.A.; Hixson, P.M.; Pursley, A.N.; Kang, S.-H.L.; Yang, Y.; Wiszniewska, J.; Nowakowska, B.A.; et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010, 31, 1326–1342. [Google Scholar] [CrossRef]
- Heru Sumarsono, S.; Wilson, T.J.; Tymms, M.J.; Venter, D.J.; Corrick, C.M.; Kola, R.; Lahoud, M.H.; Papas, T.S.; Seth, A.; Kola, I. Down’s syndrome-like skeletal abnormalities in Ets2 transgenic mice. Nature 1996, 379, 534–537. [Google Scholar] [CrossRef]
- Zimmermann, M.; Arachchige-Don, A.P.S.; Donaldson, M.S.; Patriarchi, T.; Horne, M.C. Cyclin G2 promotes cell cycle arrest in breast cancer cells responding to fulvestrant and metformin and correlates with patient survival. Cell Cycle 2016, 15, 3278–3295. [Google Scholar] [CrossRef]
- Greifenberg, A.K.; Hönig, D.; Pilarova, K.; Düster, R.; Bartholomeeusen, K.; Bösken, C.A.; Anand, K.; Blazek, D.; Geyer, M. Structural and Functional Analysis of the Cdk13/Cyclin K Complex. Cell Rep. 2016, 14, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Kandaswamy, K.K.; Werber, M.; Weiss, M.E.R.; Oprea, G.; Kishore, S.; Lohmann, K.; Rolfs, A. Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. J. Neurodev. Disord. 2019, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- van den Akker, W.M.R.; Brummelman, I.; Martis, L.M.; Timmermans, R.N.; Pfundt, R.; Kleefstra, T.; Willemsen, M.H.; Gerkes, E.H.; Herkert, J.C.; van Essen, A.J.; et al. De novo variants in CDK13 associated with syndromic ID/DD: Molecular and clinical delineation of 15 individuals and a further review. Clin. Genet. 2018, 93, 1000–1007. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Caswell, R.C.; Canham, N.; Cole, T.; Firth, H.V.; Foulds, N.; Heimdal, K.; Hobson, E.; Houge, G.; Joss, S.; et al. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J. Med. Genet. 2018, 55, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.J.; Suri, M. Chapter Five—CDK13-related disorder. In Advances in Genetics; Kumar, D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 163–182. [Google Scholar]
- Fan, Y.; Yin, W.; Hu, B.; Kline, A.D.; Zhang, V.W.; Liang, D.; Sun, Y.; Wang, L.; Tang, S.; Powis, Z.; et al. De Novo Mutations of CCNK Cause a Syndromic Neurodevelopmental Disorder with Distinctive Facial Dysmorphism. Am. J. Hum. Genet. 2018, 103, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Lei, T.; Zhao, C.; Zhong, J.; Tang, Y.-z.; Chen, B.; Yang, J.; Li, C.; Wang, S.; Song, X.; et al. Cyclin K-containing kinase complexes maintain self-renewal in murine embryonic stem cells. J. Biol. Chem. 2012, 287, 25344–25352. [Google Scholar] [CrossRef]
- Chen, H.-R.; Lin, G.-T.; Huang, C.-K.; Fann, M.-J. Cdk12 and Cdk13 regulate axonal elongation through a common signaling pathway that modulates Cdk5 expression. Exp. Neurol. 2014, 261, 10–21. [Google Scholar] [CrossRef]
- Lahiri, D.K. An Integrated Approach to Genome Studies. Science 2011, 331, 147. [Google Scholar] [CrossRef]
- Debomoy, K.L.; Martin, R.F.; Kumar, S.; Nigel, H.G.; Ezio, G.; Lon, S.S. A Critical Analysis of New Molecular Targets and Strategies for Drug Developments in Alzheimers Disease. Curr. Drug Targets 2003, 4, 97–112. [Google Scholar]
- Lahiri, D.K.; Maloney, B.; Long, J.M.; Greig, N.H. Lessons from a BACE1 inhibitor trial: Off-site but not off base. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2014, 10 (Suppl. S5), S411–S419. [Google Scholar] [CrossRef]
- Lopes, J.P.; Oliveira, C.R.; Agostinho, P. Neurodegeneration in an Aβ-induced model of Alzheimer’s disease: The role of Cdk5. Aging Cell 2010, 9, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Venturin, M.; Guarnieri, P.; Natacci, F.; Stabile, M.; Tenconi, R.; Clementi, M.; Hernandez, C.; Thompson, P.; Upadhyaya, M.; Larizza, L.; et al. Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J. Med. Genet. 2004, 41, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Engmann, O.; Hortobágyi, T.; Pidsley, R.; Troakes, C.; Bernstein, H.-G.; Kreutz, M.R.; Mill, J.; Nikolic, M.; Giese, K.P. Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain J. Neurol. 2011, 134 Pt 8, 2408–2421. [Google Scholar] [CrossRef]
- Patel, L.S.; Wenzel, H.J.; Schwartzkroin, P.A. Physiological and morphological characterization of dentate granule cells in the p35 knock-out mouse hippocampus: Evidence for an epileptic circuit. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 9005–9014. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Julien, J.P. Cyclin-Dependent Kinase 5 in Amyotrophic Lateral Sclerosis. Neurosignals 2003, 12, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, R.; Strazielle, C. Neurobehavioral Characteristics of Mice with Modified Intermediate Filament Genes. Rev. Neurosci. 2003, 14, 369–385. [Google Scholar] [CrossRef]
- Kesavapany, S.; Li, B.S.; Pant, H.C. Cyclin-Dependent Kinase 5 in Neurofilament Function and Regulation. Neurosignals 2003, 12, 252–264. [Google Scholar] [CrossRef]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef]
- Patzke, H.; Tsai, L.-H. Cdk5 sinks into ALS. Trends Neurosci. 2002, 25, 8–10. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Larivière, R.C.; Julien, J.-P. Deregulation of Cdk5 in a Mouse Model of ALS: Toxicity Alleviated by Perikaryal Neurofilament Inclusions. Neuron 2001, 30, 135–148. [Google Scholar] [CrossRef]
- Miguel, S.; Erik De, C.; Jan, B. The regulation of HIV-1 transcription: Molecular targets for chemotherapeutic intervention. Med. Res. Rev. 2006, 26, 595–625. [Google Scholar]
- Zhou, M.; Deng, L.; Kashanchi, F.; Brady, J.N.; Shatkin, A.J.; Kumar, A. The Tat/TAR-dependent phosphorylation of RNA polymerase II C-terminal domain stimulates cotranscriptional capping of HIV-1 mRNA. Proc. Natl. Acad. Sci. USA 2003, 100, 12666–12671. [Google Scholar] [CrossRef] [PubMed]
- Barboric, M.; Peterlin, B.M. A New Paradigm in Eukaryotic Biology: HIV Tat and the Control of Transcriptional Elongation. PLoS Biol. 2005, 3, e76. [Google Scholar] [CrossRef]
- Karn, J. Tackling tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef]
- Mancebo, H.S.Y.; Lee, G.; Flygare, J.; Tomassini, J.; Luu, P.; Zhu, Y.; Peng, J.; Blau, C.; Hazuda, D.; Price, D.; et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997, 11, 2633–2644. [Google Scholar] [CrossRef]
- Wei, P.; Garber, M.E.; Fang, S.-M.; Fischer, W.H.; Jones, K.A. A Novel CDK9-Associated C-Type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Zhou, M.; Halanski, M.A.; Radonovich, M.F.; Kashanchi, F.; Peng, J.; Price, D.H.; Brady, J.N. Tat Modifies the Activity of CDK9 To Phosphorylate Serine 5 of the RNA Polymerase II Carboxyl-Terminal Domain during Human Immunodeficiency Virus Type 1 Transcription. Mol. Cell. Biol. 2000, 20, 5077–5086. [Google Scholar] [CrossRef]
- Ivanov, D.; Kwak, Y.T.; Guo, J.; Gaynor, R.B. Domains in the SPT5 Protein That Modulate Its Transcriptional Regulatory Properties. Mol. Cell. Biol. 2000, 20, 2970–2983. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of Human Immunodeficiency Virus Transcription: P-TEFb Phosphorylates RD and Dissociates Negative Effectors from the Transactivation Response Element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Barboric, M.; Yik, J.H.N.; Czudnochowski, N.; Yang, Z.; Chen, R.; Contreras, X.; Geyer, M.; Matija Peterlin, B.; Zhou, Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007, 35, 2003–2012. [Google Scholar] [CrossRef]
- Fraldi, A.; Varrone, F.; Napolitano, G.; Michels, A.; Majello, B.; Bensaude, O.; Lania, L. Inhibition of Tat activity by the HEXIM1 protein. Retrovirology 2005, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.A.; Sano, M.; Schneider, M.D. Phosphorylation of RNA Polymerase II in Cardiac Hypertrophy: Cell Enlargement Signals Converge on Cyclin T/Cdk9. Recent Prog. Horm. Res. 2004, 59, 125–139. [Google Scholar] [CrossRef]
- Sano, M.; Abdellatif, M.; Oh, H.; Xie, M.; Bagella, L.; Giordano, A.; Michael, L.H.; DeMayo, F.J.; Schneider, M.D. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat. Med. 2002, 8, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.; Stiegler, P.; Bagella, L.; Pucci, B.; Bellan, C.; De Falco, G.; De Luca, A.; Guanti, G.; Puri, P.L.; Giordano, A. Activation of MyoD-dependent transcription by cdk9/cyclin T2. Oncogene 2002, 21, 4137–4148. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Hong, C.; Chen, I.-Y.; Lypowy, J.; Abdellatif, M. MicroRNAs Play an Essential Role in the Development of Cardiac Hypertrophy. Circ. Res. 2007, 100, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Wang, S.C.; Shirai, M.; Scaglia, F.; Xie, M.; Sakai, S.; Tanaka, T.; Kulkarni, P.A.; Barger, P.M.; Youker, K.A.; et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004, 23, 3559–3569. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review. Int. J. Mol. Sci. 2021, 22, 2935. https://doi.org/10.3390/ijms22062935
Łukasik P, Załuski M, Gutowska I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review. International Journal of Molecular Sciences. 2021; 22(6):2935. https://doi.org/10.3390/ijms22062935
Chicago/Turabian StyleŁukasik, Paweł, Michał Załuski, and Izabela Gutowska. 2021. "Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review" International Journal of Molecular Sciences 22, no. 6: 2935. https://doi.org/10.3390/ijms22062935
APA StyleŁukasik, P., Załuski, M., & Gutowska, I. (2021). Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review. International Journal of Molecular Sciences, 22(6), 2935. https://doi.org/10.3390/ijms22062935