
Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action


Abstract
1. Cyclin-Dependent Kinases (CDKs)
2. Cyclin-Dependent Kinase (CDK) Inhibitors in Drug Development
3. Type I Inhibitors

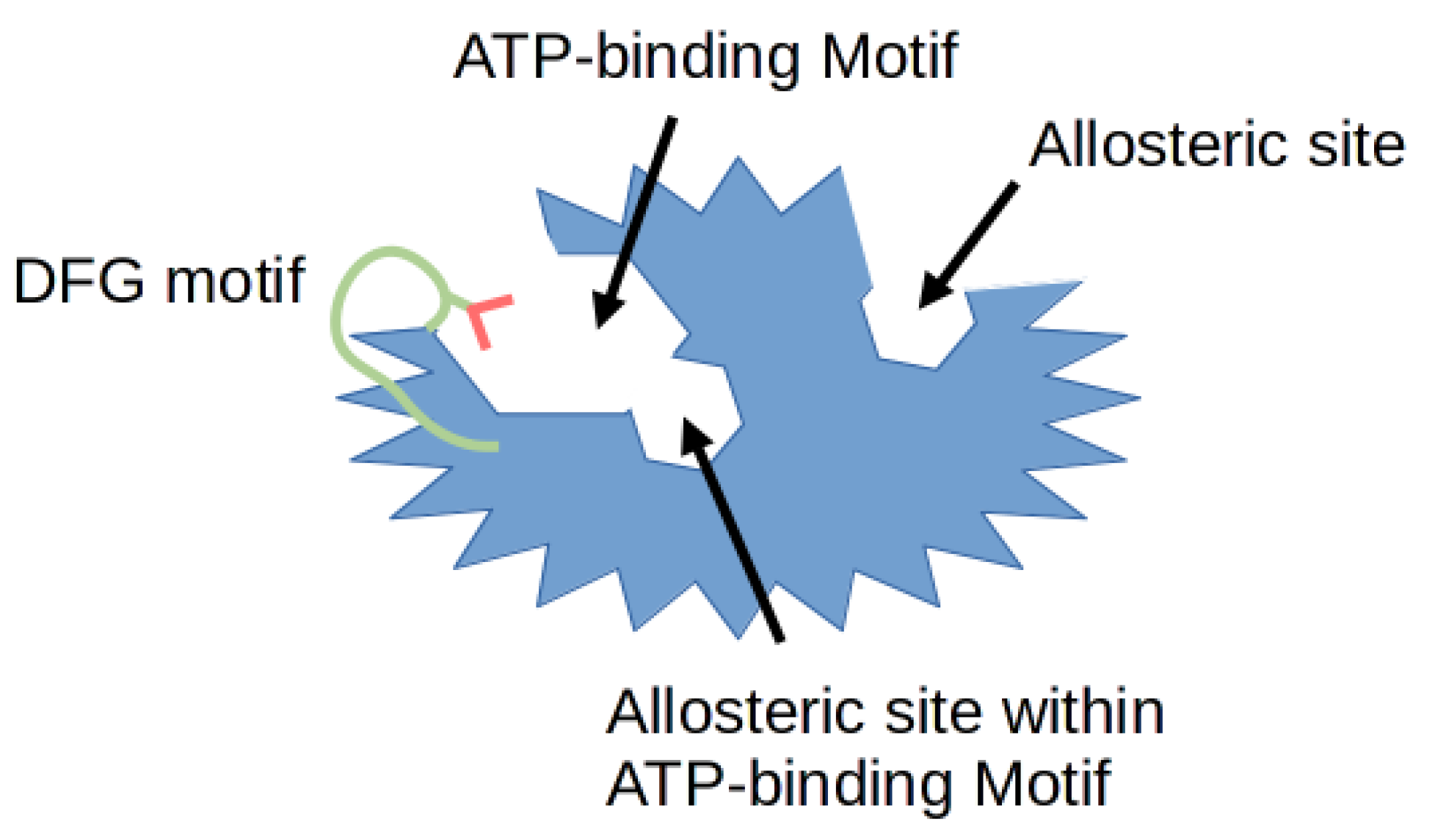{kind=link}
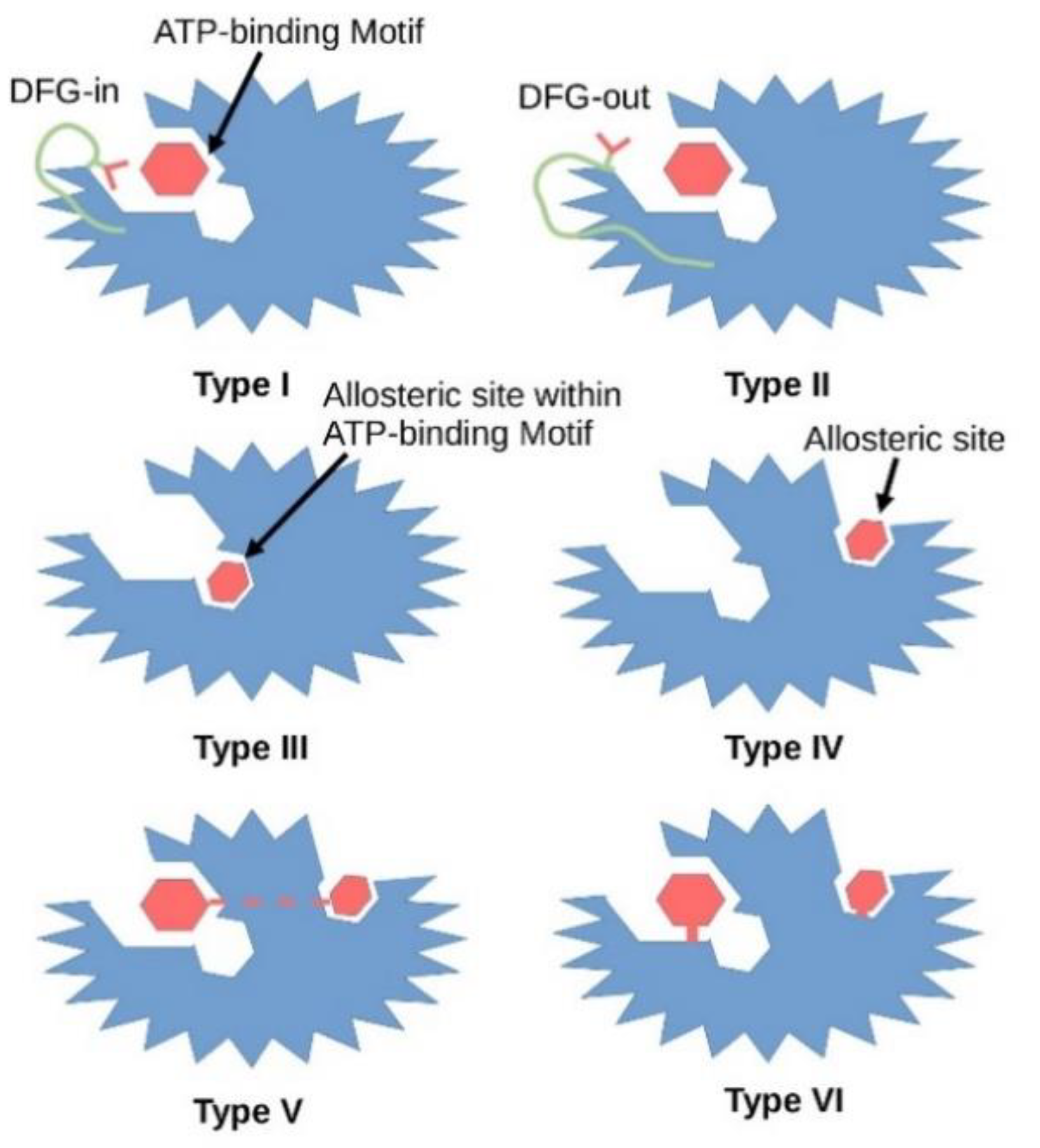{kind=link}
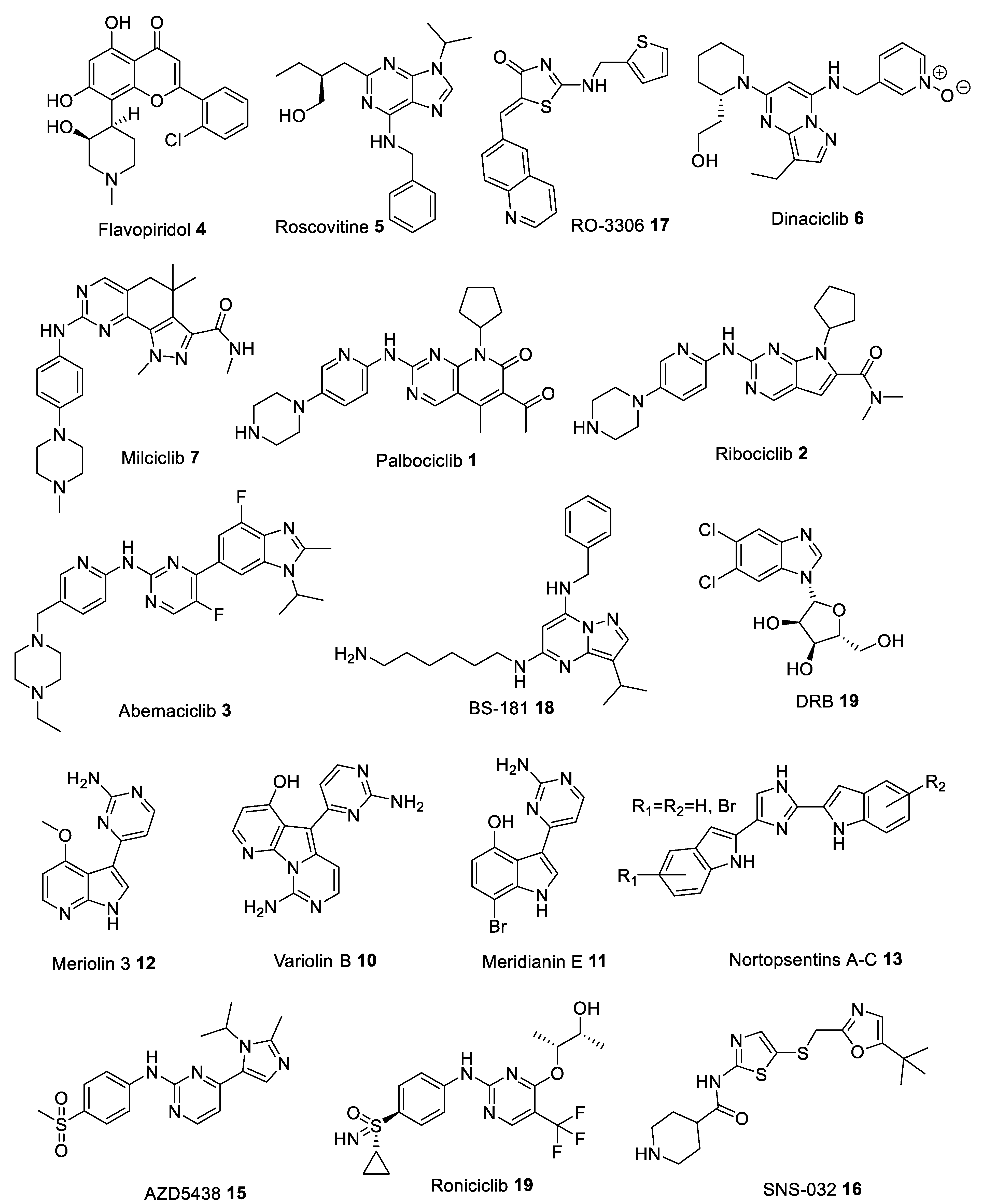{kind=link}
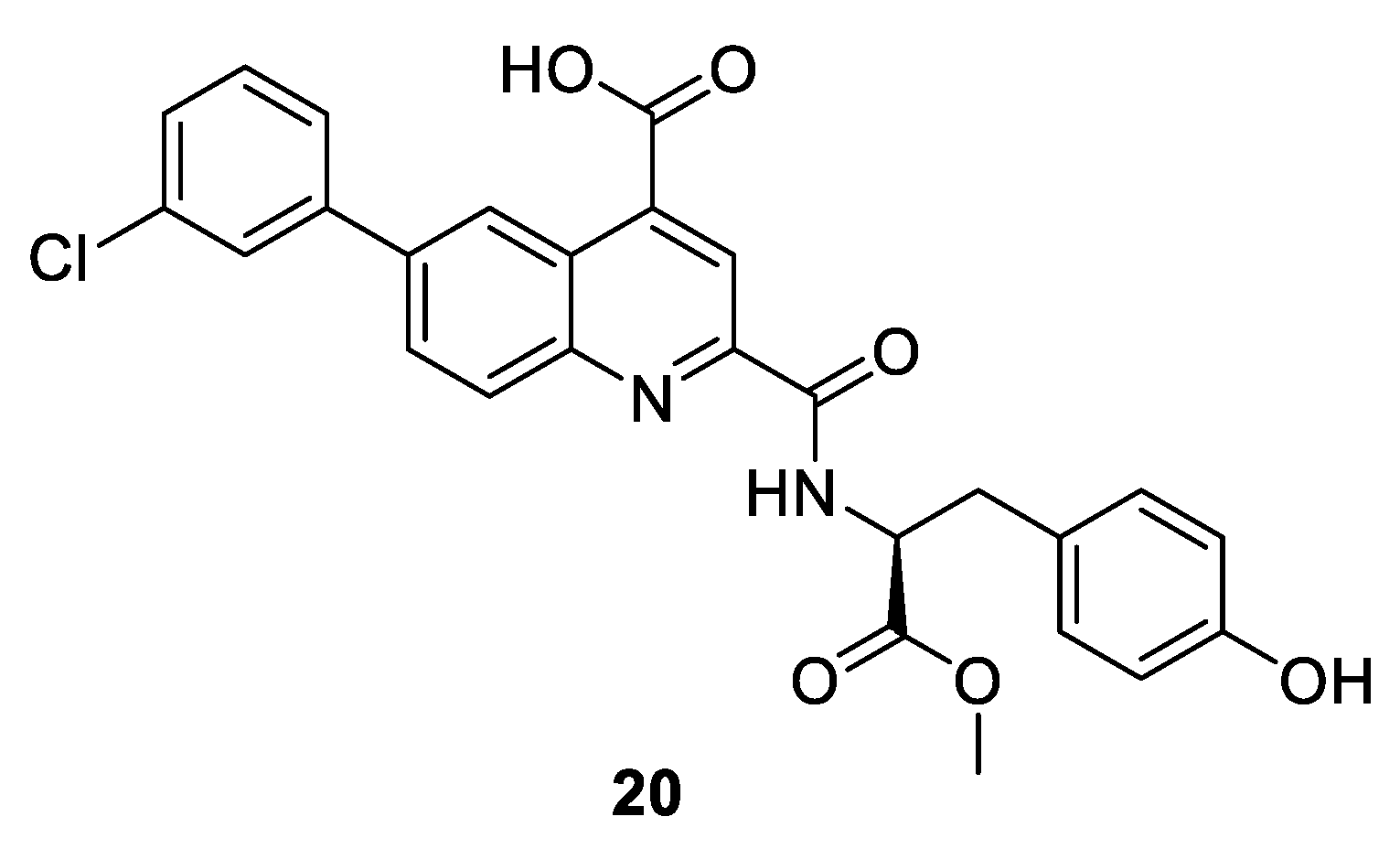{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Kinase IC50 [nM] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CDK1/B | CDK2/A | CDK2/E | CDK4/D | CDK5/p25 | CDK6/D | CDK7/H | CDK8/C | CDK9/T1 | |
| Flavopiridol 4 [38,39] | 30 | 100 | 100 | 20–40 | - | 60 | 110 | - | 20 |
| Roscovitine 5 [40] | 650 | 700 | 700 | >100,000 | 160 | >100,000 | 460 | >100,000 | 600 |
| RO-3306 17 [41] | 35 | - | 340 | >2000 | - | - | - | - | - |
| Dinaciclib 6 [42] | 3 | 1 | 1 | 100 | 1 | - | - | - | 4 |
| Milciclib 7 [28] | 398 | 45 | 363 | 160 | 265 | - | 150 | - | - |
| Palbociclib 1 [43] | >10,000 | >10,000 | >10,000 | 11 | >10,000 | 15 | - | - | - |
| Ribociclib 2 [44] | 113,000 | 76,000 | 76,000 | 10 | 43,900 | 39 | - | - | - |
| Abemaciclib 3 [45] | 1627 | - | 504 | 2 | 355 | 10 | 3910 | - | 57 |
| BS-181 18 [46] | 8100 | 730 | 880 | 33,000 | 3000 | 47,000 | 21 | - | 4200 |
| DRB 9 [47] | 17,000 | - | >10,000 | >10,000 | - | - | >10,000 | >10,000 | 340 |
| Meriolin 3 12 [48] | 170 | 11 | - | >100,000 | 170 | >100,000 | >100,000 | - | 6 |
| Variolin B 10 [49] | 60 | 80 | - | >10,000 | 90 | >10,000 | >1000 | - | 26 |
| Meridianin E 11 [50] | 180 | 800 | 1800 | 3000 | 150 | - | - | - | 18 |
| Nortopsentins 13 [51] | 310–900 | - | - | - | - | - | - | - | - |
| AZD5438 15 [52] | 16 | 45 | 6 | 449 | 14 | 21 | 821 | - | 20 |
| Roniciclib 19 [53] | 7 | - | 9 | 11 | - | - | 25 | - | 5 |
| SNS-032 16 [54] | 480 | 38 | 48 | 925 | 340 (CDK5/p35) | - | 62 | - | 4 |
| Inhibitor | Main Targets | Condition or Disease | Phase | Status | Identifier |
|---|---|---|---|---|---|
| Flavopiridol 4 | CDK1, CDK2, CDK4, CDK6, CDK9 | Acute Myeloid Leukemia (AML) | on the market | “orphan drug” | - |
| Roscovitine 5 | CDK2, CDK7, CDK9 | Pituitary Cushing Disease | II | active | NCT02160730 NCT03774446 |
| Cystic Fibrosis | II | terminated | NCT02649751 | ||
| Advanced Solid Tumors | I | terminated | NCT00999401 | ||
| Lung Cancer | II | terminated | NCT00372073 | ||
| RO-3306 17 [41] | CDK1 | Acute Myeloid Leukemia (AML) | pre-clinical | - | - |
| Dinaciclib 6 | CDK1, CDK2, CDK5, CDK9 | Chronic Lymphocytic Leukemia (CLL) | on the market | “orphan drug” | - |
| Breast and Lung Cancers | II | terminated | NCT00732810 | ||
| Milciclib 7 | CDK1, CDK2, CDK4, CDK7 | Hepatocellular Carcinoma (HCC) | II | active | NCT03109886 |
| Thymic Carcinoma | II | terminated | NCT01301391 NCT01011439 | ||
| Palbociclib 1 | CDK4, CDK6 | HR+/HER2- Breast Cancer | on the market | used in combination with Letrozole | - |
| III | active, to be used with other drugs like Fulvestrant | NCT02692755 | |||
| Head and Neck, Brain, Colon, and other Solid Cancers | II | active, to be used alone and in combination with different drugs | NCT02255461 NCT03446157 NCT02896335 NCT03965845 | ||
| Ribociclib 2 | CDK4, CDK6 | HR+/HER2- Breast Cancer | on the market | used in combination with Letrozole | - |
| III | active, to be used with other drugs like Fulvestrant | NCT02422615 NCT03439046 NCT03294694 | |||
| Prostate, and other Solid Cancers | II | active, to be used alone and in combination with different drugs | NCT02555189 NCT01543698 NCT02934568 | ||
| Abemaciclib 3 | CDK4, CDK6 | HR+/HER2- Breast Cancer | on the market | used in combination with Fulvestrant | - |
| III | active, to be used with other drugs like Letrozole | NCT02763566 | |||
| Lung, Brain, Colon, and other Solid Cancers | II or III | active, to be used alone and in combination with different drugs | NCT04545710 NCT02152631 NCT03220646 NCT04616183 NCT03310879 | ||
| BS-181 18 [46] | CDK7 | Breast, Lung, Prostate and Colorectal Cancers | pre-clinical | - | - |
| DRB 9 [55] | CDK7, CDK8, CDK9 | HIV Transcription | pre-clinical | - | - |
| Meriolin 3 12 [48] | CDK1, CDK2, CDK5, CDK9 | Neuroblastoma, Glioma, Myeloma, Colon Cancer | pre-clinical | - | - |
| Variolin B 10 [56] | CDK1, CDK2, CDK5, CDK9 | Murine Leukemia | pre-clinical | - | - |
| Meridianin E 11 [57] | CDK1, CDK5, CDK9 | Larynx Carcinoma, Myeloid Leukemia | pre-clinical | - | - |
| Nortopsentins 13 [58] | CDK1 | Malignant Pleural Mesothelioma (MPM) | pre-clinical | - | - |
| AZD5438 15 | CDK1, CDK2, CDK5, CDK6, CDK9 | Advanced Solid Malignancies | I | terminated | NCT00088790 |
| Roniciclib 19 | CDK1, CDK2, CDK4, CDK7, CDK9 | Lung and Advanced Solid Cancers | II | terminated | NCT02161419 NCT01573338 NCT02656849 |
| SNS-032 16 | CDK2, CDK7, CDK9 | Chronic Lymphocytic Leukemia and other Solid Cancers | I | terminated | NCT00446342 NCT00292864 |
4. Type 1.5 Inhibitors
5. Type II Inhibitors
6. Type III Inhibitors
7. Type IV Inhibitors
8. Type V Inhibitors
9. Type VI Inhibitors
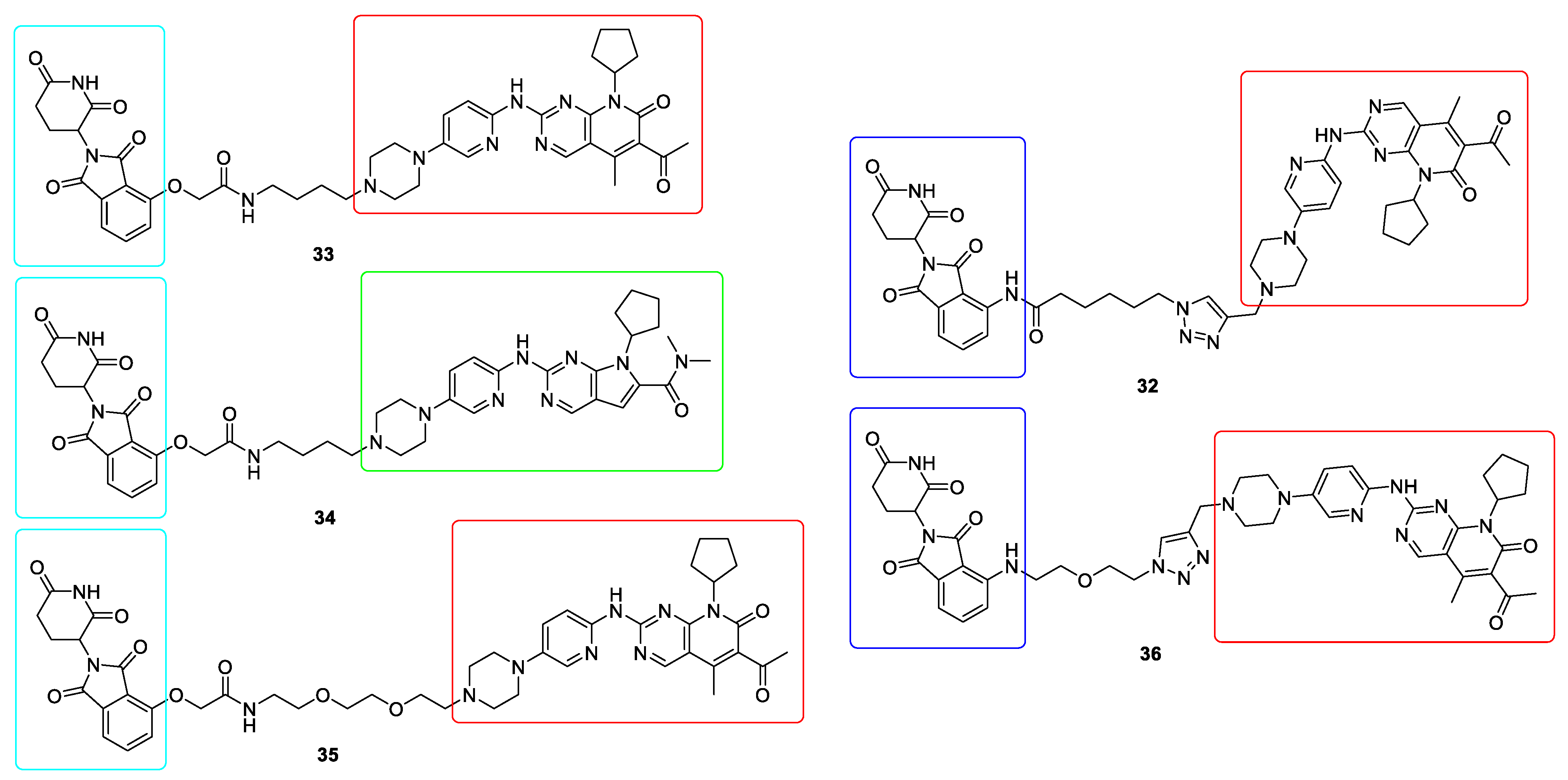
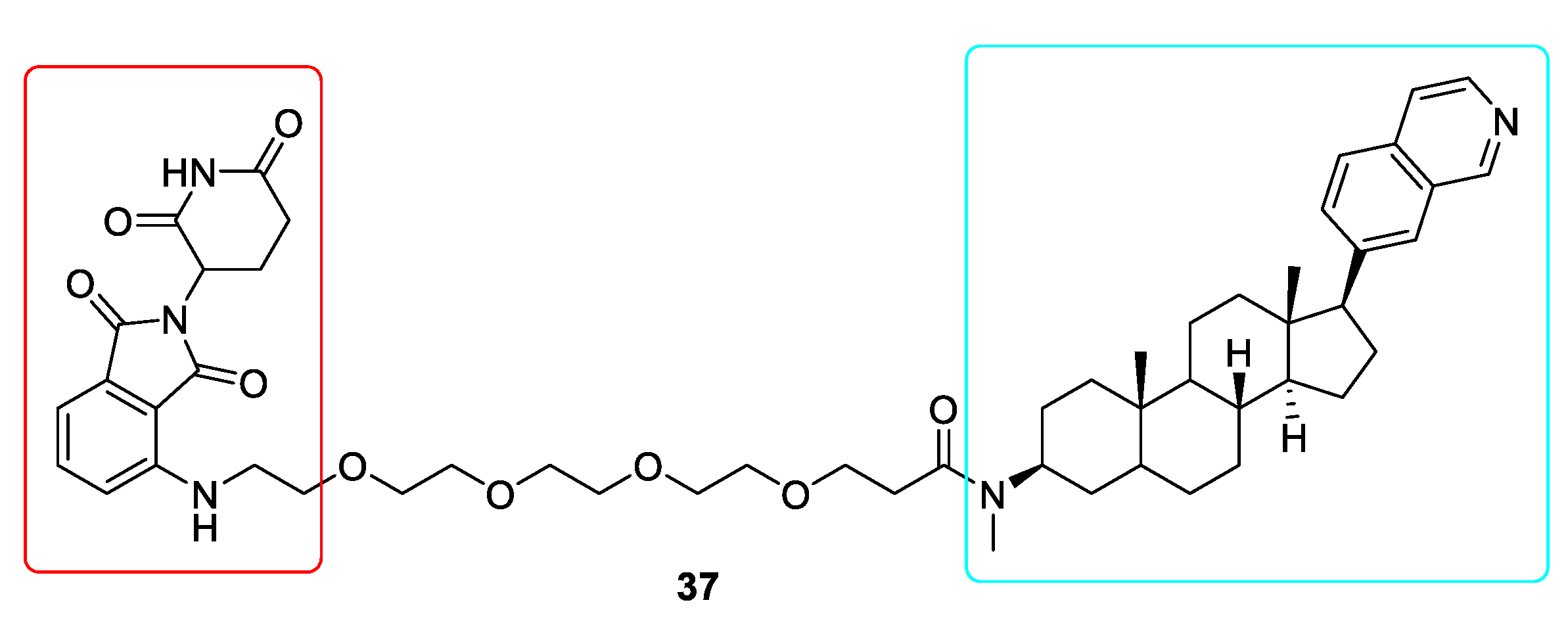
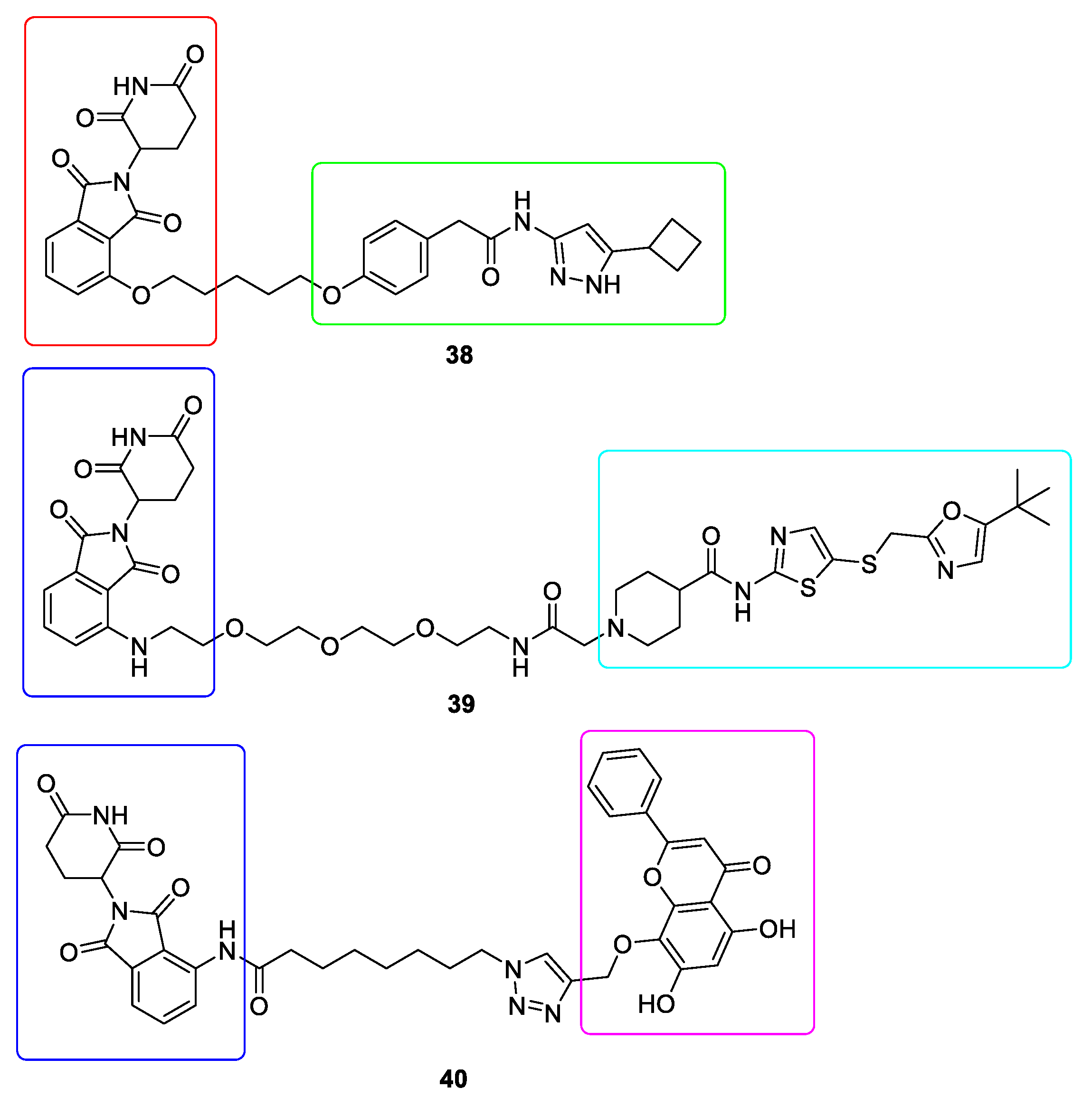
10. Targeted Protein Degradation (TPD)
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethical Approval
References
- Pavletich, N.P. Mechanisms of cyclin-dependent kinase regulation: Structures of cdks, their cyclin activators, and cip and INK4 inhibitors. J. Mol. Biol. 1999, 287, 821–828. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Marra, A.; Curigliano, G. Are all cyclin-dependent kinases 4/6 inhibitors created equal? NPJ Breast Cancer 2019, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Clausen, M.H.; Nielsen, T.E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. The Cell Cycle: Principles of Control, 1st ed.; New Science Press: London, UK, 2007. [Google Scholar]
- Patrick, G.N.; Zukerberg, L.R.; Nikolic, M.; De La Monte, S.; Dikkes, P.; Tsai, L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nat. Cell Biol. 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Osuga, H.; Osuga, S.; Wang, F.; Fetni, R.; Hogan, M.J.; Slack, R.S.; Hakim, A.M.; Ikeda, J.-E.; Park, D.S. Cyclin-dependent kinases as a therapeutic target for stroke. Proc. Natl. Acad. Sci. USA 2000, 97, 10254–10259. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, H.; Wang, L.; Liu, Y.; Knapp, S.; Liu, Q.; Gray, N.S. Exploration of Type II Binding Mode: A Privileged Approach for Kinase Inhibitor Focused Drug Discovery? ACS Chem. Biol. 2014, 9, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Rabiller, M.; Getlik, M.; Klüter, S.; Richters, A.; Tückmantel, S.; Simard, J.R.; Rauh, D. Proteus in the World of Proteins: Conformational Changes in Protein Kinases. Arch. Pharm. 2010, 343, 193–206. [Google Scholar] [CrossRef]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.É.; Knapp, S.; Johnson, L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Chao, S.-H.; Fujinaga, K.; Marion, J.E.; Taube, R.; Sausville, E.A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol Inhibits P-TEFb and Blocks HIV-1 Replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar] [CrossRef] [PubMed]
- A Carlson, B.; Dubay, M.M.; A Sausville, E.; Brizuela, L.; Worland, P.J. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996, 56, 2973–2978. [Google Scholar] [PubMed]
- De Azevedo, W.F.; Mueller-Dieckmann, H.J.; Schulzegahmen, U.; Worland, P.J.; A Sausville, E.; Kim, S.H. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 2735–2740. [Google Scholar] [CrossRef]
- E Kahn, M.; Senderowicz, A.; A Sausville, E.; E Barrett, K. Possible mechanisms of diarrheal side effects associated with the use of a novel chemotherapeutic agent, flavopiridol. Clin. Cancer Res. 2001, 7, 343. [Google Scholar]
- Stadler, W.M.; Vogelzang, N.J.; Amato, R.; Sosman, J.; Taber, D.; Liebowitz, D.; Vokes, E.E. Flavopiridol, A Novel Cyclin-Dependent Kinase Inhibitor, in Metastatic Renal Cancer: A University of Chicago Phase II Consortium Study. J. Clin. Oncol. 2000, 18, 371. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R. Chronic Lymphocytic Leukemia: A Niche for Flavopiridol? Clin. Cancer Res. 2005, 11, 3971–3973. [Google Scholar] [CrossRef]
- De Azevedo, W.F., Jr.; Gaspar, R.T.; Canduri, F.; Camera, J.C., Jr.; da Silveira, N.J.F.J.B. Molecular model of cyclin-dependent kinase 5 complexed with roscovitine. Biochem. Biophy. Res. Commun. 2002, 297, 1154–1158. [Google Scholar] [CrossRef]
- Blachly, J.S.; Byrd, J.C. Emerging drug profile: Cyclin-dependent kinase inhibitors. Leuk. Lymphoma 2013, 54, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Pippin, J.W.; Qu, Q.; Meijer, L.; Shankland, S.J. Direct in vivo inhibition of the nuclear cell cycle cascade in experimental mesangial proliferative glomerulonephritis with Roscovitine, a novel cyclin-dependent kinase antagonist. J. Clin. Investig. 1997, 100, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, A.J.; Roelofs, J.J.T.H.; Duitman, J.; Van Lieshout, M.H.P.; Blok, D.C.; Van Der Poll, T.; Wieland, C.W. R-roscovitine Reduces Lung Inflammation Induced by Lipoteichoic Acid and Streptococcus pneumoniae. Mol. Med. 2012, 18, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Schang, L.M.; Bantly, A.; Knockaert, M.; Shaheen, F.; Meijer, L.; Malim, M.H.; Gray, N.S.; Schaffer, P.A. Pharmacological Cyclin-Dependent Kinase Inhibitors Inhibit Replication of Wild-Type and Drug-Resistant Strains of Herpes Simplex Virus and Human Immunodeficiency Virus Type 1 by Targeting Cellular, Not Viral, Proteins. J. Virol. 2002, 76, 7874–7882. [Google Scholar] [CrossRef] [PubMed]
- Patrick, C.; Crews, L.; Desplats, P.; Dumaop, W.; Rockenstein, E.; Achim, C.L.; Everall, I.P.; Masliah, E. Increased CDK5 Expression in HIV Encephalitis Contributes to Neurodegeneration via Tau Phosphorylation and Is Reversed with Roscovitine. Am. J. Pathol. 2011, 178, 1646–1661. [Google Scholar] [CrossRef]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.; Jones, J.; Johnson, A.J.; Andritsos, L.; Maddocks, K.; Jaglowski, S.; Hessler, J.; Grever, M.R.; Im, E.; Zhou, H.; et al. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 2015, 29, 1524–1529. [Google Scholar] [CrossRef]
- Yakisich, J.S.; Vita, M.F.; Siden, Å.; Tasat, D.R.; Cruz, M. Strong inhibition of replicative DNA synthesis in the developing rat cerebral cortex and glioma cells by roscovitine. Investig. New Drugs 2009, 28, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250. [Google Scholar] [CrossRef]
- Brasca, M.G.; Amboldi, N.; Ballinari, D.; Cameron, A.; Casale, E.; Cervi, G.; Colombo, M.; Colotta, F.; Croci, V.; D’Alessio, R.; et al. Identification of N,1,4,4-Tetramethyl-8-{[4-(4-methylpiperazin-1-yl)phenyl]amino}-4,5-dihydro-1H-pyrazolo [4,3-h]quinazoline-3-carboxamide (PHA-848125), a Potent, Orally Available Cyclin Dependent Kinase Inhibitor. J. Med. Chem. 2009, 52, 5152–5163. [Google Scholar] [CrossRef] [PubMed]
- Milciclib. Available online: http://www.tizianalifesciences.com/drug-pipeline/milciclib/ (accessed on 20 January 2021).
- Rocca, A.; Farolfi, A.; Bravaccini, S.; Schirone, A.; Amadori, D. Palbociclib (PD 0332991): Targeting the cell cycle machinery in breast cancer. Expert Opin. Pharmacother. 2014, 15, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Samson, K. LEE011 CDK Inhibitor Showing Early Promise in Drug-Resistant Cancers. Oncol. Times 2014, 36, 39–40. [Google Scholar] [CrossRef]
- Diéras, V.; Harbeck, N.; Joy, A.A.; Gelmon, K.; Ettl, J.; Verma, S.; Lu, D.R.; Gauthier, E.; Schnell, P.; Mori, A.; et al. Palbociclib with Letrozole in Postmenopausal Women with ER+/HER2− Advanced Breast Cancer: Hematologic Safety Analysis of the Randomized PALOMA-2 Trial. Oncologist 2019, 24, 1514. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kittaneh, M.; Lolkema, M.P.J.K.; Postow, M.A.; Schwartz, G.; Franklin, C.; Matano, A.; Bhansali, S.; Parasuraman, S.; Kim, K. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: Early encouraging clinical activity. J. Clin. Oncol. 2014, 32, 9009. [Google Scholar] [CrossRef]
- Corbel, C.; Zhang, B.; Le Parc, A.; Baratte, B.; Colas, P.; Couturier, C.; Kosik, K.S.; Landrieu, I.; Le Tilly, V.; Bach, S. Tamoxifen Inhibits CDK5 Kinase Activity by Interacting with p35/p25 and Modulates the Pattern of Tau Phosphorylation. Chem. Biol. 2015, 22, 472–482. [Google Scholar] [CrossRef]
- Maccallum, D.E. Seliciclib (CYC202, R-Roscovitine) Induces Cell Death in Multiple Myeloma Cells by Inhibition of RNA Polymerase II-Dependent Transcription and Down-regulation of Mcl-1. Cancer Res. 2005, 65, 5399–5407. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.; Maupin, M.K. 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole inhibits a HeLa protein kinase that phosphorylates an RNA polymerase II-derived peptide. Biochem. Biophys. Res. Commun. 1989, 159, 508–515. [Google Scholar] [CrossRef]
- Zhu, Y.; Pe’Ery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef]
- Sedlacek, H. Mechanisms of action of flavopiridol. Crit. Rev. Oncol. 2001, 38, 139–170. [Google Scholar] [CrossRef]
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008, 4, 357–365. [Google Scholar] [CrossRef]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine Targets, Protein Kinases and Pyridoxal Kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Mita, M.; Shapiro, G.I.; Poon, J.; Small, K.; Tzontcheva, A.; Kantesaria, B.; Zhu, Y.; Bannerji, R.; Statkevich, P. Effect of aprepitant on the pharmacokinetics of the cyclin-dependent kinase inhibitor dinaciclib in patients with advanced malignancies. Cancer Chemother. Pharmacol. 2012, 70, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer 2004, 3, 1427. [Google Scholar]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef]
- Ali, S.; Heathcote, D.A.; Kroll, S.H.B.; Jogalekar, A.S.; Scheiper, B.; Patel, H.; Brackow, J.; Siwicka, A.; Fuchter, M.J.; Periyasamy, M.; et al. The Development of a Selective Cyclin-Dependent Kinase Inhibitor That Shows Antitumor Activity. Cancer Res. 2009, 69, 6208–6215. [Google Scholar] [CrossRef]
- Wang, S.; Fischer, P. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef]
- Echalier, A.; Bettayeb, K.; Ferandin, Y.; Lozach, O.; Clément, M.; Valette, A.; Liger, F.; Marquet, B.; Morris, J.C.; Endicott, J.A.; et al. Meriolins (3-(Pyrimidin-4-yl)-7-azaindoles): Synthesis, Kinase Inhibitory Activity, Cellular Effects, and Structure of a CDK2/Cyclin A/Meriolin Complex†. J. Med. Chem. 2008, 51, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Tirado, O.M.; Marionneau-Lambot, S.; Ferandin, Y.; Lozach, O.; Morris, J.C.; Mateo-Lozano, S.; Drueckes, P.; Schächtele, C.; Kubbutat, M.H.; et al. Meriolins, a New Class of Cell Death–Inducing Kinase Inhibitors with Enhanced Selectivity for Cyclin-Dependent Kinases. Cancer Res. 2007, 67, 8325–8334. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, Y.A.; Taylor, M.A.; Napoleon, J.V.; Rana, S.; Contreras, J.I.; Natarajan, A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J. Med. Chem. 2016, 59, 8667–8684. [Google Scholar] [CrossRef]
- Parrino, B.; Attanzio, A.; Spanò, V.; Cascioferro, S.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Diana, P.; Cirrincione, G.; Carbone, A. Synthesis, antitumor activity and CDK1 inhibiton of new thiazole nortopsentin analogues. Eur. J. Med. Chem. 2017, 138, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Byth, K.F.; Thomas, A.; Hughes, G.; Forder, C.; McGregor, A.; Geh, C.; Oakes, S.; Green, C.; Walker, M.; Newcombe, N.; et al. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol. Cancer Ther. 2009, 8, 1856–1866. [Google Scholar] [CrossRef]
- Siemeister, G.; Lücking, U.; Wengner, A.M.; Lienau, P.; Steinke, W.; Schatz, C.; Mumberg, D.; Ziegelbauer, K. BAY 1000394, a Novel Cyclin-Dependent Kinase Inhibitor, with Potent Antitumor Activity in Mono- and in Combination Treatment upon Oral Application. Mol. Cancer Ther. 2012, 11, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wierda, W.G.; Chubb, S.; Hawtin, R.E.; Fox, J.A.; Keating, M.J.; Gandhi, V.; Plunkett, W. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 2009, 113, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Mediouni, S.; Valente, S.T. Targeting HIV transcription: The quest for a functional cure. Curr. Top. Microbiol. Immunol. 2015, 389, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Carter, E.J.; Huff, B.C.; Morris, J.C. Variolins and Related Alkaloids. Chem. Rev. 2009, 109, 3080–3098. [Google Scholar] [CrossRef]
- Gompel, M.; Leost, M.; Joffe, E.B.D.K.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the Ascidian Aplidium meridianum. Bioorganic Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef]
- Carbone, A.; Pennati, M.; Parrino, B.; Lopergolo, A.; Barraja, P.; Montalbano, A.; Spanò, V.; Sbarra, S.; Doldi, V.; De Cesare, M.; et al. Novel 1H-Pyrrolo[2,3-b]pyridine Derivative Nortopsentin Analogues: Synthesis and Antitumor Activity in Peritoneal Mesothelioma Experimental Models. J. Med. Chem. 2013, 56, 7060–7072. [Google Scholar] [CrossRef]
- Perry, N.B.; Ettouati, L.; Litaudon, M.; Blunt, J.W.; Munro, M.H.; Parkin, S.; Hope, H. Alkaloids from the antarctic sponge Kirkpatrickia varialosa. Tetrahedron 1994, 50, 3987–3992. [Google Scholar] [CrossRef]
- Trimurtulu, G.; Faulkner, D.; Perry, N.B.; Ettouati, L.; Litaudon, M.; Blunt, J.W.; Munro, M.H.; Jameson, G.B. Alkaloids from the antarctic sponge Kirkpatrickia varialosa. Part 2: Variolin A and N(3′)-methyl tetrahydrovariolin B. Tetrahedron 1994, 50, 3993–4000. [Google Scholar] [CrossRef]
- Simone, M.; Erba, E.; Damia, G.; Vikhanskaya, F.; Di Francesco, A.M.; Riccardi, R.; Bailly, C.; Cuevas, C.; Sousa-Faro, J.M.F.; D’Incalci, M. Variolin B and its derivate deoxy-variolin B: New marine natural compounds with cyclin-dependent kinase inhibitor activity. Eur. J. Cancer 2005, 41, 2366–2377. [Google Scholar] [CrossRef]
- Bharate, S.B.; Yadav, R.R.; Battula, S.; Vishwakarma, R.A. Meridianins: Marine-derived potent kinase inhibitors. Mini-Reviews Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef]
- Li, B.; Chonghaile, T.N.; Fan, Y.; Madden, S.F.; Klinger, R.; O’Connor, A.E.; Walsh, L.; O’Hurley, G.; Udupi, G.M.; Joseph, J.; et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3834–3845. [Google Scholar] [CrossRef] [PubMed]
- Walsby, E.; Lazenby, M.; Pepper, C.; Burnett, A.K. The cyclin-dependent kinase inhibitor SNS-032 has single agent activity in AML cells and is highly synergistic with cytarabine. Leukemia 2011, 25, 411–419. [Google Scholar] [CrossRef]
- Reck, M.; Horn, L.; Novello, S.; Barlesi, F.; Albert, I.; Juhász, E.; Kowalski, D.; Robinet, G.; Cadranel, J.; Bidoli, P.; et al. Phase II Study of Roniciclib in Combination with Cisplatin/Etoposide or Carboplatin/Etoposide as First-Line Therapy in Patients with Extensive-Disease Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Boss, D.; Schwartz, G.; Middleton, M.; Amakye, D.; Swaisland, H.; Midgley, R.; Ranson, M.; Danson, S.; Calvert, H.; Plummer, R.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclin-dependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann. Oncol. 2010, 21, 884–894. [Google Scholar] [CrossRef]
- Sundaram, J.R.; Poore, C.P.; Bin Sulaimee, N.H.; Pareek, T.; Asad, A.B.M.A.; Rajkumar, R.; Cheong, W.F.; Wenk, M.R.; Dawe, G.S.; Chuang, K.-H.; et al. Specific Inhibition of p25/Cdk5 Activity by the Cdk5 Inhibitory Peptide Reduces Neurodegeneration In Vivo. J. Neurosci. 2013, 33, 334–343. [Google Scholar] [CrossRef]
- Deng, Y.; Shipps, G.W.; Zhao, L.; Siddiqui, M.A.; Popovici-Muller, J.; Curran, P.J.; Duca, J.S.; Hruza, A.W.; Fischmann, T.O.; Madison, V.S.; et al. Modulating the interaction between CDK2 and cyclin A with a quinoline-based inhibitor. Bioorganic Med. Chem. Lett. 2014, 24, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; McNae, I.; Kontopidis, G.; McClue, S.J.; McInnes, C.; Stewart, K.J.; Wang, S.; Zheleva, D.I.; Marriage, H.; Lane, D.P.; et al. Discovery of a Novel Family of CDK Inhibitors with the Program LIDAEUS: Structural Basis for Ligand-Induced Disordering of the Activation Loop. Structure 2003, 11, 399–410. [Google Scholar] [CrossRef]
- Martin, M.P.; Endicott, J.A.; Noble, M.E. Structure-based discovery of cyclin-dependent protein kinase inhibitors. Essays Biochem. 2017, 61, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Hari, S.B.; Merritt, E.A.; Maly, D.J. Sequence Determinants of a Specific Inactive Protein Kinase Conformation. Chem. Biol. 2013, 20, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic Control in the EGF Receptor and Its Connection to General Kinase Regulatory Mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, L.; Robert, L.J.-L.J.A.A.C. Targeting Protein Multiple Conformations: A Structure-Based Strategy for Kinase Drug Design. Curr. Top. Med. Chem. 2007, 7, 1394–1407. [Google Scholar] [CrossRef]
- Wohlbold, L.; Merrick, K.A.; De, S.; Amat, R.; Kim, J.H.; LaRochelle, S.; Allen, J.J.; Zhang, C.; Shokat, K.M.; Petrini, J.H.J.; et al. Chemical Genetics Reveals a Specific Requirement for Cdk2 Activity in the DNA Damage Response and Identifies Nbs1 as a Cdk2 Substrate in Human Cells. PLoS Genet. 2012, 8, e1002935. [Google Scholar] [CrossRef]
- Hasegawa, M.; Nishigaki, N.; Washio, Y.; Kano, K.; Harris, P.A.; Sato, H.; Mori, I.; West, R.I.; Shibahara, M.; Toyoda, H.; et al. Discovery of Novel Benzimidazoles as Potent Inhibitors of TIE-2 and VEGFR-2 Tyrosine Kinase Receptors. J. Med. Chem. 2007, 50, 4453–4470. [Google Scholar] [CrossRef]
- Emrick, M.A.; Lee, T.; Starkey, P.J.; Mumby, M.C.; Resing, K.A.; Ahn, N.G. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc. Natl. Acad. Sci. USA 2006, 103, 18101–18106. [Google Scholar] [CrossRef]
- Azam, M.; A Seeliger, M.; Gray, N.S.; Kuriyan, J.; Daley, G.Q. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 2008, 15, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Tong, W.; Manshouri, T.; Vega, F.; A Lennon, P.; Cools, J.; Gilliland, D.G.; Lee, F.; Cortés, J.; Kantarjian, H.; et al. Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 2008, 22, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L.; Levy, R.M. Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The Structural Basis for Autoinhibition of FLT3 by the Juxtamembrane Domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Wang, Z.; Harkins, P.C.; Ulevitch, R.J.; Han, J.; Cobb, M.H.; Goldsmith, E.J. The structure of mitogen-activated protein kinase p38 at 2.1-Å resolution. Proc. Natl. Acad. Sci. USA 1997, 94, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Seeliger, M.A.; Eastwood, M.P.; Frank, F.; Xu, H.; Jensen, M.Ø.; Dror, R.O.; Kuriyan, J.; Shaw, D.E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Adams, J.M.; Badger, J.; Buchanan, M.D.; Feil, I.K.; Froning, K.J.; Gao, X.; Hendle, J.; Keegan, K.; Leon, B.C.; et al. A Novel Mode of Gleevec Binding Is Revealed by the Structure of Spleen Tyrosine Kinase. J. Biol. Chem. 2004, 279, 55827–55832. [Google Scholar] [CrossRef]
- Liao, J.J.-L. Molecular Recognition of Protein Kinase Binding Pockets for Design of Potent and Selective Kinase Inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Alexander, L.T.; Möbitz, H.; Drueckes, P.; Savitsky, P.; Fedorov, O.; Elkins, J.M.; Deane, C.M.; Cowan-Jacob, S.W.; Knapp, S. Type II Inhibitors Targeting CDK2. ACS Chem. Biol. 2015, 10, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Gurbani, D.; Weisberg, E.L.; Hunter, J.C.; Li, L.; Jones, D.S.; Ficarro, S.B.; Mowafy, S.; Tam, C.-P.; Rao, S.; et al. Structure-guided development of covalent TAK1 inhibitors. Bioorganic Med. Chem. 2017, 25, 838–846. [Google Scholar] [CrossRef]
- Russo, A.A.; Jeffrey, P.D.; Patten, A.K.; Massagué, J.; Pavletich, N.P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inibitor bound to the cyclin A–Cdk2 complex. Nature 1996, 382, 325–331. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Tong, L.; Pavletich, N.P. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 2000, 14, 3115–3125. [Google Scholar] [CrossRef] [PubMed]
- McInnes, C.; Andrews, M.J.I.Z.; Daniella, I.; Lane, D.P.; Fischer, P.M. Peptidomimetic Design of CDK Inhibitors Targeting theRecruitment Site of the Cyclin Subunit. Curr. Med. Chem. Anti-Cancer Agents 2003, 3, 57–69. [Google Scholar]
- Kontopidis, G.; Andrews, M.J.; McInnes, C.; Plater, A.; Innes, L.; Renachowski, S.; Cowan, A.; Fischer, P.M. Truncation and Optimisation of Peptide Inhibitors of Cyclin-Dependent Kinase 2-Cyclin A Through Structure-Guided Design. ChemMedChem 2009, 4, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorganic Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- MM D37K. Available online: https://adisinsight.springer.com/drugs/800040966 (accessed on 20 January 2021).
- Schneider, E.V.; Böttcher, J.; Blaesse, M.; Neumann, L.; Huber, R.; Maskos, K. The Structure of CDK8/CycC Implicates Specificity in the CDK/Cyclin Family and Reveals Interaction with a Deep Pocket Binder. J. Mol. Biol. 2011, 412, 251–266. [Google Scholar] [CrossRef]
- Bergeron, P.; Koehler, M.F.T.; Blackwood, E.M.; Bowman, K.; Clark, K.; Firestein, R.; Kiefer, J.R.; Maskos, K.; McCleland, M.L.; Orren, L.; et al. Design and Development of a Series of Potent and Selective Type II Inhibitors of CDK8. ACS Med. Chem. Lett. 2016, 7, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK Pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef]
- Dale, T.C.; Clarke, P.A.; Esdar, C.; Waalboer, D.; Adeniji-Popoola, O.; Ortiz-Ruiz, M.-J.; Mallinger, A.; Samant, R.S.; Czodrowski, P.; Musil, D.; et al. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat. Chem. Biol. 2015, 11, 973–980. [Google Scholar] [CrossRef]
- Nagaria, T.S.; Williams, J.L.; LeDuc, C.; A Squire, J.; A Greer, P.; Sangrar, W. Flavopiridol Synergizes with Sorafenib to Induce Cytotoxicity and Potentiate Antitumorigenic Activity in EGFR/HER-2 and Mutant RAS/RAF Breast Cancer Model Systems. Neoplasia 2013, 15, 939-IN27. [Google Scholar] [CrossRef]
- Mancini, M.; Yarden, Y. Mutational and network level mechanisms underlying resistance to anti-cancer kinase inhibitors. Semin. Cell Dev. Biol. 2016, 50, 164–176. [Google Scholar] [CrossRef]
- Adrián, F.J.; Ding, Q.; Sim, T.; Velentza, A.V.; Sloan, C.; Liu, Y.; Zhang, G.; Hur, W.; Ding, S.; Manley, P.W.; et al. Allosteric inhibitors of Bcr-abl–dependent cell proliferation. Nat. Chem. Biol. 2006, 2, 95–102. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S.; Dudley, D.T.; Herrera, R.; Van Becelaere, K.; Wiland, A.; Gowan, R.C.; Tecle, H.; Barrett, S.D.; Bridges, A.; Przybranowski, S.; et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999, 5, 810–816. [Google Scholar] [CrossRef]
- Gavrin, L.K.; Saiah, E. Approaches to discover non-ATP site kinase inhibitors. MedChemComm 2013, 4, 41–51. [Google Scholar] [CrossRef]
- Yueh, C.; Rettenmaier, J.; Xia, B.; Hall, D.R.; Alekseenko, A.; Porter, K.A.; Barkovich, K.; Keseru, G.M.; Whitty, A.; Wells, J.A.; et al. Kinase Atlas: Druggability Analysis of Potential Allosteric Sites in Kinases. J. Med. Chem. 2019, 62, 6512–6524. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; E Mottarella, S.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- Betzi, S.; Alam, R.; Martin, M.; Lubbers, D.J.; Han, H.; Jakkaraj, S.R.; Georg, G.I.; Schönbrunn, E. Discovery of a Potential Allosteric Ligand Binding Site in CDK2. ACS Chem. Biol. 2011, 6, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Parang, K.; Till, J.H.; Ablooglu, A.J.; A Kohanski, R.; Hubbard, S.; A Cole, P. Mechanism-based design of a protein kinase inhibitor. Nat. Genet. 2001, 8, 37–41. [Google Scholar] [CrossRef]
- Poot, A.J.; Van Ameijde, J.; Slijper, M.; Berg, A.V.D.; Hilhorst, R.; Ruijtenbeek, R.; Rijkers, D.T.S.; Liskamp, R.M.J. Development of Selective Bisubstrate-Based Inhibitors Against Protein Kinase C (PKC) Isozymes By Using Dynamic Peptide Microarrays. ChemBioChem 2009, 10, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Gower, C.M.; Chang, M.E.K.; Maly, D.J. Bivalent inhibitors of protein kinases. Crit. Rev. Biochem. Mol. Biol. 2013, 49, 102–115. [Google Scholar] [CrossRef]
- Hill, Z.B.; Perera, B.G.K.; Maly, D.J. A Chemical Genetic Method for Generating Bivalent Inhibitors of Protein Kinases. J. Am. Chem. Soc. 2009, 131, 6686–6688. [Google Scholar] [CrossRef] [PubMed]
- Kedika, S.R.; Udugamasooriya, D.G. Converting a weaker ATP-binding site inhibitor into a potent hetero-bivalent ligand by tethering to a unique peptide sequence derived from the same kinase. Org. Biomol. Chem. 2018, 16, 6443–6449. [Google Scholar] [CrossRef]
- Wong, M.L.; Murphy, J.; Harrington, E.; Gower, C.M.; Jain, R.K.; Schirle, M.; Thomas, J.R. Examining the influence of specificity ligands and ATP-competitive ligands on the overall effectiveness of bivalent kinase inhibitors. Proteome Sci. 2016, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.; Botting, R. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- McCrae, J.C.; Morrison, E.E.; MacIntyre, I.M.; Dear, J.W.; Webb, D.J. Long-term adverse effects of paracetamol—A review. Br. J. Clin. Pharmacol. 2018, 84, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.P.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nat. Cell Biol. 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Marineau, J.J.; Rajagopal, N.; Hamman, K.B.; Choi, Y.J.; Schmidt, D.R.; Ke, N.; Johannessen, L.; Bradley, M.J.; Orlando, D.A.; et al. Discovery and Characterization of SY-1365, a Selective, Covalent Inhibitor of CDK7. Cancer Res. 2019, 79, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; E Dixon-Clarke, S.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, T.; Terai, H.; Ficarro, S.B.; Kwiatkowski, N.; Hao, M.-F.; Sharma, B.; Christensen, C.L.; Chipumuro, E.; Wong, K.-K.; et al. Overcoming Resistance to the THZ Series of Covalent Transcriptional CDK Inhibitors. Cell Chem. Biol. 2018, 25, 135–142.e5. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.-H.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Liu, R.; Yue, Z.; Tsai, C.-C.; Shen, J. Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2019, 141, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Coll-Martínez, B.; Delgado, A.; Crosas, B. The Potential of Proteolytic Chimeras as Pharmacological Tools and Therapeutic Agents. Molecules 2020, 25, 5956. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Wakeling, A.; I Nicholson, R. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Chopra, R.; Sadok, A.; Collins, I. A critical evaluation of the approaches to targeted protein degradation for drug discovery. Drug Discov. Today Technol. 2019, 31, 5–13. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Fouad, S.; Wells, O.S.; Hill, M.A.; D’Angiolella, V. Cullin Ring Ubiquitin Ligases (CRLs) in Cancer: Responses to Ionizing Radiation (IR) Treatment. Front. Physiol. 2019, 10, 1144. [Google Scholar] [CrossRef]
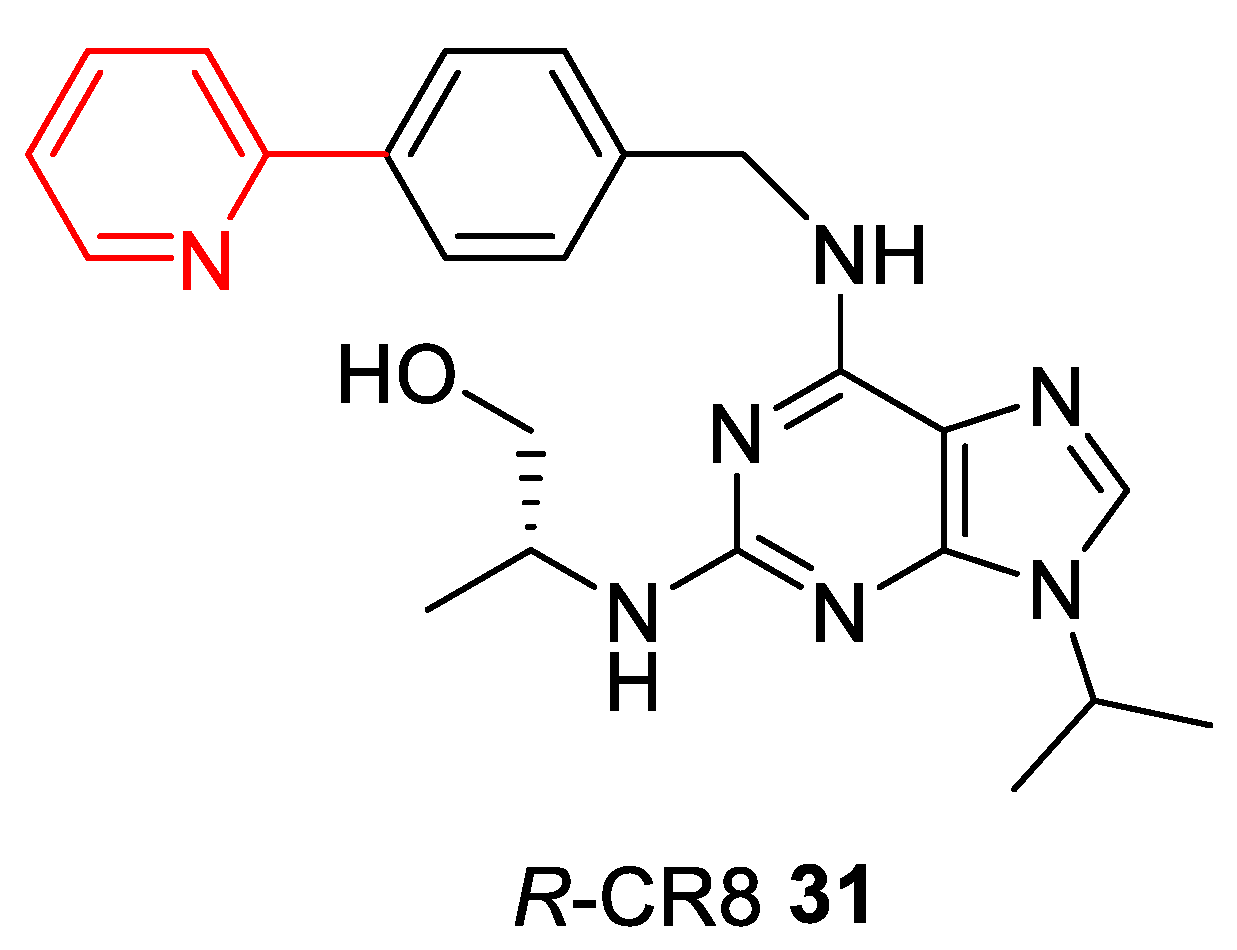
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; A Endicott, J.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef]
- Słabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.-D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Ohoka, N.; Shibata, N.; Tsukumo, Y. Targeted Protein Degradation by Chimeric Small Molecules, PROTACs and SNIPERs. Front. Chem. 2019, 7, 849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Burgess, K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem. Commun. 2019, 55, 2704–2707. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, E.S.; Donovan, K.A.; Liang, Y.; Fischer, E.S.; Zhang, T.; Gray, N.S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. 2019, 58, 6321–6326. [Google Scholar] [CrossRef]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e9. [Google Scholar] [CrossRef]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Rzymski, T.; Mikula, M.; Wiklik, K.; Brzózka, K. CDK8 kinase—An emerging target in targeted cancer therapy. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2015, 1854, 1617–1629. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Wang, E.S.; Johannessen, L.; Kwiatkowski, N.; Sim, T.; Gray, N.S. Development of Highly Potent and Selective Steroidal Inhibitors and Degraders of CDK8. ACS Med. Chem. Lett. 2018, 9, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Bagella, L.; MacLachlan, T.K.; Buono, R.J.; Pisano, M.M.; Giordano, A.; De Luca, A. Cloning of murine CDK9/PITALRE and its tissue-specific expression in development. J. Cell. Physiol. 1998, 177, 206–213. [Google Scholar] [CrossRef]
- Vladimir, K.; Stjepan, U. Cyclin-Dependent Kinase Inhibitors as Anticancer Drugs. Curr. Drug Targets 2010, 11, 291–302. [Google Scholar] [CrossRef]
- Petzold, G.; Fischer, E.S.; Thomä, G.P.E.S.F.N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nat. Cell Biol. 2016, 532, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Robb, C.M.; Contreras, J.I.; Kour, S.; Taylor, M.A.; Abid, M.; Sonawane, Y.A.; Zahid, M.; Murry, D.J.; Natarajan, A.; Rana, S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Ren, J.; Li, Y.; Wang, J.; Xu, X.; Feng, Y.; Tang, H.; Wang, Y.; Li, Z. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorganic Chem. 2018, 81, 373–381. [Google Scholar] [CrossRef]











| Compound 2 20 [nM] [68] | |||
|---|---|---|---|
| CDK2/A IC50 | FP Ki | TdCD Kd | Clinical phase |
| >10,000 | 140 | 300 | pre-clinical |
| Kinase IC50 [nM] | Clinical Phase | |||||
|---|---|---|---|---|---|---|
| CDK2/A | Cyclin-Free CDK2 | CDK4/D | CDK6/D | CDK8/C | ||
| H-His-Ala-Lys-Arg-Arg-Leu-Ile-Phe-NH2 21 [90] | 140 | - | - | - | - | pre-clinical |
| H-Ala-Ala-Abu-Arg-Arg-Leu-Ile-pFPhe-NH2 22 [90] | 80 | - | - | - | - | pre-clinical |
| MM-D37K 23 [91,92] | - | - | active | active | - | phase I/II for Bladder, Gastrointestinal, Glioblastoma, and Malignant Melanoma |
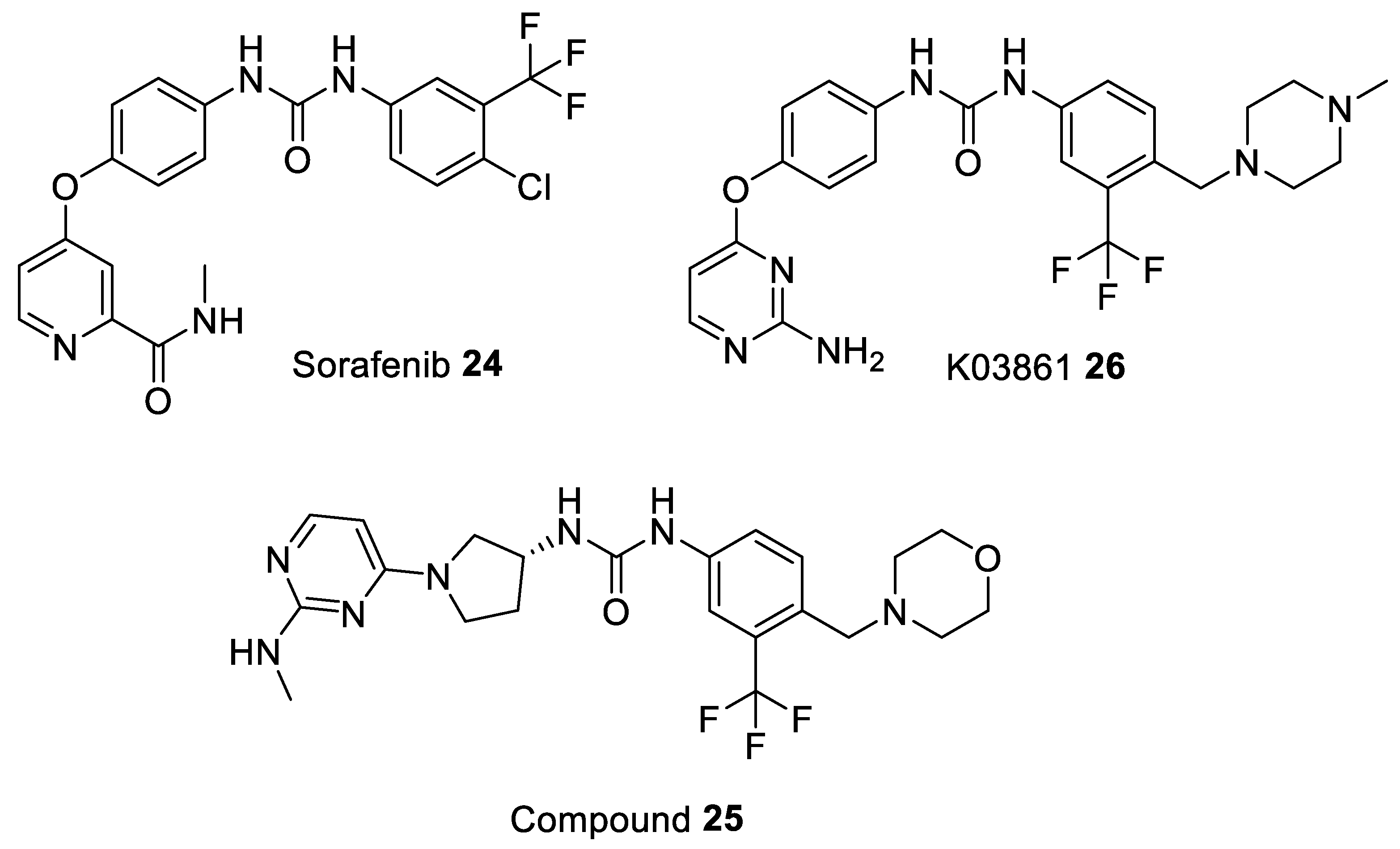
| Sorafenib 24 [93] | - | - | - | - | 74 | drug approved for Renal Cell Carcinoma, Hepatocellular Carcinoma, AML, and Advanced Thyroid Carcinoma |
| Compound 25 [94] | - | - | - | - | 17.4 | pre-clinical |
| K03861 26 [85] | 10,000 | 9.7–50 | - | - | - | pre-clinical |
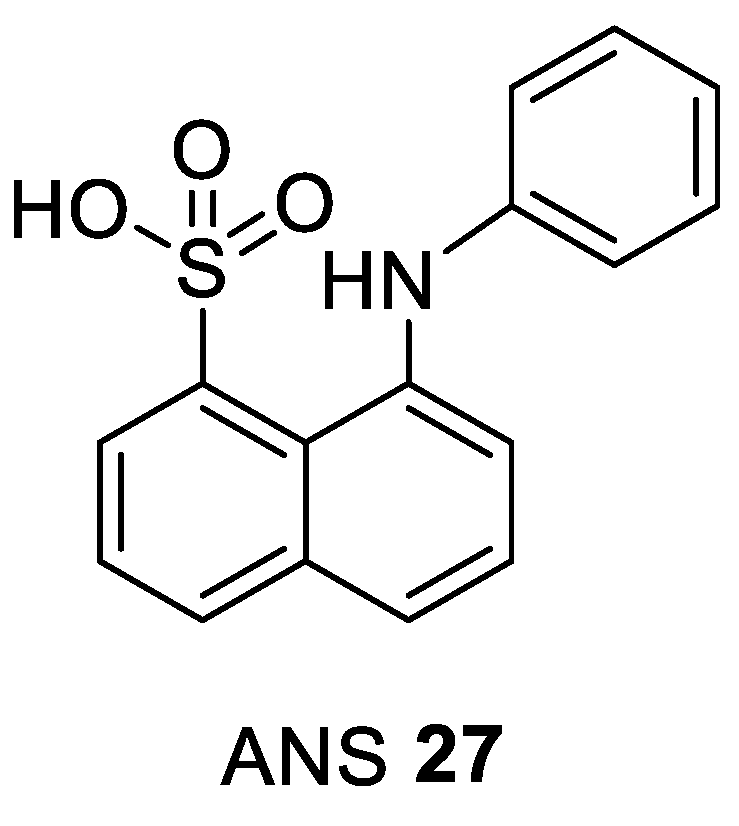
| ANS 27 [nM] [106] | Clinical Phase | ||
|---|---|---|---|
| CDK2/A IC50 | Cyclin-Free CDK2 Kd | ANS Displacement EC50 | |
| 91,000 | 37,000 | 600 | pre-clinical |
| Kinase IC50 [nM] | Clinical Phase | |||||
|---|---|---|---|---|---|---|
| CDK2/A | CDK7/H/MAT1 | CDK9/T1 | CDK12/K | CDK13/K | ||
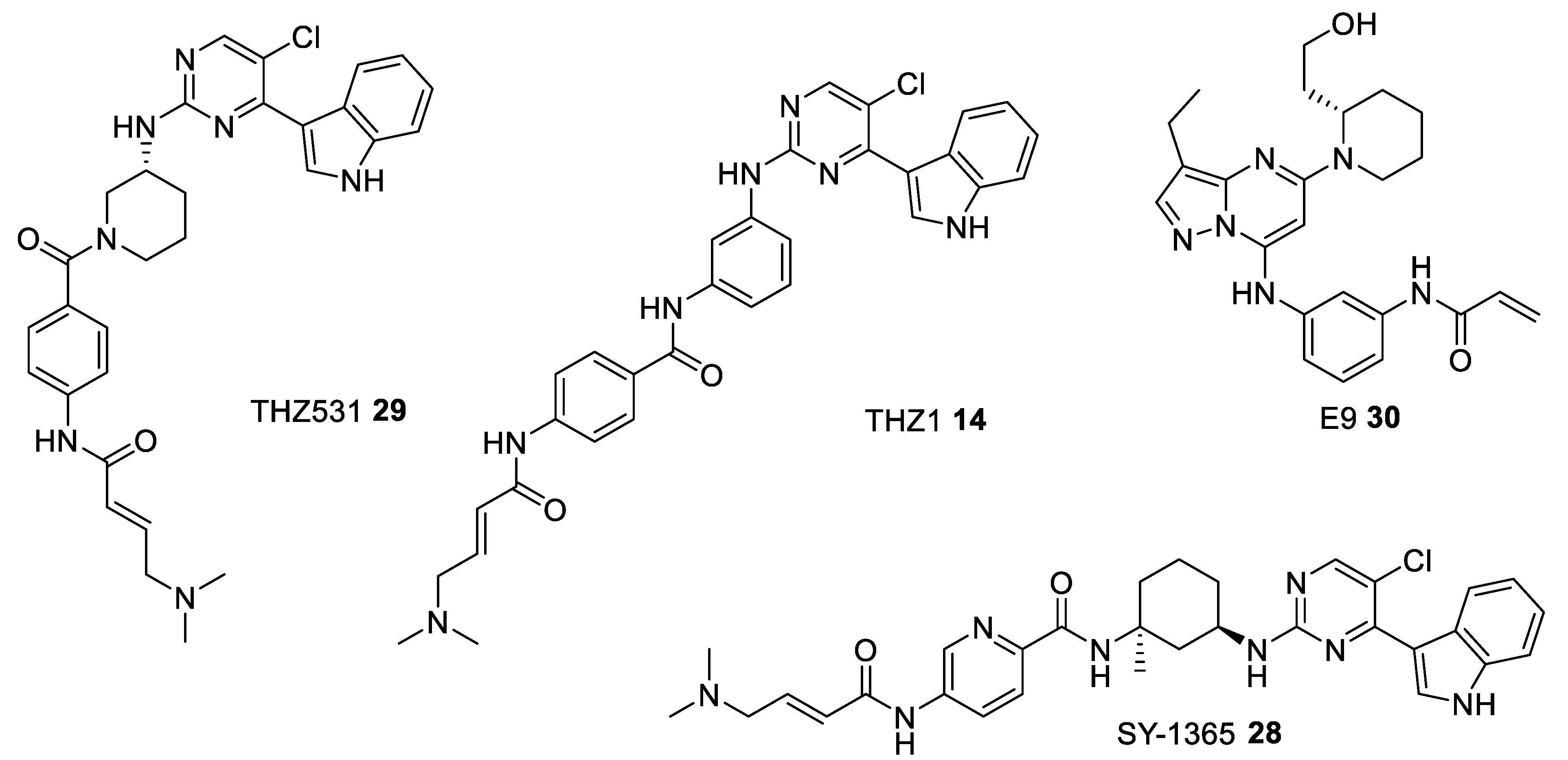
| THZ531 29 [118] | - | 8500 | 10500 | 158 | 69 | phase II—observational study for the patients-derived High Grade Serous Ovarian Cancer (HGSOC) organoids NCT04555473 |
| THZ1 14 [116] | - | 3.2 | - | >1000 | >1000 | pre-clinical |
| SY-1365 28 [117] | - | 20 | - | - | - | phase I for Advanced Solid Tumors, Ovarian, and Breast Cancer NCT03134638 |
| E9 30 [119] | 932 | 1210 | 23.9 | - | - | pre-clinical |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łukasik, P.; Baranowska-Bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 2806. https://doi.org/10.3390/ijms22062806
Łukasik P, Baranowska-Bosiacka I, Kulczycka K, Gutowska I. Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. International Journal of Molecular Sciences. 2021; 22(6):2806. https://doi.org/10.3390/ijms22062806
Chicago/Turabian StyleŁukasik, Paweł, Irena Baranowska-Bosiacka, Katarzyna Kulczycka, and Izabela Gutowska. 2021. "Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action" International Journal of Molecular Sciences 22, no. 6: 2806. https://doi.org/10.3390/ijms22062806
APA StyleŁukasik, P., Baranowska-Bosiacka, I., Kulczycka, K., & Gutowska, I. (2021). Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. International Journal of Molecular Sciences, 22(6), 2806. https://doi.org/10.3390/ijms22062806